

Abstract
We have demonstrated previously that spores of the non-pathogenic Clostridial strain C. sporogenes genetically engineered to express the E. coli-derived cytosine deaminase (CD) gene are effective in converting systemically injected nontoxic 5-fluorocytosine (5-FC) into the toxic anticancer drug 5-fluorouracil (5-FU), thereby producing tumor-specific antitumor activity. To improve expression of E. coli-derived genes with this system we first replaced the original fdP promoter in the vector with one of two powerful endogenous Clostridial promoters: that of the thiolase gene (thlP) and that for the Clostridial transcription factor abrB310, abrBP. These substitutions improved protein expression levels of the prodrug activating genes by 2–3 fold in comparison with fdP-driven expression. However, despite these strong promoters we found much higher expression of the nitroreductase (NTR) protein in the E. coli host compared to the Clostridial host, which we hypothesized could be the result of different codon use between the two organisms. To test this we constructed new expression vectors with an artificially synthesized NTR gene using optimized Clostridial codons (sNTR). Results from both enzymatic assays and Western blots of cell extracts from Clostridial transformants harboring plasmid constructs of thlP-sNTR and abrBP-sNTR demonstrated that the expression and activity of the NTR gene product was increased by some 20-fold compared to the original construct. In vivo studies with intravenously administrated sNTR-expressing C. sporogenes spores in SiHa tumor-bearing mice demonstrated significantly improved antitumor efficacy when combined with either 5-aziridinyl-2,4-dinitrobenzamide (CB1954) or the novel dinitrobenzamide mustard prodrug, PR-104.
Introduction
The presence of necrotic areas is a common feature of human solid tumors (1). Because necrotic regions do not usually occur in normal tissues and are generally anaerobic, we have proposed that they could be used to target anticancer drugs selectively to tumors using genetically engineered anaerobic bacteria of the genus Clostridium (2, 3). This genus comprises a large and heterogeneous group of gram-positive, spore-forming bacteria that become vegetative and grow only in the absence (or at low levels) of oxygen and can be genetically engineered to produce a prodrug activating enzyme to specifically target anticancer drugs to tumors, an approach we have termed Clostridia-Directed Enzyme Prodrug Therapy (CDEPT) (4).
Initially, efforts to use the tumor targeting properties of non pathogenic strains of Clostridia focused on the fact that following intravenous injection, spores of these strains localized and germinated in the necrotic areas of experimental tumors and produced extensive tumor lysis. (5–10). These animal experiments were followed by clinical studies with cancer patients, which demonstrated that spores of non pathogenic strains of Clostridia could be given safely, that the spores germinate in the necrotic regions of tumors, and that lysis of the tumors can occur (11–13). However, these clinical experiments did not improve patient outcome and were discontinued.
More recently there has been renewed interest in the use of Clostridia, and impressive preclinical results have been obtained with a detoxified strain of C. novyi in combination with chemotherapy (1, 14), radiation therapy (15) and immunotherapy (16). Preclinical toxicology has been performed with this strain (17) and a phase 1 trial is underway. Meanwhile we, and others, have proposed using genetically engineered Clostridia (2–4, 18, 19) or Bifidobacterium longum (20, 21). In a related approach, attenuated Salmonella genetically engineered to produce cytosine deaminase (CD), have been used both in rodents (22) and in a phase 1 clinical trials (23). However, the basis for tumor selective targeting by Salmonella is unclear, and in patients it appears that tolerable doses of the attenuated Salmonella fail to germinate in the tumors at sufficiently high density.
In previous studies we demonstrated very high tumor colonization levels with the clinically used Clostridium C. sporogenes and that we could express the E. coli enzyme cytosine deaminase in this organism to convert the non toxic prodrug 5-fluorocytosine (5-FC) to the toxic anticancer drug 5-fluorouracil (5-FU) both in vitro and in vivo and produce significant antitumor activity with 5-FC combined with the recombinant Clostridial spores (4). We also showed that intravenous injection of spores of recombinant C. sporogenes expressing the E. coli enzyme nitroreductase (NTR) into tumor bearing mice produced NTR protein solely in the tumors (3). However, we also noted that the expression level of prodrug-activating enzymes from our vector in C. sporogenes was many fold lower than that of the same vector in E. coli implying that antitumor activity from prodrug activating enzymes from C. sporogenes was considerably less than optimal.
Our goal in this study was to significantly improve the antitumor activity of CDEPT. Since the expression level of a prodrug activating enzyme is critical for the efficacy of CDEPT, our goal was to maximize the expression level of prodrug activating enzymes in C. sporogenes. We did this in two ways: i) by replacing the currently used ferredoxin promoter from C. perfringens with a stonger constitutively expressed, endogenous Clostridial promoter, and ii) by replacing the native E. coli gene coding NTR with an artificially synthesized gene using Clostridia preferred codon usage. E. coli genes usually are very poorly expressed in the Clostridial host because of the large differences in the codon usage and the lack of the corresponding tRNAs in Clostridia (24, 25). Thus, in order to overcome the codon usage bias for an E. coli gene expressed in a Clostridial host, we artificially synthesized the open-reading frame of E. coli NTR, maintaining the peptide sequence but optimizing the DNA sequence according to known Clostridial codon usage preference.
We tested the new CDEPT system by assessing its antitumor activity using two clinically used dinitrobenzamide mustard prodrugs, both substrates for E. coli NTR; 5-aziridinyl-2,4-dinitrobenzamide (CB1954), and the novel hypoxia-activated drug PR-104 [2-((2-bromoethyl)-2-{[(2-hydroxyethyl)amino]carbonyl}-4,6-dinitroanilino)ethyl methanesulfonate phosphate ester] (26). We demonstrate that the new construct is highly active in vivo.
Materials and Methods
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table 1 Supplemental data. E. coli strains were grown aerobically at 37ºC in LB broth. For recombinant strains, antibiotics were added to the medium after cooling down to below 55ºC at the following final concentrations: 100 μg/ml ampicillin, 10 μg/ml chloramphenicol and 250 μg/ml erythromycin when needed. Both wild-type and recombinant E. coli strains were maintained at −80°C in LB broth supplemented with 15% glycerol.
