

Abstract
VLA-4 plays a critical role in T cell trafficking into inflammatory sites. Our recent studies have suggested that VLA-4 expression on CD8+ T cells is negatively controlled by IL-4 and serves as a functionally distinguishing variable for why Type-1, but not Type-2, CD8+ T cells are able to traffic into tumors. In this study, using in vitro culture of murine CD8+ T cells under Type-1 and Type-2 cytokine conditions, we show that IL-4-mediated down-regulation of VLA-4 expression is completely abrogated in Stat6-deficient CD8+ T cells. Conversely, CD8+ T cells expressing a constitutively active mutant form Stat6 (Stat6VT) failed to express VLA-4 even in the absence of IL-4-stimulation. Notably, Type-2 CD8+ T cells developed from Stat6−/− but not wild-type mice were competent to migrate into tumor lesions in vivo. These results suggest that Stat6-signaling is necessary and sufficient to restrict CD8+ T cell expression of VLA-4 (by IL-4), thereby serving as a regulator for CD8+ T cell infiltration into tumors.
Tumor infiltration by tumor-specific T cells is an important intermediate step associated with the clinical efficacy of cancer vaccines and adoptive immunotherapies. In our previous study using mice bearing intracranial M05 melanomas, we showed that adoptively transferred OVA-specific Tc1 cells were superior to Tc2 cells in trafficking into the intracranial tumor lesions and in mediating potent therapeutic responses (1). Furthermore, when characterizing the expression of a panel of homing receptors on Tc1 and Tc2 cells, we found that VLA-4 (CD49d/CD29) is preferentially expressed on Tc1 cells, but not on Tc2 cells, and that this integrin plays a critical role in the effective trafficking of therapeutic Tc1 cells into CNS tumors (2). Moreover, we have also recently identified IL-4 as a principal factor that negatively regulates VLA-4 expression on CD8+ T cells (2). The current study was undertaken to further delineate relevant mechanisms associated with IL-4-dependent suppression of VLA-4 on CD8+ T cells, which renders these cells clinically inert.
IL-4 is known to activate two major signaling cascades: the insulin receptor substrate (IRS)3-PI3K pathway that promotes the growth of target lymphocytes and the Jak-Stat6 pathway that induces the expression of IL-4-responsive genes (3, 4). In this study, using Stat6 knockout/transgenic mice and pharmacological inhibitor for PI3K, we clearly showed that Stat6 is a necessary as well as sufficient mediator of IL-4-mediated VLA-4 down-regulation in CD8+ T cells and restricts CTL trafficking into tumor sites.
Materials and Methods
Mice
C57BL/6 mice (5–9 wk of age) and C57BL/6 background Stat6 knockout mice were purchased from The Jackson Laboratory and maintained in a pathogen-free animal facility at the University of Pittsburgh Cancer Institute. C57BL/6 background Stat6VT transgenic mice were generated as previously described (5) and maintained in specific pathogen-free animal facility at Indiana University. Animals were handled in the Animal Facility at University of Pittsburgh per an Institutional Animal Care and Use Committee-approved protocol.
Reagents
rmIL-12 was purchased from Cell Sciences Technologies. rmIL-4 and rhIL-2 were purchased from PeproTech. Mouse VCAM-1-Ig fusion protein was purchased from R&D Systems. Purified anti-CD49d mAb (PS/2) was purchased from Southern Biotechnology Associates. Purified isotype-control rat IgG2b (RTK4530) was purchased from BioLegend. Purified mAbs against IL-12 (C15.6), IFN-γ (R4 – 6A2), IL-4 (11B11), CD3 (145-2C11), FITC-conjugated anti-CD29 mAb (HMβ1), and PE-conjugated anti-CD49d mAb were all purchased from BD Pharmingen. The PI3K inhibitor LY294002 was purchased from Calbiochem.
