

Abstract
Innate immune recognition of intracellular pathogens involves both extracellular and cytosolic surveillance mechanisms. The intracellular protozoan parasite Trypanosoma cruzi triggers a robust type I IFN response in both immune and nonimmune cell types. In this study, we report that signaling through TBK1 and IFN regulatory factor 3 is required for T. cruzi-mediated expression of IFN-β. The TLR adaptors MyD88 and TRIF, as well as TLR4 and TLR3, were found to be dispensable, demonstrating that T. cruzi induces IFN-β expression in a TLR-independent manner. The potential role for cytosolic dsRNA sensing pathways acting through RIG-I and MDA5 was ruled out because T. cruzi was shown to trigger robust expression of IFN-β in macrophages lacking the MAVS/IPS1/VISA/CARDif adaptor protein. The failure of T. cruzi to activate HEK293-IFN-β-luciferase cells, which are highly sensitive to cytosolic triggers of IFN-β expression including Listeria, Sendai virus, and transfected dsRNA and dsDNA, further indicates that the parasite does not engage currently recognized cytosolic surveillance pathways. Together, these findings identify the existence of a novel TLR-independent pathogen-sensing mechanism in immune and nonimmune cells that converges on TBK1 and IFN regulatory factor 3 for activation of IFN-β gene expression.
Type I IFNs, traditionally regarded as mediators of antiviral defense, have recently emerged as important immuno-modulatory cytokines that influence disease progression in a variety of nonviral pathogen infections (1). Pathogen triggers of type I IFN expression include those that engage cytosolic surveillance molecules (2) and those that activate TLRs, well-described pattern recognition receptors capable of recognizing a diverse range of pathogen products and nucleic acids (3, 4). TLRs that signal type I IFN expression include TLRs 3, 4, 7, and 9 and utilize the adaptor proteins TRIF and/or MyD88 (reviewed in Ref. 4). Nucleic acids in the host cytoplasm can also trigger type I IFN expression in a TLR-independent manner. Triphosphate RNA and dsRNA are recognized by the RIG-like receptors, a family of RNA helicases that include retinoic acid-inducible gene (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) (5, 6). RIG-I and MDA5 both signal through the adaptor molecule MAVS (also called IPS1/VISA/CARDif) (7–10). Cytosolic DNA can also induce type I IFNs in a TLR-independent manner (11–13). A candidate sensor DNA-dependent activator of IFN regulatory factors (IRFs)3 (DAI) has been identified; however, gene targeting of DAI suggests it may not function as a cytosolic DNA receptor upstream of type I IFN expression (14–16). Cytosolic bacteria such as Listeria monocytogenes stimulate type I IFNs independently of TLRs and RIG-like receptors (17, 18) by a mechanism that requires the pore-forming molecule listeriolysin O to escape from the vacuole and enter the host cell cytosol, but the receptor for this pathway has not yet been identified (2, 13).
Members of the IRF family of transcription factors are central regulators of the type I IFN response (19). IRF3, which is sufficient to drive IFN-β expression, is phosphorylated on C-terminal serine and threonine residues by the noncanonical IκB kinases TANK-binding kinase 1 (TBK1) or inducible IKKε, which leads to the formation of IRF3 dimers, nuclear translocation, and binding to the IFN-β promoter (20–22). Secreted IFN-β binds to the type I IFN receptor (IFNAR), signaling an autocrine or paracrine response, resulting in the expression of a multitude of IFN-stimulated genes (ISGs) as well as additional IFN-β (23).
The obligate intracellular protozoan pathogen Trypanosoma cruzi causes human Chagas’ disease, a leading cause of heart failure in Latin America. To initiate infection, T. cruzi trypomastigotes activate a number of host cell signaling pathways, resulting in plasma membrane invagination and entry into the endocytic pathway or targeted exocytosis of host cell lysosomes at the parasite attachment site (reviewed in Ref. 24). Consequently, invading parasites are rapidly trafficked to the host cell lysosomal compartment where they transiently reside before taking up residence in the host cell cytoplasm (25). Similar to Listeria, T. cruzi secretes a pore-forming molecule into the vacuole, which is thought to facilitate egress into the host cell cytosol (26). During the course of infection, T. cruzi triggers a strong type I IFN response in a variety of infected cell types (27–29) and in vivo at the site of intradermal infection of mice (A.-D. C. Chessler, M. Unnikrishnan, and B. A. Burleigh, manuscript in preparation), however, the mechanistic basis for T. cruzi-mediated induction of IFN-β is not well understood. In addition there is some indication that the type I IFN response influences the outcome of T. cruzi infection in vivo (28–30). In this study, we present evidence that T. cruzi induces expression of IFN-β in macrophages and fibroblasts independently of TLRs and known cytosolic sensors of nucleic acids with kinetics that suggest that IFN-β induction occurs before vacuole egress.