Wild type C. sporogenes strains were grown at 37ºC in a BacII anaerobic chamber (Shel Lab, Cornelius, OR) in TPGY media (trypticase peptone, 2%; peptone, 0.5%; glucose, 0.1%; yeast extract, 0.5%; cysteine HCl, 0.1%; agar was added at 2% when needed). Media was brought quickly into the anaerobic chamber after autoclaving (which drives off the dissolved oxygen) and was equilibrated with the chamber atmosphere for at least 24 hrs before use. For recombinant Clostridial strains, erythromycin at 10 μg/ml was added to the liquid medium after atmospheric equilibrium with anaerobic gas (90% N2, 5% H2 and 5% CO2) inside the hypoxic chamber. For maintenance of both wild-type and recombinant Clostridial strains, cells were first sporulated, as describe below, and the spores were aliquoted into phosphate buffered saline (PBS) solution and maintained at −80ºC until use.
Sporulation Procedure
Sporulation was induced as previously described (3). Briefly cultures were incubated at a suboptimal growth temperature of 30ºC for 5 to 7 days, after which maximum sporulation (usually 40–60%) was obtained. After extensive washing and purification the spores were aliquoted to cryogenic tubes and stored at −80ºC until use. Immediately before use, spores were subjected to a 72ºC 15-min heat shock, which simultaneously kills all vegetative rods and activates the spores to the vegetative form. To assess the spore concentration and plasmid retention we plated a sample of this on both selective and non-selective media.
Plasmid DNA isolation and manipulation
Plasmids isolation from recombinant E. coli strains were performed with Purelink Quick plasmid minipreps kit (Invitrogen, Carlsbad, CA) or Wizard Plus Minipreps, DNA Purification System (Promega, Madison, WI) by following the manufacturers instructions. The plasmid DNA was further purified first by using a standard phenol-chloroform extraction procedure then by differential precipitation with polyethylene Glycol (PEG 8000). All of the commercial enzymes utilized in this study (i.e., restriction endonucleases, T4 DNA ligase, Calf intestine alkaline phosphatase, T4 DNA polymerase, the Klenow fragment of DNA polymerase I) were purchased from New England Biolabs (Ipswich, MA). The construction of promoter strength screening vectors was based on a promoter-less vector of pJIR2717 (Table 1, Supplemental data). Five well-studied, endogenous Clostridial promoters including adcP (coding for acetoacetate decarboxylase) (27, 28), glnAp (coding for glutamine synthetase) (29), ptbP (coding for phosphotransbutyrylase) (28), thlP (coding for thiolase) (27, 28, 30, 31), abrBP (coding for transcription factor abrB310)(32) and a hypoxia-inducible promoter (vgbP) encoding Vitreoscilla hemoglobin gene were cloned upstream of a catP gene and downstream of an Ω fragment in pJIR2717. The ferrodoxin promoter (fdP) used in our original NTR-expressing vector, pNTR540FT, was also included for comparison. These promoters were chosen as likely to be the strongest based on the literature. The Ω fragment is a 2.0-kb DNA segment consisting of an antibiotic resistance gene flanked by short inverted repeats carrying transcription and translation termination signals that terminates RNA and protein synthesis prematurely, thus allowing the definition and mapping of both transcription and translation units (33). A 0.45 kb EcoRI-BamHI fragment of pHT4, which contained the putative promoter region of ptbP, was treated with T4 DNA polymerase and the Klenow fragment to form a blunt-ended DNA fragment. This blunt-ended fragment was then inserted into the single SmaI site of pJIR2717 to create the vector pJIR2717-ptbP. Similarly, a 0.44kb EcoRI-BamHI fragment of pHT5 and a 0.32 kb EcoRI-BamHI fragment of pHTA yielded the desired putative thlP and adcP promoter regions, respectively. These fragments were also treated with T4 DNA polymerase and the Klenow fragment to form blunt-ended DNA. In order to construct pJIR2717-thlP and pJIR2717-adcP, these blunt-ended DNA fragments were then inserted into the single SmaI site of pJIR2717. For construction of pJIR2717-abrBP and pJIR2717-glnAP, appropriate primer pairs (Table 2, Supplemental data) were used and corresponding putative promoter regions were amplified from genomic DNA of C. acetobutylicum ATCC824. Amplified fragments were blunt-ended with T4 DNA polymerase and Klenow fragment and inserted into the single SmaI site of pJIR2717 to create vectors pJIR2717-abrBP and pJIR2717-glnAP. Similarly, fdP was amplified from plasmid DNA of pNTR540FT and vgbP was amplified from plasmid DNA of pUC8:16 with appropriate primer pairs (Table 2, Supplemental data). Amplified fragments were then gel purified and blunt-ended with T4 DNA polymerase and Klenow fragment and ligated with SmaI digested pJIR2717. The plasmids obtained were renamed pJIR2717-fdP and pJIR2717-vgbP, respectively. All plasmids were first constructed in E. coli and then electroporated into C. sporogenes using methods previously described (4, 34). Positive clones were identified by resistance to chloramphenicol and confirmed by restriction analysis. Promoter strength was assayed by dilution plating as described below.
Strong promoters obtained from the above analysis were PCR-amplified with appropriate template and primer pairs (Table. 2, Supplemental data). Amplified fragments contained the putative promoter regions were restriction enzyme treated and ligated with a PCR amplified NTR fragment and inserted into the backbone plasmid of pMTL540FT. The thlP and abrBP promoter fragments including their own ribosome-binding sites (RBS) were first PCR amplified from genomic DNA of C. acetobutylicum (ATCC824) with appropriate primer pairs (Table 2, Supplemental data) which contained restriction adaptors of KasI at the 5′-end and NdeI at the 3′-end. These amplified fragments of thlP and abrBP were first digested with NdeI and then ligated with PCR amplified NTR fragments pre-digested with AseI enzymes. The resulting fragments were then amplified with either abrBP-fwd, NTR-rev or thlP-fwd, NTR-rev primer pairs (Table 2, Supplemental data). These amplified promoter-NTR fragments were then digested with KasI and XbaI and ligated with pMTL540FT pre-digested with the same set of enzymes. These cloning procedures for replacing the fdP promoter in pNTR540FT with the abrBP and thlP promoters were required because of the presence of an internal NdeI site in both of the promoter regions. These amplified promoter fragments were cloned immediately in front of the coding sequence of NTR [UniProt P38489]. These ligation mixtures were introduced into E. coli GBE180 then electroporated into C. sporogenes. Positive clones were identified by restriction enzymes analysis and confirmed by DNA sequencing.