Generation of CD8+ effector T cells
Tc1 and Tc2 cells were induced from MACS-separated CD8+ splenic T cells isolated from either wild-type (WT), Stat6−/− or Stat6VT transgenic mice as previously described (2). In brief, purified CD8+ cells were stimulated with 5 μg/ml anti-CD3 mAb in the presence of irradiated (3000 rad) C57BL/6 spleen cells as feeder cells, 2 ng/ml of rmIL-12, 2 ng/ml of rmIFN-γ, 1 μg/ml of anti-IL-4 mAb, and 100 U/ml rhIL-2 for Tc1 development. Tc2 cells were generated from the same CD8+ cell precursors stimulated with 5 μg/ml anti-CD3 mAb-pulsed feeder cells in the presence of 100 ng/ml of rmIL-4, 10 μg/ml of two anti-IFN-γ mAbs (R4 – 6A2 and XMG1.2), 10 μg/ml of anti-IL-12 mAb (C15.6) and 100 U/ml of rhIL-2. When indicated, in other experiments, T cells were generated in the presence of 100 U/ml rhIL-2 with rmIL-4, or with anti-IL-4 mAb. After 48 h, cells were restimulated under the same conditions used for primary induction. rhIL-2 was maintained for the entire culture period. At day 10 post-stimulation, T cells were harvested and used for the assays.
Cell adhesion assays
Cell adhesion to VCAM-1-Ig was assessed as described previously (2). In brief, 96-well-ELISA plates were coated with 10 μg/ml of mouse VCAM-1-Ig or heat-denatured BSA. Tc1 and Tc2 cells were harvested at day 10, suspended in binding buffer (0.5% BSA, 2 mM CaCl2, 2 mM MgCl2 in PBS), and then added to the plate. For blocking experiments, cells resuspended in binding buffer were pretreated with 20 μg/ml of anti-CD49d mAbs (PS/2) for 15 min at 37°C, and then added to the plate. Plates were centrifuged at 500 rpm for 1 min and cells were allowed to adhere for 30 min at room temperature with gentle shaking. The plate was then gently washed three times using binding buffer and the number of adherent cells were enumerated by flow cytometry. Percent adhesion was calculated as the (Number of adherent cells to VCAM-1-Ig – Number of adherent cells to heat-denatured BSA)/Number of total input cells.
In vivo homing of Tc1 and Tc2 cells into s.c. M05 tumors
To investigate the in vivo trafficking of Tc1 and Tc2 cells induced from WT or Stat6−/− C57BL/6 mice, mice received s.c. injection in the right flank with 1 × 106 M05 (OVA-transfected B16 melanoma) cells. Cultured Tc1 and Tc2 cells induced from WT or Stat6−/− C57BL/6 mice were harvested at day 9 and labeled with 0.4 μM of CFSE (Vybrant CFDA SE Cell Tracer kit; Molecular Probes) according to the manufacturer’s protocol. At day 7 after tumor inoculation, mice received an i.v. injection with 4 × 107 cells of labeled Tc1 and Tc2 cells. Twenty-seven hours later, mice were sacrificed and then perfused through the left cardiac ventricle with PBS. Tumors were enzymatically digested by 1% collagenase, 1% hyaluronidase, and 0.1% DNase, and cells from each tumor were resuspended in RPMI 1640 supplemented with 10% FCS and overlaid on equal volume of lympholyte-M (Cedarlane Laboratories), and then centrifuged for 20 min at 2000 rpm. Enriched tumor-infiltrating lymphocytes (TIL) populations were recovered at the interface. TILs were stained with PE-anti-mouse CD8 mAb and the frequency of CD8+CFSE+ T cells was assessed by flow cytometry.
Statistical analyses
All intergroup comparisons of means were assessed with one-sided t tests. The p values ≤ 0.05 were considered significant.
Results
Stat6 but not PI3K plays a critical role in IL-4-mediated VLA-4-down-regulation on CD8+ T cells
IL-4 is known to activate two major signaling cascades: the IRS-PI3K pathway that promotes the growth of target lymphocytes and the Jak-Stat6 pathway that induces the expression of IL-4-responsive genes (3, 4). Identification and antagonistic targeting of the specific signaling cascade(s) responsible for IL-4-mediated integrin down-regulation could prove beneficial to enhancing Type-1 T cell trafficking into tumors. To address this possibility, CD8+ T cells derived from WT C57BL/6 or Stat6−/− splenocytes were stimulated with anti-CD3 mAb under Tc1 (IL-12, IFN-γ, anti-IL-4 mAb) or Tc2 (IL-4, anti-IFN-γ mAb, anti-IL-12 mAb)-polarizing cytokine conditions. These cultured CD8+ effector T cells were then examined for expression of VLA-4 (CD49d/CD29). In WT mice, VLA-4 is preferentially expressed on Tc1 cells and dramatically down-regulated on Tc2 cells (Fig. 1A), which is consistent with our previous report (2). Levels of VLA-4 expression on Tc1-conditioned and neutral-conditioned (IL-2 only), neutral plus anti-IL-4 mAb conditioned (IL-2 plus anti-IL-4 mAb) CD8+ T cells showed no difference between groups, and the addition of rIL-4 to neutral culture conditions was sufficient to down-regulate VLA-4 expression on CD8+ T cells (data not shown). This further confirmed the critical role of IL-4, but not IL-12 or IFN-γ, in the observed differential expression of VLA-4 by Tc1 vs Tc2 cells.