Materials and Methods
Cell culture and pathogens
IRF3−/−, TRIF−/−, MyD88−/−, and TLR3−/− mouse embryonic fibroblasts (MEF) and matched WT controls, HEK293 IFN-β-luc reporter cells, and HEK293-tlr3 cells were from K. Fitzgerald (University of Massachusetts, Worcester, MA). TBK1−/− and matched WT MEF were provided by W.-C. Yeh (University of Toronto, Toronto, Ontario, Canada) and MyD88−/−, MyD88−/−/TIRAP−/−, and TLR2−/−/TLR4−/− MEF and matched WT controls were from R. Medzhitov (Yale University, New Haven, CT). MEF, HEK293-tlr3, or HEK293-IFN-β-luc cells were maintained in DMEM containing 4.5 g/L glucose and 110 mg/L sodium pyruvate (Life Technologies 10313) supplemented with 10% FBS, 2 mM glutamine, and 100 μg/ml penicillin-streptomycin (Pen-Strep; DMEM-10).
Bone marrow-derived macrophages (BMDM) and MEFs were generated from WT, IRF3−/−, TLR3−/−, TLR4−/−, TRIF−/−, MyD88−/−, MAVS−/−, and C57BL/6 mice as previously described (31). IRF3−/− mice were a gift from T. Taniguchi (University of Tokyo, Tokyo, Japan). MyD88−/−/TRIF−/− mice were bred from MyD88−/− (32) and TRIF−/− (33) mice provided by S. Akira (Osaka University, Osaka, Japan). TLR3−/− and TLR4−/− mice were from S. Akira (Osaka University, Osaka, Japan). MAVS−/− mice were a gift from Z. Chen (University of Texas Southwestern, Dallas, TX). Mice were bred and maintained at the University of Massachusetts Medical School and all experiments were conducted with Institutional Animal Care and Use Committee approval.
BMDM were flushed from femurs and cultured in 100-mm-diameter petri dishes for 6 days in BMDM medium (DMEM supplemented with 20% FBS, 2 mM glutamine, 1 mM sodium pyruvate, 100 μg/ml Pen-Strep, 55 μM 2-ME, and 30% L929 cell supernatant).
MEF, HEK293-tlr3, or HEK293-IFN-β-luc cells were maintained in DMEM-10. Tissue culture-derived T. cruzi trypomastigotes (Y strain) were generated as previously described (34) and washed twice in DMEM containing 4.5 g/L glucose and 110 mg/L sodium pyruvate (Life Technologies 10313) supplemented with 2% FBS, 2 mM glutamine, and 100 μg/ml Pen-Strep (DMEN-2) before cell infection. Aldehyde-fixed parasites were fixed in 2% paraformaldehyde for more than 24 h and washed twice in DMEM-2 before use. Epimastigotes from the T. cruzi Y strain were maintained in liver infusion tryptose medium containing 10% FBS at 28°C (35) and washed twice in DMEM-2 before incubation with mammalian cells. Parasites were heat killed for 10 min at 56°C and washed twice in DMEM-2 before use. Overnight cultures in Luria-Bertani medium of Listeria monocytogenes strain 10403S were provided by D. Higgins (Harvard University, Boston, MA) and cells were infected with a multiplicity of infection of five bacteria per cell for 1 h in DMEM-10 without Pen-Strep, then medium was removed and fresh medium containing 50 μg/ml gentamicin was added.
Cell infection
Semiconfluent monolayers grown in 6-well plates were incubated with 5 × 107 parasites/well for 2 h at 37°C in 5% CO2. Remaining extracellular parasites were removed by washing cells three times with PBS and DMEM-2 added in the presence or absence of bafilomycin A (50 nM; Sigma-Aldrich) as indicated and incubated for the indicated total infection times.