A synthetic gene encoding NTR (sNTR) was synthesized (GenScript Corp., Piscataway, NJ, USA), incorporating Clostridium preferred codons based on a codon usage table of C. botulinum posted by Kazusa DNA Res. Inst., Chiba, Japan. This sNTR was first cloned into pSC208 (Table 1, Supplemental data). This sNTR nucleotide fragment was first cloned into pSC208 (Table 1, Supplemental data) and amplified with primer pairs of sNTR-fwd and sNTR-rev which contained restriction adaptors of the 5′-end NdeI site and 3′-end BamHI sites (Table 2, Supplemental data). Amplified sNTR fragments were then digested with NdeI and BamHI and ligated with plasmids of pNTR540FT, pSC205AT and pSC207TT pre-digested with the same pair of enzymes to create the new set of sNTR expressing plasmids of pSC213FT (fdP-sNTR), pSC214AT (abrBP-sNTR) and pSC215TT (thlP-sNTR). All plasmids were first constructed in E. coli and then electroporated into C. sporogenes.
Promoter strength assay by growth on selective media by dilution plating
E.coli hosts harboring various promoter strength reporting constructs were grown aerobically overnight in LB broth containing 250 μg/ml of erythromycin. The cultures were then transferred 1:100 into fresh non-selective LB and grown to log phase. Serial 1:10 dilutions were made from these log phase cultures, and the cell densities adjusted to 0.4 (A600) with fresh LB broth. A 15 μl aliquot of each dilution was spotted on LB agar plates supplemented with increasing concentrations of chloramphenicol ranging from 150 μg/ml to 300 μg/ml. The spotted agar plates were scored by growth of each dilution spot after incubation at 37ºC for 24 hrs. Similarly, Clostridial transformants were grown overnight in liquid TPGY medium containing 10 μg/ml of erythromycin at 37ºC in a hypoxic chamber. Next day the transformants were transferred 1:100 into fresh TPGY broth without erythromycin and grown to exponential phase. Serial dilutions of 1:5 with TPGY broth were made from these log phase cultures, the cell densities adjusted to 0.6 (A600) and 15-μl aliquots of each dilution were spotted on TPGY agar plates supplemented with various concentration of chloramphenicol ranging from 2 μg/ml to 10 μg/ml. The spotted agar plates were incubated at 37ºC in a hypoxic chamber for 48 to 72 hrs. The promoter strength was determined by the growth of each dilution spot on the selective plates based on the resistance to chloramphenicol expressed from the catP gene (35, 36). This measure of promoter strength has been used earlier with C. perfringens (37).
Immunoblot Analysis of NTR
A 15 ml overnight culture of Clostridial cells was collected by centrifugation, washed 3X with cold PBS, resuspended in 1 ml of cold PBS and lysed by three 30-second sonications (VC130 sonicator, Sonics & Materials Inc., Newtown, CT) with the samples being kept on ice for at least two minutes between each sonication. Protein concentrations were determined by a Bradford assay (Protein Assay kit, Bio-Rad Lab. Inc., Hercules, CA) with bovine serum albumin as a standard. Lysed samples were run on 10% SDS-PAGE mini-gels with a EI9001-Xcell II™ Mini Cell apparatus (Novex, San Diego, CA). Proteins were transferred to a 0.2 μm nitrocellulose membrane (Invitrogen, Carlsbad CA), and gels stained with Coomassie brilliant blue to check for uniform transfer. Nitrocellulose membranes were blocked with 5% non-fat milk in PBS-T (Dulbecco’s PBS containing 0.1% Tween-20) overnight at 4ºC and then incubated at room temperature for 2 hrs with a 1:5,000 dilution of a custom made anti-NTR polyclonal rabbit serum (Invitrogen, Carlsbad CA) using an immunogen of a synthetic peptide with the sequence of KGRKFFADMHRK corresponding to amino acids 119–130 at the N-terminus of the E. coli nfnB gene. Membranes were washed 8X in PBS-T and incubated for 1 hour at room temperature with alkaline phosphatase conjugated goat anti-rabbit IgG (Vector Lab. Inc., Burlingame, CA) diluted 1:3,000. Antibody was detected using enhanced chemifluorescence (ECF) (Amersham Biosciences, Pittsburgh, PA) with a Storm 860 imaging system and quantitatively analyzed by ImageQuant TL (Molecular Dynamics, Piscataway, NJ).
Assay of NTR activity
Quantitative NTR enzyme assays using PR-104A (2-[(2-bromoethyl)-2-{[(2-hydroxyethyl)amino]-carbonyl}-4,6-dinitroanilino]ethyl methanesulfonate) (generous gift from Professor W. Denny, University of Auckland, New Zealand) as the substrate were carried out at 37ºC by HPLC as previously described (38, 39). The substrate (initially 50–800 μM) was diluted from 2–32 mM stock in DMSO, and incubated at 37ºC with cofactor [NAD(P)H at 1.0 mM] and cell extract (0.1 – 1 mg) in 500 μl 100 mM sodium phosphate. Samples were taken at different time point and frozen immediately in liquid nitrogen and then kept in −20ºC until analyzed. The presence of nitroreductase activity was indicated by the disappearance of PR-104A. The final concentration of DMSO in the reaction mixture was kept constant at 2.5% (v/v) because this solvent is an inhibitor of enzyme activity if present at > 5% (v/v) (38).
Loss of PR-104A by nitroreduction were determined using 10–100 μl of the final product in 500 μl assay mix using a reverse phase HPLC system with a diode array detector (Agilent 1100, Agilent Technologies, Inc., Palo Alto, CA). The mobile phase was a gradient of 80 % acetonitrile/water (A) and 0.45 M ammonium formate in water, pH 6.5, with 5 % A initially for 0–2 min, increasing in linear segments to 70 % A at 15 min, then decreasing to 5 % at 17 min, and held at 5 % for a further 3 min. The column was a 3.2 × 150 mm Altima C8 column (Alltech Associates Inc., Chicago, IL) with a flow rate of 0.5 ml/min. Absorbance detection was at 330 nm (band width 4 nm), with quantification by integration of peak areas (Chemstation software) with reference to standard curves prepared with authentic compounds in the same medium. Standard curves were linear for all compounds (r2 > 0.95; GraphPad PRISM Version 4.0, GraphPad Software Inc., San Diego, CA).