FIGURE 1.
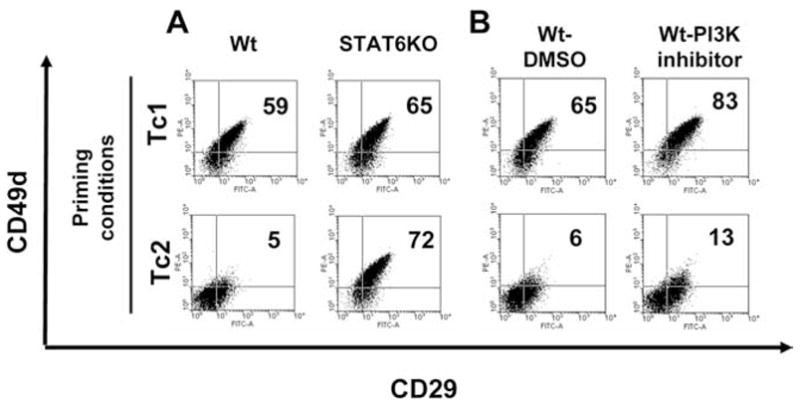
Stat6 but not PI3K is essential for IL-4-induced down-modulation of VLA-4 (CD49d/CD29) on CD8+ T cells. A, Resting T cells from WT and Stat6−/− C57BL/6 mice were stimulated with anti-CD3 mAb and cultured under Tc1 (IL-12, IFN-γ, anti-IL-4 mAb) or Tc2 (IL-4, anti-IFN-γ mAb, anti-IL-12 mAb) cytokine conditions. B, Naive T cells from WT C57BL/6 mice were stimulated with anti-CD3 mAb and cultured under Tc1 or Tc2 cytokine condition in the presence of PI3K inhibitor (LY294002) or DMSO as a control. Cells were analyzed by flow cytometry for expression of CD49d and CD29 at day 10. All data are representative of three independent experiments performed.
In contrast to WT mice, the negative effects of rIL-4 on CD8+ T cell expression of VLA-4 were totally abrogated in Stat6−/− mice, with both Tc1- and Tc2-conditioned CD8+ T cells expressing high levels of VLA-4 (Fig. 1A). To evaluate the relative contribution of the IRS-PI3K pathway in IL-4-mediated VLA-4-down-regulation, CD8+ T cells were next stimulated with either Tc1- or Tc2-cytokine conditions in the absence or presence of 10 μmol/L of LY294002, a specific inhibitor of PI3K activity, or DMSO as a control. PI3K inhibitor-treated, Tc2-conditioned CD8+ T cells exhibited a comparable reduction of VLA-4 as non-treated, or DMSO-treated, Tc2-conditioned CD8+ T cells (Fig. 1B). These data clearly demonstrated the essential role of Stat6, but not PI3K, in IL-4-mediated VLA-4 down-regulation on CD8+ T cells.
The inability of Tc2 cells to bind to VCAM-1 is recovered in Stat6−/− mice
To examine the functional significance of VLA-4 expression on these CD8+ T cell subsets derived from WT or Stat6−/− mice, Tc1- or Tc2-conditioned CD8+ T cells were tested for their ability to adhere to plate-bound VCAM-1-Ig fusion protein. In line with the differential expression of VLA-4 expression on Tc1 and Tc2 cells derived from WT mice, WT Tc1 cells exhibited specific adhesion to plate-bound VCAM-1-Ig (42 ± 2.7%), whereas, WT Tc2 cells displayed only background levels of adhesion (4.1 ± 2.3%) (Fig. 2). In contrast, both Tc1- and Tc2-conditioned CD8+ T cells derived from Stat6−/− mice bound well to immobilized VCAM-1 (62 ± 0.5% and 61 ± 2.0%, respectively). Pretreatment of CD8T+ cells with the anti-VLA-4α mAb (PS/2) virtually ablated their ability to adhere to VCAM-1, supporting the critical nature of VLA-4 in the strong binding of Stat6-deficient CD8+ T cells to this substrate (Fig. 2). These data suggested that ablation of IL-4R/Stat6 signaling prevents IL-4-mediated down-modulation of VLA-4 expression on CD8+ T cells and their coordinate ability to adhere to VCAM-1.