Western blot analysis
Infected or mock-treated control cells were washed three times in ice-cold PBS and then lysed in 100 μl of 95°C 2× Laemmli sample buffer. Lysates were separated on a 10% polyacrylamide gel and blotted onto polyvinylidene difluoride membrane (Millipore). Membranes were blocked with TBS/5% milk and probed with rabbit anti-human phospho-IRF3 Ser396 (a gift from J. Hiscott, McGill University, Montreal, Quebec, Canada) or rabbit anti-human IRF3 (Santa Cruz Biotechnology) at 4°C overnight. After washing, bound Abs were detected with anti-rabbit IgG coupled to HRP (1/10,000) at room temperature for 1 h. The signal was visualized by using ECL (Amersham Pharmacia Biosciences). A representative blot of three independent experiments is shown.
Immunofluorescence
Glass coverslips (12 mm2) with adherent cells were fixed in 2% paraformaldehyde/PBS, washed with PBS, and incubated for 10 min in 50 mM NH4Cl/PBS. Ab dilutions, incubations, and washes were conducted in TBS/BSA (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 1% BSA) for staining of external Ags or permeabilized with TBS/BSA/0.1% saponin to detect intracellular Ags. T. cruzi immunostaining was performed with a rabbit anti-T. cruzi Ab (1/1500) (36) and Lamp-1 staining was performed with mouse anti-rat lamp-1 mAb LY1C6 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Secondary Ab conjugates used were Cy5-labeled goat anti-rabbit IgG (Amersham Pharmacia) and TR-conjugated anti-mouse (Molecular Probes). Ab incubations were conducted for 40 min with extensive washes for 25 min following primary and secondary incubation steps. Parasite and mammalian DNA was stained with 1.25 μg/ml 4′,6-diamidino-2-phenylindole (Pierce) for 3 min. Coverslips were washed with PBS and mounted in 10% Mowiol (Calbiochem) containing 2.5% 1,4-diazobicyclo-[2,2,2]-octane). Fluorescence images were obtained with a Nikon TE-300 inverted epifluorescence microscope equipped with an Orca-100 CCD camera (Hamamatsu) and analyzed using MetaMorph imaging software (Universal Imaging). For quantitative analysis of infection, internal parasites per cell were counted in a minimum of 100 cells/coverslip.
RT-PCR and real-time-PCR
Total RNA was harvested at the time points indicated with a Qiagen RNeasy Mini Kit. The Superscript II RT Kit (Invitrogen) was used for cDNA synthesis. cDNA templates were used for a quantitative real-time PCR using an Applied Biosystems 7000 or Applied Biosystems 7300 real-time PCR machine. The relative level of gene expression was determined by the comparative threshold cycle method as described by the manufacturer, whereby data for each sample were normalized to gapdh and expressed as a fold change compared with untreated or uninfected controls. Primers and probes for human ifnb used were as follows: 5′-GACCAA CAAGTGTCTCCTCCAAA-3′ (forward), 5′-AGCAAGTTGTAGCTC ATGGAAAGAG-3′ (reverse), and 5′-6FAM-TCTCCTGTTGTGCTT CTCCACGA-3′ TAMRA (probe). The following TaqMan gene expression assays were used: mouse ifnb (Mm00439546_s1), human gapdh (4310884E), mouse gapdh (4308313) human il6 (Hs00174131_m1), mouse il6 (Mm00446190_m1), mouse cxcl10 (Mm99999072_m1), mouse viperin (Mm00491265_m1) (Applied Biosystems). For quantification of mouse lrg47 SYBR Green PCR Master Mix (Applied Biosystems) was used with the following primers: 5′-TGAGCTCAGCCTTCCCCTTT-3′ (forward) and 5′-TGGGACAATGTTGCCACAGT-3′ (reverse).
Quantification of luciferase expression
Quantification of luciferase expression was performed with the dual-luciferase reporter assay system (Promega) according to the manufacturer’s protocols using supplied reagents. Briefly, 24 h after treatment, cells were washed with PBS and lysed with 350 μl/well passive lysis buffer and 20 μl of sample was added to 100 μl of luciferase assay substrate in luciferase assay buffer II in a luminometer tube. Luciferase activity was measured using an AutoLumat LB953 luminometer (EG&G Berthold).
Statistics
Samples were compared using a two-tailed, unpaired t test with Welch’s correction; p < 0.05 were considered to be significant.