Mice and Tumors
Immunodeficient nude mice (Charles River) were housed in sterile cages with autoclaved bedding and free access to food and water. The human cervical carcinoma cell line, SiHa, was maintained in Dulbecco’s medium supplemented with 10% fetal bovine serum and established from mycoplasma free frozen stocks every three months. Cells were harvested from monolayer cultures with 0.05% trypsin/1.8mg/ml EDTA, centrifuged, re-suspended in medium, and inoculated s.c. at 5 – 8 × 106 cells per mouse on the back approximately 1 cm proximal to the base of the tail. Tumor volume (V) was calculated from three orthogonal Vernier caliper measurements (a, b, and c) using the formula V = π/6(abc). Experiments were initiated when tumor volume was approximately 200 mm3. Tumor growth delay was calculated in days for each tumor to reach three times tumor volume at the start of treatment. The growth delay was calculated within each group by determining the days taken for the tumors to reach the three times their initial tumor volumes. The difference between the growth curves was analyzed for statistical significance by using one-way ANOVA with Tukey post-test (PRISM Version 4.0, GraphPad, Inc., San Diego, CA). All animal procedures were approved by Stanford’s Administrative Panel on Laboratory Animal Care (APLAC).
Drug preparation and treatment
PR-104 was prepared daily in PBS containing one molar equivalent of sodium bicarbonate and used within 6 hr of preparation. CB1954 was prepared freshly in 10% N,N-dimethyl acetamide, 40% polyethylene glycol and 50% ddH2O (added just prior to injection) and used within 6 hr of preparation. PR-104 (250 mg/kg) and CB 1954 (50 mg/kg) were given i.p. 48 hours after intravenous injection of Clostridia spores (1 × 108). Treatment with CB 1954 was given only once and treatment with PR-104 was repeated once per week for 3 weeks. These protocols were chosen based on our prior published studies (26) and on data showing that these protocols equally well tolerated (30% of the MTD of each drug) and gave similar levels of antitumor activity.
In vivo imaging of NTR activity in nude mice
To image NTR activity in vivo we first performed retroviral transduction of CMV-driven green fluorescent protein (GFP) into SiHa cells. GFP expression was confirmed by fluorescent microscopy. SiHa/GFP cells (5 – 8 × 106) were suspended in 100 μl PBS and inoculated subcutaneously (s.c.) into the dorsal area approximately 1 cm proximal to the base of the tail of 6-week-old female immunodeficient nude mice. When the tumors reached a mean volume of 200–300 mm3, mice were given intravenous injections of saline or Clostridial spores transformed with sNTR. Two days later 6-chloro-9-nitro-5-oxo-5H-benzo[a]phenoxazine (CNOB), 100 μg/mouse in 100% DMSO (Invitrogen, Carlsbad, CA) was injected intraperitoneally. This nonfluorescent compound is metabolized by E.coli NTR to a highly fluorescent aminophenoxazine, with fluorescence excitation/emission maxima at 617 nm/625 nm. After 24 hr mice were imaged with a small-animal imaging Maestro system (CRi Inc, Woburn, MA). The collected images were analyzed using the spectral unmixing procedure in the Maestro software (CRi Inc).
Results
Development of improved strength promoters
The Clostridial promoters (adcP, abrBP, fdP, glnAp, ptbP, thlP and a hypoxia inducible vgbP) were cloned into a promoterless vector of pJIR2717 and transformed into the E. coli host GBE180. We assayed the promoter strength by dilution plating on LB plates contained increasing concentrations of chloramphenicol (Cm) or its analog, Thiamphenicol (Tm) for Clostridial catP expression. Our results showed the following order of promoter strength based on the catP expression in E. coli host as determined by the resistance to Cm (data not shown):
glnAP ≥ fdP > abrBP > thlP > ptbP > vgbP ≥ adcP.
However, since the promoter strength assayed in E. coli may not reflect the expression level in the Clostridial host. We electroporated these same seven promoter constructs into C. sporogenes and assayed promoter strength by dilution plating on TPGY plates containing increasing concentrations of Cm for Clostridial catP expression. Our results from this promoter strength assay were as follows (Fig. 1):
Fig. 1.
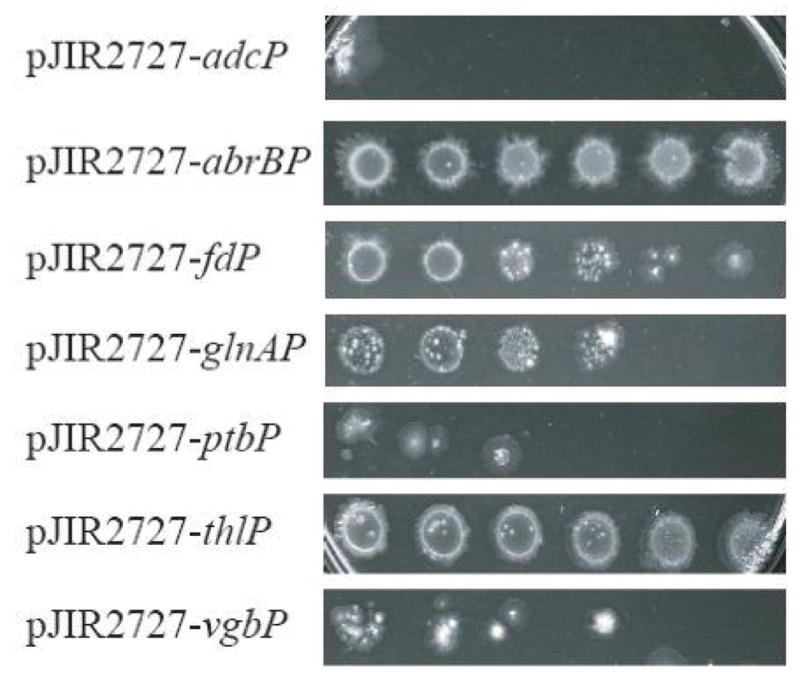
Effect of catP expression driven by various promoters on chloroamphenicol tolerance in recombinant C. sporogenes. Exponentially growing cells were harvested and washed with fresh TPGY and equilibrated to an A600 of 0.6. Five-fold serial dilutions of each strain of cells were spotted on TPGY plates supplemented with 2–10 μg/ml of chloramphenicol and incubated anaerobically for 72 hrs. A rough guide to promoter strength was ascertained by the read out of the total growth of each dilution spot on the selective plates. The image in Fig. 1 shows the data for the plate with 8 μg/ml chloramphenicol.