FIGURE 2.
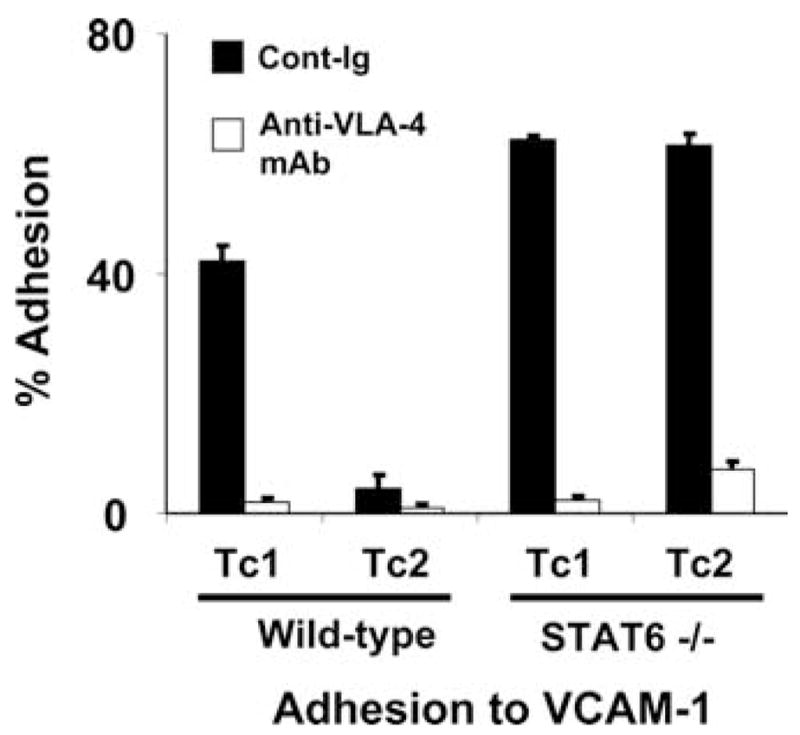
Stat6-deficient Tc2-conditioned cells express VLA-4 and bind to VCAM-1. After preincubation with either anti-VCAM-1 mAb (PS/2) (□) or control rat IgG2b mAb (■), Tc1 and Tc2 cells were incubated in 96-well plates coated with VCAM-1 for 30 min. The plates were then washed twice and the number of cells adherent to the bottom of the wells was enumerated by flow cytometry, as described in Materials and Methods. Data are representative of three independent experiments performed.
Constitutive STAT6 activation results in the silencing of VLA-4 expression by CD8+ T cells
Recently, transgenic mice expressing a constitutively active, mutant form of Stat6 (Stat6VT) under the CD2 promoter have been generated (5). In these mice, T cells over express an active form of Stat6. Taking advantage of this mouse model, we next examined if chronically activated Stat6 results in the prevention of VLA-4 expression by polarized CD8+ T cells. Splenic CD8+ T cells isolated from WT and Stat6VT transgenic mice were similarly stimulated with Tc1- or Tc2- cytokine conditions and then examined for expression of VLA-4. As shown in Fig. 3A, both Tc1- and Tc2-conditioned CD8+ T cells derived from Stat6VT Tg mice exhibited dramatically reduced expression levels of VLA-4 when compared with WT Tc1 cells, or Tc1/Tc2 cells generated from Stat6−/− mice. Predictably, these T cells were also defective in their ability to adhere to VCAM-1-coated surfaces (Fig. 3B). These results further support our hypothesis that IL-4R/Stat6 signaling is both necessary and sufficient for (the IL-4-mediated) down-regulation of VLA-4 on CD8+ T cells.
FIGURE 3.