Results
T. cruzi induces a delayed IFN-β response in immune and nonimmune cells
T. cruzi infection of isolated human foreskin fibroblasts is known to trigger robust up-regulation of a group of ISGs by 24 h after infection, correlating with synthesis and secretion of IFN-β from infected cells (27). In this study, we examine the kinetics of IFN-β mRNA expression in T. cruzi-infected MEF and BMDM by quantitative RT-PCR (qRT-PCR; Fig. 1). As compared with infection with Sendai virus, which strongly induces ifnb in both fibroblasts and macrophages with peak responses between 4 and 8 h after infection (Fig. 1, A and B, second panels), the peak response to T. cruzi was significantly delayed (Fig. 1, A and B, first panels). In T. cruzi-infected fibroblasts, ifnb transcript levels gradually increased over the first 12 h of infection, followed by a dramatic rise in ifnb expression, peaking at 16 h after infection (Fig. 1A). A similar trend was observed in BMDM, although parasites triggered an initial peak of ifnb expression at 4 h after infection, followed by a large peak at 16 h (Fig. 1B).
FIGURE 1.

Kinetics of ifnb and ISG induction in T. cruzi-infected cells. Kinetics of IFN-β mRNA induction in WT or IFNAR−/− MEF (a) and BMDM (b) in response to T. cruzi or Sendai virus infection as determined by qRT-PCR. Induction of the ISGs cxcl10, viperin, and lrg47 in response to 16 h of T. cruzi infection was confirmed in BMDM and MEF by qRT-PCR (c). Values reported represent the mean fold induction relative to mock-infected controls.
T. cruzi infection of IFNAR−/− MEF revealed the absence of the 16-h peak of ifnb expression observed in WT MEF that corresponds to the IFN-dependent autocrine activation loop (Fig. 1A, third panel). In the absence of this loop, it is clear that the parasite-driven ifnb response increases steadily and peaks at ~12 h after infection (Fig. 1B, third panel). Overall, these findings demonstrate that primary signals elicited by T. cruzi in fibroblasts and professional phagocytes trigger induction of ifnb expression with strikingly different kinetics as compared with virus-infected cells. In addition, IFNAR-dependent amplification of ifnb expression, as well as the induction of the ISGs cxcl10, viperin, and lrg47 (Fig. 1C) is consistent with the production and secretion of active IFN-β from T. cruzi-infected cells, as previously reported (27).
Following T. cruzi entry into cells, parasites reside in a lysosomal vacuole (36, 37) for several hours before taking up residence in the host cell cytoplasm (25). We demonstrate that cytoplasmic localization of T. cruzi is a gradual process where loss of Lamp-1 staining of parasite vacuoles begins at ~8 h and is complete by 16 h after entry (Fig. 2, A and B). Electron microscopic examination of intracellular T. cruzi over this time course demonstrates that loss of Lamp-1 correlates with loss of vacuole membrane (data not shown). Given that the initial induction of IFN-β mRNA in T. cruzi-infected cells precedes onset of egress from the parasitophorous vacuole, these data suggest that the trigger for ifnb expression is not linked to cytosolic localization of the parasite. Further support for this was obtained using bafilomycin A to block T. cruzi exit from the vacuole (25). Although bafilomycin A treatment exerted a strong inhibitory effect on T. cruzi vacuole egress (Fig. 2B), drug treatment did not block the initial rise in ifnb expression observed over the first 8 h of infection. Instead, bafilomycin A treatment attenuated ifnb expression at time points >10 h after infection, suggesting an impact on the autocrine activation loop, perhaps due to inhibition of IFNAR signaling from an endosomal compartment. Together, these data demonstrate that induction of ifnb by T. cruzi occurs before cytosolic localization and is likely to be initiated via extracellular receptors or from within the enodosomal compartment in a manner that is not pH dependent.
FIGURE 2.

Intracellular localization is not required for ifnb induction in T. cruzi-infected cells. Kinetics of egress from the parasitophorous vacuole was examined by immunofluorescence staining (A). Extracellular parasites were stained with T. cruzi-specific Abs (green), vacuole membrane with Lamp-1 (red), host and parasite DNA (blue) in T. cruzi-infected MEF at 4, 8, 16, and 24 h after infection. A subset of parasites in Lamp-1-positive vacuoles is highlighted by arrowheads and arrows are used to indicate cytosolic parasites. Fibroblasts treated with (gray triangles) or without (black squares) 50 nM bafilomycin A (BafA) were infected with T. cruzi and the percentage of Lamp-1-positive parasites (B, first panel) or IFN-β mRNA (B, second panel) was quantitated. MEF were exposed to infective T. cruzi trypomastigotes (live trypos), heat-killed trypomastigotes (hk), aldehyde-fixed trypomastigotes (fixed), parasite-conditioned medium (pcm), live noninfective T. cruzi epimastigotes (epi), and heat-killed epimastigotes (hk epi) to induce ifnb. Transcription was determined by qRT-PCR at 6 and 16 h after infection (C). Values reported represent the mean fold induction relative to mock-infected controls ± SD.