abrBP ≥ thlP > fdP > glnAP > vgbP > ptbP > adcP
Thus two strong promoters, abrBP and thlP, with similar strengths in driving the expression of catP in C. sporogenes were i dentified. These two promoters were then selected for the next round of cloning into vectors for NTR expression.
NTR expression in E. coli and Clostridial transformants
For optimized NTR expression vectors we substituted thlP or abrBP for fdP in our original NTR expression vector pNTR540FT. The resulting plasmids were designated pSC205AT (with abrBP-NTR) and pSC207TT (with thlP-NTR). Positive clones were identified by restriction enzyme analysis and confirmed by DNA sequencing. In Western blot analysis, expression of NTR driven by either thlP or abrBP were found to be approximately 3-fold higher than that driven by fdP (Fig. 2). Similar results were obtained in the enzymatic assays (Fig. 4). However, despite these strong promoters there was still approximately 50-fold higher NTR expression in the E. coli host compared to the Clostridial host transformed with identical plasmids (Fig. 2), which we hypothesized was because of very different codon uses between the two organisms (Fig. 3A).
Fig. 2.

Western blot analysis of NTR expression driven by various promoters in recombinant E. coli and C. sporogenes. Cell extracts were generated from overnight cultures of recombinant E. coli and C. sporogenes by sonication, Cultures of wild type C. sporogenes are also included as a negative control.
Fig. 4.

(A). Western blot analysis of NTR and sNTR expression driven by various promoters in recombinant E. coli and C. sporogenes. Total protein was obtained from overnight cultures of recombinant E. coli and C. sporogenes harboring pNTR540FT (NTR driven by fdP), pSC205AT (NTR driven by abrBP), pSC207TT (NTR driven by thlP), pSC213FT (sNTR driven by fdP), pSC214AT (sNTR driven by abrBP) and pSC215TT (sNTR driven by thlP). (B). Specific enzyme activity of NTR (mmol/min/mg of protein) plotted against the initial prodrug (PR-104) concentration (μM) with total protein generated from recombinant C. sporogenes harboring pNTR540FT, pSC205AT, pSC207TT, pSC213FT, pSC214AT and pSC215TT. Cell extracts of wild type C. sporogenes as the negative control and total protein from recombinant E. coli harboring pSC214AT as a positive control were also included.
Fig. 3.

(A) Plot of codon usage frequencies between E. coli and C. sporogenes (left) and C. botulinum and C. sporogenes (right). The greater the similarity in codon usage frequency of a given codon in both strains the higher the correlation coefficient will be. Of all the genome-sequenced Clostridial species, C. botulinum possessed the highest correlation coefficient in codon usage frequency when plotting against that of C. sporogenes. (B) Alignment of nucleotides sequence of optimized NTR (sNTR) and original NTR. Nucleotides not matched to the original NTR are shown in red. Of note, there is only a 34% similarity in DNA sequence between sNTR and the original NTR.
To test this hypothesis we synthesized a novel gene encoding NTR (sNTR) preserving the peptides sequence of E. coli nfnB (NTR) but having a Clostridium preferred codon scheme based on a codon usage table of C. botulinum posted by Kazusa DNA Res. Inst., Chiba, Japan. The C. botulinum sequence was chosen as this gave the highest correlation (R2 = 0.96) of codon use frequency for all known genes of C. sporogenes when plotted against that of all the Clostridial species with a sequenced genome (Fig. 3A). The close relationship of these two species is also indicated by the greater than 95% sequence identity of nucleotide and protein level with the fldAIBC gene cluster in C. sporogenes and that of C. botulinum Hall strain A (40). Fig 3B shows a comparison of the nucleotide sequences of the native and synthetic NTR genes with the changed codons shown in red.
Superiority of the synthetic gene for expression in C. sporogenes.
In both Western and enzyme assays with cell extracts from Clostridial transformants harboring plasmid constructs of thlP-sNTR and abrBP-sNTR we found an improvement by some 20–30 fold compared to the original construct of pNTR540FT (Fig. 4). The constructs thlP-sNTR and abrBP-sNTR performed equally well with in both functional assays and Western analysis. However, we chose the abrBP-sNTR/C. sporogenes construct for the in vivo studies because of its superior sporulation efficiency than the thlP-sNTR/C. sporogenes construct.
In vivo antitumor activity following systemic injection of recombinant Clostridial spores and dinitrobenzamide mustard prodrugs
We first determined if there was an effect of spore transformation on tumor colonization and found no difference in the number of C. sporogenes/gm of tumor between transformed and non transformed spores in SiHa human tumor xenografts (data not shown). To compare the antitumor activities of CDEPT with the new and prior constructs we injected mice bearing the SiHa tumor with Clostridial spores with or without the prodrugs CB1954 or PR-104. The data show that either sNTR alone, CB1954 or PR-104 alone resulted in a small but statistically insignificant delay in tumor growth (Fig. 5A & B). However when sNTR was combined with either CB1954 or with PR-104, we observed superior antitumor activity that was statistically significant (tumor growth delay >13 days; p < 0.05). Treatment with E. coli NTR alone resulted in growth delay similar to that with sNTR, and the combination with CB1954 or with PR-104 did not show a significant increase in antitumor activity.
Fig. 5.