Constitutively active Stat6 in CD8+ T cells is associated with low VLA-4 expression levels and the inability to bind to immobilized VCAM-1. Naive T cells from WT and Stat6VT transgenic C57BL/6 mice were stimulated with anti-CD3 mAb and cultured under Tc1 or Tc2 cytokine conditions, as described in Materials and Methods. A, Two-color flow cytometric analysis of the VLA-4 (CD49d/CD29) complex. B, T cells induced under the indicated conditions were harvested at day 10 and assessed for their ability to bind to immobilized VCAM-1-Ig, as described in Materials and Methods. All data are representative of three independent experiments performed.
Tc2 cells developed from Stat6−/− mice are competent to traffic into tumor sites
In our previous study, we have demonstrated that adoptively transferred Tc2 cells are defective in their ability to traffic into intracranial tumor sites (1, 2). Therefore, we next examined whether Tc2 cells generated from Stat6−/− mice splenocytes exhibiting a VLA-4+ phenotype could traffic into peripheral tumor sites after adoptive transfer. WT, tumor-bearing mice received i.v. transfers of CFSE-labeled 2 × 107 Tc1 or Tc2 cells developed from WT or Stat6−/− mice. One day later, TIL were isolated and analyzed by flow cytometry. Consistent with our previous study (2), a high percentage (8.8%) of TILs represented adoptive-transferred CD8+CFSE+ T cells following WT Tc1 infusion, whereas very few (0.6%) CD8+CFSE+ T cells were identified in TILs following WT Tc2 infusion (Fig. 4). In contrast, a high frequency of both Tc1 and Tc2 cells were recovered in the TILs after adoptive-transfer if the transferred T cells were of Stat6−/− origin (Tc1; 7.6%, Tc2; 6.7%; Fig. 4). In contrast, only a very small fraction of adoptively transferred T cells were recovered in the spleen (0.5–1.5%) for any of these groups, with no statistically significant difference between cohorts observed (data not shown). Collectively, these results demonstrated that Stat6-signaling coordinately regulates both VLA-4 expression by CD8+ T cells and their capacity to traffic into s.c. tumor lesions.
FIGURE 4.

Tc2 cells developed from Stat6−/− mice can effectively migrate into tumor lesions in vivo. Mice bearing established, day 10 s.c. M05 melanomas received i.v. adoptive transfer of 4 × 107 Tc1 or Tc2 cells induced from WT or Stat6−/− mice. TILs were harvested 27 h after the adoptive-transfer of CFSE-labeled cells and stained with PE-conjugated anti-CD8 mAb, and then analyzed by flow cytometry to enumerate the percentage of CD8+CFSE+ populations. Left panel, Representative flow cytometry data from one of three mice per group are shown. Right panel, Mean ± SD of the frequency of CSFE+ cells/1000 TIL for three mice analyzed per cohort. P = 0.003 vs Tc1 WT.
Discussion
In the current study, we found that Stat6 activation is required for mediating the down-regulation of VLA-4 expression and trafficking of CD8+ T cells induced by IL-4. We initially showed that IL-4-induced VLA-4 down-regulation was completely abrogated on Tc2-conditioned Stat6−/− CD8+ T cells and VLA-4 expression was highly up-regulated in these cells. Interestingly, we have recently noted a similar pattern of Stat6-dependent VLA-4-down-regulation for CD4+ T cells (our unpublished data), suggesting that a common signaling cascade may exist downstream of the IL-4 receptor that negatively regulates VLA-4 expression on both CD4+ and CD8+ T cells.
Consistent with the critical role of Stat6 in Type-2 T cell development (6, 7), Tc2-conditioned Stat6−/− CD8+ T cells failed to produce IL-4 and secreted large amounts of IFN-γ, suggesting that these cells are of Tc1-like (data not shown). This raises the question as to whether IL-4R/Stat6-mediated VLA-4 down-regulation is accompanied by a Type-2 skewing of T cells. Interestingly, we observed that IL-4 treatment of mature, polarized Tc1 cells resulted in their down-regulation of VLA-4, without changing the profile of cytokines produced by these cells, suggesting that IL-4R/Stat6-mediated VLA-4-down-regulation is independently regulated from the functional Type-2 polarization of treated T cells (our unpublished data). Considering that GATA-3 and c-Maf serve as central regulators of Type-2 cytokine genes in Th2 cells (8, 9), these data may suggest that Stat6 down-regulates VLA-4 on CD8+ T cells without affecting the levels or function of GATA-3 or c-Maf. We are currently evaluating this possibility.