To determine whether the ability of T. cruzi to trigger ifnb expression is strictly associated with intracellular infection, MEF were exposed to live or heat-killed trypomastigotes, the invasive life cycle stage, or epimastigotes, the insect stage of the parasite that does not infect mammalian cells. Heat-killed trypomastigotes were found to elicit strong induction of ifnb with more rapid kinetics than the response to live parasites (Fig. 2C). In contrast, a minimal IFN response to noninfective live or heat-killed epimastigotes was observed at both 6 and 16 h after treatment (Fig. 2C). These findings indicate that the ability of T. cruzi to trigger ifnb expression in mammalian cells is associated with the infective forms of the parasite, but that active invasion by live trypomastigotes is not necessary to trigger the response. Interestingly, neither aldehyde-fixed trypomastigotes or secreted/released trypo-mastigote-associated material present in “parasite-conditioned medium” triggers ifnb expression in MEF. Presently, it is not known whether live and heat-killed trypomastigotes trigger ifnb expression by engaging the same “receptors”/signaling pathways or whether the heat-killing process exposes a different set of ligands that causes this response.
TBK1 and IRF3 are required for T. cruzi-dependent induction of IFN-β
Transcriptional activation of the IFN-β promoter by IRF3 is central to IFN-β expression triggered by a diverse set of signals. Induction of ifnb in response to T. cruzi was completely abolished in IRF3−/− and TBK1−/− fibroblasts (Fig. 3A) as well as in IRF3−/− macrophages (Fig. 3B), confirming the critical role of this signaling axis in T. cruzi-dependent IFN-β expression. Comparable infection levels were observed in WT cells and IRF3- or TBK1-deficient MEF and no differences were observed in the kinetics of vacuole egress by T. cruzi in these cells (data not shown). Activation of IRF3 involves phosphorylation of multiple serine and threonine residues located in the C terminus, including Ser396 which can be phosphorylated by TBK1 (38, 39). Using an Ab specific for phospho-Ser396, we demonstrate elevated levels of phospho-IRF3 in T. cruzi-infected fibroblasts at 2 h after infection (Fig. 3C). The observation that IRF3 is phosphorylated at this early time point is consistent with our findings that T. cruzi triggers for IFN-β expression by engaging a signaling pathway that requires both TBK1 and IRF3 before cytosolic localization of the parasite.
FIGURE 3.

TBK1 and IRF3 are required for T. cruzi-induced IFN-β expression. ifnb transcript levels were measured by qRT-PCR in WT and IRF-3−/− or TBK1−/− MEF (A) and in IRF3−/− BMDM (B) following infection with T. cruzi for 6 and 16 h (MEF) or 6 h (BMDM). Values reported represent the mean fold induction relative to mock-infected controls ± SD. C, IRF3 is phosphorylated (Ser396) following a 2-h exposure of MEF to live T. cruzi trypomastigotes.