Superior antitumor activity of CDEPT with sNTR compared to the native NTR. All the tumors were treated with 108 spores on day 0 when they reached a mean volume of 200 mm3. A. Effect of combinations of recombinant spores with the prodrug CB1954 (50 mg/kg given once 48 hours after intravenous injection of Clostridia spores. B. Effect of combinations of recombinant spores with the prodrug PR-104 (250 mg/kg given once per week for 3 weeks starting 48 hr after spore injection). C. Western analysis of tumor homogenates taken 2 days after iv. injection of the NTR expressing spores. The upper band is a non specific band from SiHa tumors and is proportional to the total amount of protein loaded. As can be seen from the two right hand lanes, it is present in tumors from mice that did not receive an injection of spores (Control #1 and #2).
We also performed Western blot analysis of tumor homogenates on tumors treated in parallel to those in the growth delay experiments and sacrificed two days following injection of recombinant spores. The data (Fig. 5C) demonstrate much higher levels of NTR protein in the tumors of mice injected with C. sporogenes with the sNTR compared to the native NTR.
In vivo imaging of the activity of Clostridial spores transformed with sNTR
As a test of whether it would be possible to monitor in real time the activity of sNTR in the tumor we injected mice bearing the SiHa tumor transfected with CMV-driven GFP with spores of C. sporogenes transformed with the sNTR bearing plasmid. Two days later we injected the non-fluorescent NTR substrate 6-chloro-9-nitro-5-oxo-5H-benzo[a]phenoxazine (CNOB) intravenously and 24 hr later imaged both the GFP and red fluorescent signal from the metabolized CNOB using a small-animal imaging Maestro system (Fig. 6). Preferential metabolism of CNOB localized to the tumor can be clearly seen with no evidence of normal tissue expression. In addition inhomogeneity of both signals is evident (insert), consistent with maximum CNOB metabolism around an area of central necrosis in the tumor.
Fig. 6.

Imaging of activity of transformed C. sporogenes using the nonfluorescent substrate CNOB (6-chloro-9-nitro-5-oxo-5H-benzo[a]phenoxazine). SiHa tumors transformed with CMV-driven green fluorescent protein (GFP) were implanted into immunodeficient nude mice and when they reached a mean diameter of 200mm3 108 spores of C. sporogenes transformed with the abrBP/sNTR plasmid were injected intravenously. Two days later the non fluorescent CNOB (6-chloro-9-nitro-5-oxo-5H-benzo[a]phenoxazine) was injected intraperitoneally. This nonfluorescent compound is metabolized by E. coli NTR to a highly fluorescent aminophenoxazine, with fluorescence excitation/emission maxima at 617 nm/625 nm. Mice were imaged with a small-animal imaging Maestro system and the collected images were analyzed using the spectral unmixing procedure in the Maestro software. The inset shows that the metabolism of CNOB is preferentially metabolized around a central necrotic core in the tumor. The mouse on the left in each panel is a control (non tumor-bearing) mouse. This study was performed with three tumors and all three showed similar metabolism of CNOB to its fluorescent metabolite.
Discussion
We have proposed that the unique presence in solid tumors of necrotic and hypoxic regions could be exploited in cancer therapy using genetically engineered non-pathogenic strain of the bacterial genus Clostridium (2, 41). The goal of the present study was to develop an optimized heterologous gene expression system in C. sporogenes for use in CDEPT. As a first step we tested promoters from five key metabolic pathway genes of Clostridium and one hypoxia inducible promoter from Vitreoscilla. We found two promoters (abrBP and thlP) that were superior to the C. perfringens ferredoxin promoter in the construct used to drive the E. coli-derived cytosine deaminase gene in the original CDEPT studies (4). However, even with these promoters there was much lower expression (by approximately 50-fold) of the E. coli derived NTR gene in C. sporogenes compared to E. coli. We hypothesized that this was the result of the very different codon usage between Clostridia and E. coli and tested this by constructing a synthetic gene (sNTR) with the same amino acid sequence as the E. coli NTR gene but with the preferred Clostridial codon usage. This produced a significant improvement in gene expression such that the combination of the strong promoter (abrBP or thlP) with the synthetic NTR gene, gave approximately 20–30 fold higher expression levels than the original NTR expression vector pNTR540FT. Thus, this study demonstrates significant progress in optimizing the enzyme expression of E. coli-derived NTR in Clostridial transformants used in CDEPT to deliver prodrug-activating enzymes specifically to tumors. With this system we were able to show that ineffective doses of the prodrugs CB1954 and PR-104, both currently in clinical testing, could be converted into effective antitumor therapy by the addition of recombinant C. sporogenes expressing the metabolizing enzyme NTR (Fig. 5).
Though the improvements that we report here are significant, there are potentially others that could further improve the efficacy of CDEPT. One such would be to replace the E. coli NTR with a more active enzyme either naturally occurring or genetically manipulated. Theys and colleagues, for example, isolated an NTR-like protein from Haemophilus influenzae (NTR-H) with slightly better characteristics than that of original E .coli NTR gene (NfnB) in metabolizing CB1954 to its toxic species (42). In addition, methods for genetic manipulation of the native enzyme have been employed to increase reduction efficacy. For example, Grove and colleagues have generated mutants of NfnB by amino acid substitutions around the active site of the protein to produce a strain with a 5-fold improvement in enzyme kinetics for reducing CB1954 (43). Similarly, Barak and colleagues used directed evolution that introduces random mutations in NfnB with error-prone PCR, to improve the activity of the enzyme (44). In a complementary manner, novel NTR prodrugs with enhanced bystander effects such as the dinitrobenzamide mustard SN 28343 (45), may also provide improvements given that the tumor germination of Clostridial vectors is constrained to necrotic and peri-necrotic regions.
In conclusion, we report significant improvement in the heterologous expression of the E. coli-derived NTR gene in a Clostridial host and much improved antitumor activity of iv injected recombinant spores combined with two prodrugs in clinical use that are converted by NTR to cytotoxic species. In addition, should it prove necessary, other optimization strategies are available for use with this system. We believe that this optimization process could maximize the likely success of CDEPT in the clinic.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health grants P01 CA067166 and P01 CA082566. We thank Dr. Eleftherios T. Papoutsakis for providing plasmids of pHTA, pHT4 and pHT5 and Dr. Julian I. Rood for donating plasmid pJIR2717 for this study. We also thank Drs. Julian I. Rood and George Bennett for many insightful discussions on this research.