The α4β1 (VLA-4) integrin, and its counterreceptor VCAM-1 have been previously characterized as key mediators of T cell entry into sites of inflammation, such as delayed-type hypersensitivity skin (10), experimental autoimmune encephalomyelitis (11, 12) and other CNS inflammatory (13) lesions. Our finding that Stat6 is a major mediator of IL-4-dependent VLA-4 down-regulation provides additional insights regarding the mechanism(s) underlying pathogenesis and the success or failure of therapeutic intervention in the setting of various inflammatory diseases. Indeed, in mouse models, the IL-4/Stat6 signaling pathway has been shown to play critical roles in ameliorating inflammatory diseases such as experimental autoimmune encephalomyelitis (14) and rheumatoid arthritis (15). Moreover, systemic administration of rIL-4 has also been shown to effectively treat patients with psoriasis (16). However, because IL-4 is a pleiotropic cytokine that regulates a variety of cellular functions including Th2 differentiation (3), further in vivo studies will be needed to verify that IL-4R/Stat6 signaling-mediated VLA-4 down-regulation in CD8+ T cells contributes to the anti-inflammatory actions associated with IL-4 administration.
Data from our group and others indicate that Type-1 T cell responses are favorable for antitumor immunity (17–19), with cancer cells frequently being found to secrete numerous Type-2 cytokines (20–22). Type-2 cytokines secreted from tumor cells support tumor cell proliferation (22, 23), immune escape (24, 25), and the skewing of T cell response toward the Type-2 (25–27), that is commonly correlated with poor prognosis in cancer patients (28, 29). In this study, we have demonstrated that the ability of adoptively transferred Type-2 CD8+ T cells (developed in WT mice) to traffic into s.c. tumor lesions was markedly impaired (in a Stat6-dependent manner) when compared with that of Type-1 CD8+ T cells. In this experiment, we used bulk (i.e., not tumor-specific) T cells in our adoptive transfers. We feel that this is an appropriate model for the analysis of acute infiltration events. Although the ability of T cells to migrate throughout tumor lesions has been shown to exhibit a dependency on “signal 1” (Ag-specificity; Ref. 1, 30), the early stages of T cell infiltration into tumors (i.e., transmigration through tumor blood vessels into the stroma) is believed to be largely independent of Ag specificity (31–33).
Numerous studies have shown that Stat6−/− mice exhibit enhanced immunity against tumors and are inherently resistant to the establishment/development of various types of tumors (34). When considered in the context of our current work, we believe that IL-4 produced within the tumor microenvironment (or systemically) may reduce expression of VLA-4 on CD8+ T effector cells, thus preventing these T cells from being able to be recruited into tumor sites in vivo. Hence, it would interesting to know whether VLA-4-expressing Tc1 cells generated in vitro are also susceptible to alterations in their tissue trafficking capacities based on exposure to IL-4 and STAT6 activation in vivo after adoptive transfer. Indeed, as discussed above, we have recently noted that committed Tc1 cells remain susceptible to IL-4-mediated VLA-4 down-regulation in vitro (our unpublished data). Under such conditions, the antagonistic targeting of Stat6 signaling in T cells in vivo (or ex vivo when considered in the context of adoptive therapy approaches) would be predicted to enhance the trafficking (and antitumor efficacy) of polarized Type-1, tumor-specific CD8+ T cells into tumor lesions in situ. Moreover, Stat6-deficient CD8+ T cells have been shown to produce elevated levels of IFN-γ (35), a critical Type-1 mediator of therapeutic antitumor immune responses (36), and to mediate enhanced antitumor cytotoxic functions on a cell-per-cell basis when compared with control Tc1 cells (37). Overall, these observations suggest that (pharmacologic) antagonism of Stat6 activation would likely improve the antitumor action of specific Tc1 cells at multiple levels, thereby enhancing therapeutic benefit. Clearly, further in vivo studies designed to elucidate and counteract the regulatory effects of the IL-4R/Stat6 signaling axis on the therapeutic efficacy of Tc1 (endogenous or adoptively transferred) are well-warranted.
Footnotes
This work was supported by National Institutes of Health Grants P01 CA100327 and R01 CA63350 (to W.J.S.), and R01 NS055140 (to H.O.).