T. cruzi induction of IFN-β is independent of TLR signaling
A variety of pathogen-associated molecular patterns can activate cells, triggering expression of type I IFNs through TLR-dependent and -independent pathways that converge on TBK1 and IRF3. In this study, we tested the ability of T. cruzi to induce ifnb expression in macrophages and fibroblasts that lack expression of the TLR adaptors MyD88 and TRIF. We find that T. cruzi-induced ifnb expression is not abrogated in MEF lacking MyD88 (Fig. 4A) or in MyD88−/− and TRIF−/− BMDM (Fig. 4B). As a control, IL-6 mRNA levels were measured in WT, MyD88−/−, and TRIF−/− BMDM following infection with T. cruzi, which triggers proinflammatory cytokine expression in a MyD88-dependent manner (Fig. 4C) (40). As expected, the IL-6 response triggered by T. cruzi or by stimulation with the TLR2 ligand, Pam2Csk4, is attenuated in MyD88−/− macrophages (Fig. 4C) and TRIF−/− BMDM fail to respond to exogenous poly(I:C) (Fig. 4C). Robust induction of il6 (Fig. 4C) and ifnb (Fig. 4B) was observed in response to T. cruzi in TRIF-deficient cells. In fact, parasite-induced expression of ifnb and il6 was ~3- to 5-fold higher in TRIF−/− BMDM when compared with WT cells (Fig. 4, B and C). Although the significance of the increased induction of ifnb in TRIF−/− BMDM is unknown, these data clearly indicate that the TLR adaptors TRIF and MyD88 are not required for ifnb induction in response to T. cruzi. This assertion is further confirmed by demonstrating that T. cruzi-dependent induction of downstream ISGs such as cxcl10, viperin, and lrg47 occurs in MyD88/TRIF double knockout BMDM (Fig. 4D). Finally, we demonstrated that both live and heat-killed T. cruzi trypomastigotes trigger expression of ifnb in a TLR-independent manner (Fig. 4E), suggesting that a similar signaling pathway is engaged by live and heat-killed parasites.
FIGURE 4.

MyD88 and TRIF are not required for T. cruzi induction of IFN-β. Relative IFN-β mRNA levels were measured by qRT-PCR in T. cruzi-infected WT, MyD88−/−, and MyD88−/−/TIRAP−/− MEF 6 and/or 16 h after infection (A) or WT, MyD88−/−, and TRIF−/− BMDM 6 h after infection (B). Relative IL-6 mRNA levels were measured by qRT-PCR in WT, MyD88−/−, and TRIF−/− BMDM 6 h after infection with T. cruzi or treatment with Pam2Csk4 (PAM) or poly(I:C) (C). Relative induction of ifnb by live parasites or heat-killed T. cruzi trypomastigotes (D) and of the ISGs genes cxcl10, viperin, and lrg47 by live T. cruzi trypomastigotes (E) was determined by qRT-PCR in WT or MyD88−/−/TRIF−/− BMDM 16 h after exposure. Values reported represent the mean fold induction relative to mock-infected controls ± SD.
Given that these data contradict a recent report indicating that T. cruzi activates expression of ifnb in BMDM in a TRIF-dependent manner (28), we investigated the role of TLR3 and TLR4, which are the only TLRs known to signal through TRIF to activate IFN-β gene expression (41). Consistent with data presented in Fig. 4, the absence of TLR3 or TLR4 does not inhibit T. cruzi-triggered ifnb expression in BMDM (Fig. 5A) or MEF (Fig. 5, B and C). Because a decrease in ifnb expression was observed in T. cruzi-infected TLR3-deficient MEF that approaches significance (p > 0.09; Fig. 5C), we sought to further test the potential role of TLR3 in T. cruzi-mediated ifnb expression. HEK293 stably expressing TLR3 become responsive to exogenous poly(I:C); however, T. cruzi failed to elicit ifnb expression in these cells (Fig. 5D). Together, these data exclude known MyD88- and TRIF-dependent TLR signaling pathways and directly rule out a role for TLR3 and TLR4 as mediators of ifnb expression in response to T. cruzi. The data further suggest that T. cruzi may interact with an uncharacterized extracellular receptor or one of a growing number of putative cytosolic surveillance molecules to activate IFN-β.
FIGURE 5.