References
- 1.Dang LH, Bettegowda C, Huso DL, Kinzler KW, Vogelstein B. Combination bacteriolytic therapy for the treatment of experimental tumors. Proc Natl Acad Sci U S A. 2001;98:15155–60. doi: 10.1073/pnas.251543698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fox ME, Lemmon MJ, Mauchline ML, et al. Anaerobic bacteria as a delivery system for cancer gene therapy: activation of 5-fluorocytosine by genetically engineered clostridia. Gene Therapy. 1996;3:173–8. [PubMed] [Google Scholar]
- 3.Lemmon ML, Van Zijl P, Fox ME, et al. Anaerobic bacteria as a gene delivery system that is controlled by the tumor microenvironment. Gene Thrapy. 1997;4:791–6. doi: 10.1038/sj.gt.3300468. [DOI] [PubMed] [Google Scholar]
- 4.Liu SC, Minton NP, Giaccia AJ, Brown JM. Anticancer efficacy of systemically delivered anaerobic bacteria as gene therapy vectors targeting tumor hypoxia/necrosis. Gene Therapy. 2002;9:291–6. doi: 10.1038/sj.gt.3301659. [DOI] [PubMed] [Google Scholar]
- 5.Mose JR, Mose G. Onkolyseversuche mit apathogenen anaeroben Sporenbildern am Ehrlich Tumor des Maus. Z Krebsforsch. 1959;63:63–74. [Google Scholar]
- 6.Mose JR, Mose G. Oncolysis by clostridia. I. Activity of Clostridium butyricum (M-55) and other nonpathogenic clostridia against the Ehrlich carcinoma. Cancer Res. 1964;24:212–6. [PubMed] [Google Scholar]
- 7.Brantner H. The clostridia-tumor phenomenon: fundamentals in oncolytic tumour research. In: Dring GJ, Ellar DJ, Gould GW, editors. Fundamental and applied aspects of bacterial spores. London: Academic Press; 1985. pp. 463–73. [Google Scholar]
- 8.Engelbart K, Gericke D. Oncolysis by clostridia V. Transplanted tumors of the hamster. Cancer Research. 1964;24:239–43. [PubMed] [Google Scholar]
- 9.Szmigielski S, Dworecka B, Lipski S, Jeljaszewicz J, Pulverer G. Oncolytic clostridia. In: Jeljaszewicz J, Pulverer G, Roszkowski W, editors. Bacteria and Cancer. London: Academic Press; 1982. pp. 231–53. [Google Scholar]
- 10.Thiele EH, Arison RN, Boxer GE. Oncolysis by clostridia. IV. Effect of nonpathogenic clostridial spores in normal and pathological tissues. Cancer Research. 1964;24:234–8. [PubMed] [Google Scholar]
- 11.Carey RW, Holland JF, Whang HY, Neter E, Bryant B. Clostridial oncolysis in man. Europ J Cancer. 1967;3:37–46. [Google Scholar]
- 12.Heppner F, Mose JR. The liquefaction (oncolysis) of malignant gliomas by a non pathogenic clostridium. Acta Neuro. 1978;12:123–5. doi: 10.1007/BF01406639. [DOI] [PubMed] [Google Scholar]
- 13.Heppner F, Mose J, Ascher PW, Walter G. Oncolysis of malignant gliomas of the brain. 13th Int Cong Chemother. 1983;226:38–45. [Google Scholar]
- 14.Dang LH, Bettegowda C, Agrawal N, et al. Targeting vascular and avascular compartments of tumors with C. novyi-NT and anti-microtubule agents. Cancer Biol Ther. 2004;3:326–37. doi: 10.4161/cbt.3.3.704. [DOI] [PubMed] [Google Scholar]
- 15.Bettegowda C, Dang LH, Abrams R, et al. Overcoming the hypoxic barrier to radiation therapy with anaerobic bacteria. Proc Natl Acad Sci U S A. 2003;100:15083–8. doi: 10.1073/pnas.2036598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agrawal N, Bettegowda C, Cheong I, et al. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc Natl Acad Sci U S A. 2004;101:15172–7. doi: 10.1073/pnas.0406242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz LA, Jr, Cheong I, Foss CA, et al. Pharmacologic and toxicologic evaluation of C. novyi-NT spores Toxicol Sci. 2005;88:562–75. doi: 10.1093/toxsci/kfi316. [DOI] [PubMed] [Google Scholar]
- 18.Nuyts S, Theys J, Landuyt W, van Mellaert L, Lambin P, Anne J. Increasing specificity of anti-tumor therapy: cytotoxic protein delivery by non-pathogenic clostridia under regulation of radio-induced promoters. Anticancer Res. 2001;21:857–61. [PubMed] [Google Scholar]
- 19.Nuyts S, Van Mellaert L, Theys J, Landuyt W, Lambin P, Anne J. Clostridium spores for tumor-specific drug delivery. Anticancer Drugs. 2002;13:115–25. doi: 10.1097/00001813-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Yazawa K, Fujimori M, Amano J, Kano Y, Taniguchi S. Bifidobacterium longum as a delivery system for cancer gene therapy: selective localization and growth in hypoxic tumors. Cancer Gene Ther. 2000;7:269–74. doi: 10.1038/sj.cgt.7700122. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura T, Sasaki T, Fujimori M, et al. Cloned cytosine deaminase gene expression of Bifidobacterium longum and application to enzyme/pro-drug therapy of hypoxic solid tumors. Biosci Biotechnol Biochem. 2002;66:2362–6. doi: 10.1271/bbb.66.2362. [DOI] [PubMed] [Google Scholar]
- 22.King I, Bermudes D, Lin S, et al. Tumor-targeted salmonella expressing Cytosine deaminase as an anticancer agent. Hum Gene Ther. 2002;13:1225–33. doi: 10.1089/104303402320139005. [DOI] [PubMed] [Google Scholar]
- 23.Cunningham C, Nemunaitis J. A phase I trial of genetically modified Salmonella typhimurium expressing cytosine deaminase (TAPET-CD, VNP20029) administered by intratumoral injection in combination with 5-fluorocytosine for patients with advanced or metastatic cancer. Protocol no: CL-017. Version: April 9, 2001. Hum Gene Ther. 2001;12:1594–6. [PubMed] [Google Scholar]