Abbreviations used in this paper: IRS, insulin receptor substrate; WT, wild type; TIL, tumor-infiltrating lymphocyte.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Nishimura F, Dusak JE, Eguchi J, Zhu X, Gambotto A, Storkus WJ, Okada H. Adoptive transfer of type 1 CTL mediates effective anti-central nervous system tumor response: critical roles of IFN-inducible protein-10. Cancer Res. 2006;66:4478–4487. doi: 10.1158/0008-5472.CAN-05-3825. [DOI] [PubMed] [Google Scholar]
- 2.Sasaki K, Zhu X, Vasquez C, Nishimura F, Dusak JE, Huang J, Fujita M, Wesa A, Potter DM, Walker PR, et al. Preferential expression of very late antigen-4 on type 1 CTL cells plays a critical role in trafficking into central nervous system tumors. Cancer Res. 2007;67:6451–6458. doi: 10.1158/0008-5472.CAN-06-3280. [DOI] [PubMed] [Google Scholar]
- 3.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 4.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol. 2000;105:1063–1070. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- 5.Bruns HA, Schindler U, Kaplan MH. Expression of a constitutively active Stat6 in vivo alters lymphocyte homeostasis with distinct effects in T and B cells. J Immunol. 2003;170:3478–3487. doi: 10.4049/jimmunol.170.7.3478. [DOI] [PubMed] [Google Scholar]
- 6.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 8.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 9.Ho IC, Lo D, Glimcher LH. c-maf promotes T helper cell Type 2 (Th2) and attenuates Th1 differentiation by both interleukin 4-dependent and -independent mechanisms. J Exp Med. 1998;188:1859–1866. doi: 10.1084/jem.188.10.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chisholm PL, Williams CA, Lobb RR. Monoclonal antibodies to the integrin α-4 subunit inhibit the murine contact hypersensitivity response. Eur J Immunol. 1993;23:682–688. doi: 10.1002/eji.1830230317. [DOI] [PubMed] [Google Scholar]
- 11.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of α4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4 β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 13.Christensen JP, Andersson EC, Scheynius A, Marker O, Thomsen AR. α4 integrin directs virus-activated CD8+ T cells to sites of infection. J Immunol. 1995;154:5293–5301. [PubMed] [Google Scholar]
- 14.Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finnegan A, Grusby MJ, Kaplan CD, O’Neill SK, Eibel H, Koreny T, Czipri M, Mikecz K, Zhang J. IL-4 and IL-12 regulate proteoglycan-induced arthritis through Stat-dependent mechanisms. J Immunol. 2002;169:3345–3352. doi: 10.4049/jimmunol.169.6.3345. [DOI] [PubMed] [Google Scholar]
- 16.Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, van der Zee R, Biedermann T, Prinz J, Mack M, et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. 2003;9:40–46. doi: 10.1038/nm804. [DOI] [PubMed] [Google Scholar]
- 17.Kemp RA, Ronchese F. Tumor-specific Tc1, but not Tc2, cells deliver protective antitumor immunity. J Immunol. 2001;167:6497–6502. doi: 10.4049/jimmunol.167.11.6497. [DOI] [PubMed] [Google Scholar]
- 18.Hu HM, Urba WJ, Fox BA. Gene-modified tumor vaccine with therapeutic potential shifts tumor-specific T cell response from a Type 2 to a Type 1 cytokine profile. J Immunol. 1998;161:3033–3041. [PubMed] [Google Scholar]
- 19.Shurin MR, Lu L, Kalinski P, Stewart-Akers AM, Lotze MT. Th1/Th2 balance in cancer, transplantation and pregnancy. Springer Semin Immunopathol. 1999;21:339–359. doi: 10.1007/BF00812261. [DOI] [PubMed] [Google Scholar]
- 20.Bellone G, Turletti A, Artusio E, Mareschi K, Carbone A, Tibaudi D, Robecchi A, Emanuelli G, Rodeck U. Tumor-associated transforming growth factor-β and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155:537–547. doi: 10.1016/s0002-9440(10)65149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang M, Wang J, Lee P, Sharma S, Mao JT, Meissner H, Uyemura K, Modlin R, Wollman J, Dubinett SM. Human non-small cell lung cancer cells express a Type 2 cytokine pattern. Cancer Res. 1995;55:3847–3853. [PubMed] [Google Scholar]