- 24.Makoff AJ, Oxer MD, Romanos MA, Fairweather NF, Ballantine S. Expression of tetanus toxin fragment C in E. coli: high level expression by removing rare codons. Nucleic Acids Res. 1989;17:10191–202. doi: 10.1093/nar/17.24.10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zdanovsky AG, Zdanovskaia MV. Simple and efficient method for heterologous expression of clostridial proteins. Appl Environ Microbiol. 2000;66:3166–73. doi: 10.1128/aem.66.8.3166-3173.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patterson AV, Ferry DM, Edmunds SJ, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res. 2007;13:3922–32. doi: 10.1158/1078-0432.CCR-07-0478. [DOI] [PubMed] [Google Scholar]
- 27.Bermejo LL, Welker NE, Papoutsakis ET. Expression of Clostridium acetobutylicum ATCC 824 genes in Escherichia coli for acetone production and acetate detoxification. Appl Environ Microbiol. 1998;64:1079–85. doi: 10.1128/aem.64.3.1079-1085.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tummala SB, Welker NE, Papoutsakis ET. Development and characterization of a gene expression reporter system for Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol. 1999;65:3793–9. doi: 10.1128/aem.65.9.3793-3799.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quixley KW, Reid SJ. Construction of a reporter gene vector for Clostridium beijerinckii using a Clostridium endoglucanase gene. J Mol Microbiol Biotechnol. 2000;2:53–7. [PubMed] [Google Scholar]
- 30.Stim-Herndon KP, Petersen DJ, Bennett GN. Characterization of an acetyl-CoA C-acetyltransferase (thiolase) gene from Clostridium acetobutylicum ATCC 824. Gene. 1995;154:81–5. doi: 10.1016/0378-1119(94)00838-j. [DOI] [PubMed] [Google Scholar]
- 31.Tomas CA, Welker NE, Papoutsakis ET. Overexpression of groESL in Clostridium acetobutylicum results in increased solvent production and tolerance, prolonged metabolism, and changes in the cell’s transcriptional program. Appl Environ Microbiol. 2003;69:4951–65. doi: 10.1128/AEM.69.8.4951-4965.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scotcher MC, Rudolph FB, Bennett GN. Expression of abrB310 and SinR, and effects of decreased abrB310 expression on the transition from acidogenesis to solventogenesis, in Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol. 2005;71:1987–95. doi: 10.1128/AEM.71.4.1987-1995.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prentki P, Krisch HM. In vitro insertional mutagenesis with a selectable DNA fragment. Gene. 1984;29:303–13. doi: 10.1016/0378-1119(84)90059-3. [DOI] [PubMed] [Google Scholar]
- 34.Brown JM, Liu SC. Use of anaerobic bacteria for cancer therapy. In: Nakano MM, Zuber P, editors. Strict and Facultative Anaerobes: Medical and Environmental Aspects. Horizon Scientific Press; 2004. [Google Scholar]
- 35.Yamada A, Tsutsumi K, Tanimoto S, Ozeki Y. Plant RelA/SpoT homolog confers salt tolerance in Escherichia coli and Saccharomyces cerevisiae. Plant Cell Physiol. 2003;44:3–9. doi: 10.1093/pcp/pcg001. [DOI] [PubMed] [Google Scholar]
- 36.Toussaint M, Levasseur G, Gervais-Bird J, Wellinger RJ, Elela SA, Conconi A. A high-throughput method to measure the sensitivity of yeast cells to genotoxic agents in liquid cultures. Mutat Res. 2006;606:92–105. doi: 10.1016/j.mrgentox.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Matsushita C, Matsushita O, Koyama M, Okabe A. A Clostridium perfringens vector for the selection of promoters. Plasmid. 1994;31:317–9. doi: 10.1006/plas.1994.1035. [DOI] [PubMed] [Google Scholar]
- 38.Anlezark GM, Melton RG, Sherwood RF, et al. Bioactivation of dinitrobenzamide mustards by an E. Coli B nitroreductase Biochemical Pharmacology. 1995;50:609–18. doi: 10.1016/0006-2952(95)00187-5. [DOI] [PubMed] [Google Scholar]
- 39.Helsby NA, Ferry DM, Patterson AV, Pullen SM, Wilson WR. 2-amino metabolites are key mediatiors of CB 1954 and SN 23862 bystander effects in nitroreductase GDEPT. British Journal of Cancer. 2004;90:1084–92. doi: 10.1038/sj.bjc.6601612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dickert S, Pierik AJ, Buckel W. Molecular characterization of phenyllactate dehydratase and its initiator from Clostridium sporogenes. Mol Microbiol. 2002;44:49–60. doi: 10.1046/j.1365-2958.2002.02867.x. [DOI] [PubMed] [Google Scholar]
- 41.Minton NP, Mauchline ML, Lemmon MJ, et al. Chemotherapeutic tumour targeting using clostridial spores. FEMS Microbiol Rev. 1995;17:357–64. doi: 10.1111/j.1574-6976.1995.tb00219.x. [DOI] [PubMed] [Google Scholar]
- 42.Theys J, Pennington O, Dubois L, et al. Repeated cycles of Clostridium-directed enzyme prodrug therapy result in sustained antitumour effects in vivo. Br J Cancer. 2006;95:1212–9. doi: 10.1038/sj.bjc.6603367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grove JI, Lovering AL, Guise C, et al. Generation of Escherichia coli nitroreductase mutants conferring improved cell sensitization to the prodrug CB1954. Cancer Res. 2003;63:5532–7. [PubMed] [Google Scholar]
- 44.Barak Y, Thorne SH, Ackerley DF, Lynch SV, Contag CH, Matin A. New enzyme for reductive cancer chemotherapy, YieF, and its improvement by directed evolution. Mol Cancer Ther. 2006;5:97–103. doi: 10.1158/1535-7163.MCT-05-0365. [DOI] [PubMed] [Google Scholar]
- 45.Singleton DC, Li D, Bai SY, et al. The nitroreductase prodrug SN 28343 enhances the potency of systemically administered armed oncolytic adenovirus ONYX-411(NTR) Cancer Gene Ther. 2007;14:953–67. doi: 10.1038/sj.cgt.7701088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.