- 22.Prokopchuk O, Liu Y, Henne-Bruns D, Kornmann M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: evidence for autocrine and paracrine actions. Br J Cancer. 2005;92:921–928. doi: 10.1038/sj.bjc.6602416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 24.Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, Kuniyasu Y, Nomura T, Toda M, Takahashi T. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 25.Seo N, Hayakawa S, Takigawa M, Tokura Y. Interleukin-10 expressed at early tumour sites induces subsequent generation of CD4+ T-regulatory cells and systemic collapse of antitumour immunity. Immunology. 2001;103:449–457. doi: 10.1046/j.1365-2567.2001.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tatsumi T, Herrem CJ, Olson WC, Finke JH, Bukowski RM, Kinch MS, Ranieri E, Storkus WJ. Disease stage variation in CD4+ and CD8+ T-cell reactivity to the receptor tyrosine kinase EphA2 in patients with renal cell carcinoma. Cancer Res. 2003;63:4481–4489. [PubMed] [Google Scholar]
- 27.Tatsumi T, Kierstead LS, Ranieri E, Gesualdo L, Schena FP, Finke JH, Bukowski RM, Mueller-Berghaus J, Kirkwood JM, Kwok WW, Storkus WJ. Disease-associated bias in T helper Type 1 (Th1)/Th2 CD4+ T cell responses against MAGE-6 in HLA-DRB10401+ patients with renal cell carcinoma or melanoma. J Exp Med. 2002;196:619–628. doi: 10.1084/jem.20012142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito N, Nakamura H, Tanaka Y, Ohgi S. Lung carcinoma: analysis of T helper Type 1 and 2 cells and T cytotoxic Type 1 and 2 cells by intracellular cytokine detection with flow cytometry. Cancer. 1999;85:2359–2367. [PubMed] [Google Scholar]
- 29.Goto S, Sato M, Kaneko R, Itoh M, Sato S, Takeuchi S. Analysis of Th1 and Th2 cytokine production by peripheral blood mononuclear cells as a parameter of immunological dysfunction in advanced cancer patients. Cancer Immunol Immunother. 1999;48:435–442. doi: 10.1007/s002620050620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med. 2007;204:345–356. doi: 10.1084/jem.20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kjaergaard J, Shu S. Tumor infiltration by adoptively transferred T cells is independent of immunologic specificity but requires down-regulation of L-selectin expression. J Immunol. 1999;163:751–759. [PubMed] [Google Scholar]
- 32.Joncker NT, Marloie MA, Chernysheva A, Lonchay C, Cuff S, Klijanienko J, Sigal-Zafrani B, Vincent-Salomon A, Sastre X, Lantz O. Antigen-independent accumulation of activated effector/memory T lymphocytes into human and murine tumors. Int J Cancer. 2006;118:1205–1214. doi: 10.1002/ijc.21472. [DOI] [PubMed] [Google Scholar]
- 33.Palmer DC, Balasubramaniam S, Hanada K, Wrzesinski C, Yu Z, Farid S, Theoret MR, Hwang LN, Klebanoff CA, Gattinoni L, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ostrand-Rosenberg S, Sinha P, Clements V, Dissanayake SI, Miller S, Davis C, Danna E. Signal transducer and activator of transcription 6 (Stat6) and CD1: inhibitors of immunosurveillance against primary tumors and metastatic disease. Cancer Immunol Immunother. 2004;53:86–91. doi: 10.1007/s00262-003-0446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaplan MH, Wurster AL, Smiley ST, Grusby MJ. Stat6-dependent and -independent pathways for IL-4 production. J Immunol. 1999;163:6536–6540. [PubMed] [Google Scholar]
- 36.Dobrzanski MJ, Reome JB, Hollenbaugh JA, Dutton RW. Tc1 and Tc2 effector cell therapy elicit long-term tumor immunity by contrasting mechanisms that result in complementary endogenous Type 1 antitumor responses. J Immunol. 2004;172:1380–1390. doi: 10.4049/jimmunol.172.3.1380. [DOI] [PubMed] [Google Scholar]
- 37.Kachan AK, Fallarino F, Markiewicz MA, Gajewski TF. Cutting edge: spontaneous rejection of poorly immunogenic P1.HTR tumors by Stat6-deficient mice. J Immunol. 2000;165:6024–6028. doi: 10.4049/jimmunol.165.11.6024. [DOI] [PubMed] [Google Scholar]