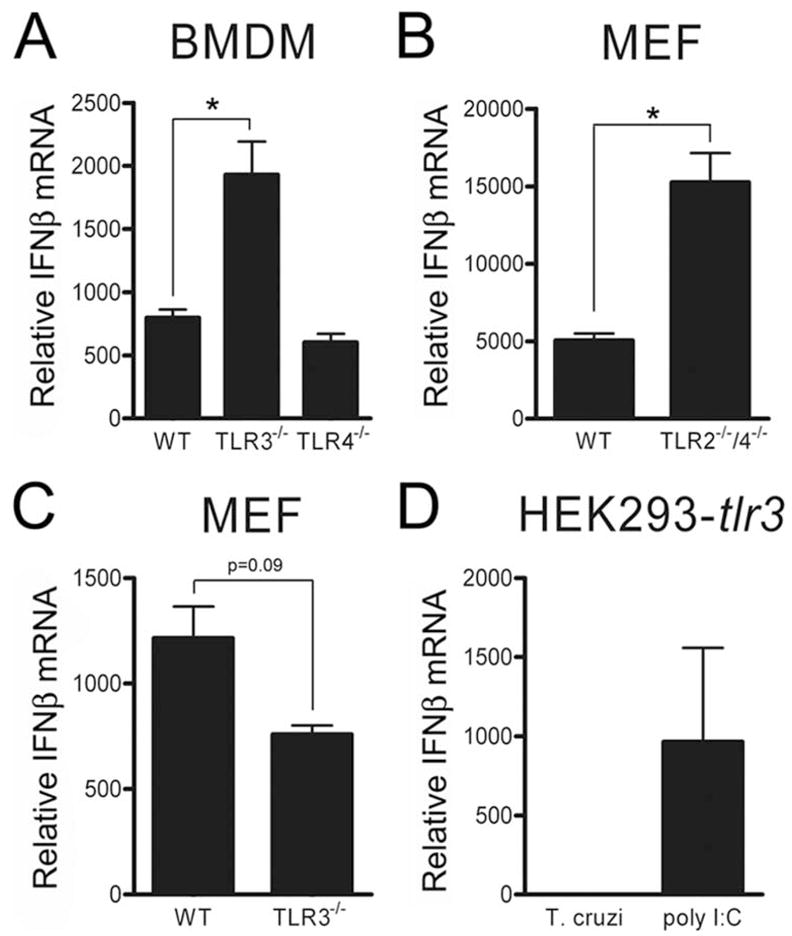
TLR3 and TLR4 are not required for T. cruzi induction of ifnb. Relative ifnb transcript abundance was measured by qRT-PCR in WT, TLR3−/−, and TLR4−/− BMDM (A), WT and TLR2−/−/TLR4−/− MEF (B), or WT and TLR3−/− MEF (C) following infection with T. cruzi for 16 h (MEF) or 6 h (BMDM). D, HEK293 cells stably expressing TLR3 HEK293-tlr3 were infected with T. cruzi or activated with the TLR3 ligand poly(I:C) for 16 h and relative IFN-β mRNA levels were measured by qRT-PCR. Values reported represent the mean fold induction relative to mock-infected controls ± SD.
T. cruzi fails to induce IFN-β expression in HEK293 cells that respond to other cytosolic pathogen triggers
HEK293 cells exhibit negligible TLR expression but remain responsive to a variety of cytosolic bacterial, viral, and nucleic acid triggers of IFN-β. Using HEK293-IFN-β-luc reporter cells, we were able to show that cytosolic delivery of dsRNA (poly(I:C)), or dsDNA (poly(dA:dT)) by lipofection lead to robust luciferase reporter activation (Fig. 6A). Similarly, infection of HEK293-IFN-β-luc cells with Sendai virus or L. monocytogenes caused reporter activation within 6 h (data not shown), with high levels of luciferase expression observed at 24 h (Fig. 6A). In stark contrast, T. cruzi failed to activate reporter expression at 6 h (data not shown) or 24 h after infection (Fig. 6A). To exclude the possibility that parasite infection interferes with the ability to assay luciferase activity, we also examined endogenous IFN-β mRNA by qRT-PCR and found that T. cruzi failed to induce ifnb gene expression in this cell type (Fig. 6B) despite achieving a high level of intracellular infection (data not shown). These data suggest that induction of ifnb expression by T. cruzi does not occur through the characterized dsRNA-sensing pathways involving RIG-I/MDA5 or through the less characterized dsDNA-sensing pathway. To directly test the role of dsRNA- sensing RIG-like receptors in the IFN response to T. cruzi, MAVS-deficient BMDM were infected and shown to mount a response that was similar to WT macrophages (Fig. 6C). Given that HEK293 cells responded to both cytosolic dA:dT and Listeria infection, which may be triggered by bacterial DNA in the host cell cytoplasm (13), our data strongly suggest that the previously described cytosolic DNA-sensing mechanisms do not play a role in eliciting a type I IFN response to T. cruzi. However, it is possible that T. cruzi DNA is recognized by a DNA recognition molecule/pathway that is not expressed in HEK cells. Coupled with data showing that known TLR-dependent pathways are not required for T. cruzi-induced ifnb expression, our results provide evidence that the intracellular protozoan pathogen T. cruzi engages a unique pathway to trigger expression of IFN-β in fibroblasts and macrophages.
FIGURE 6.
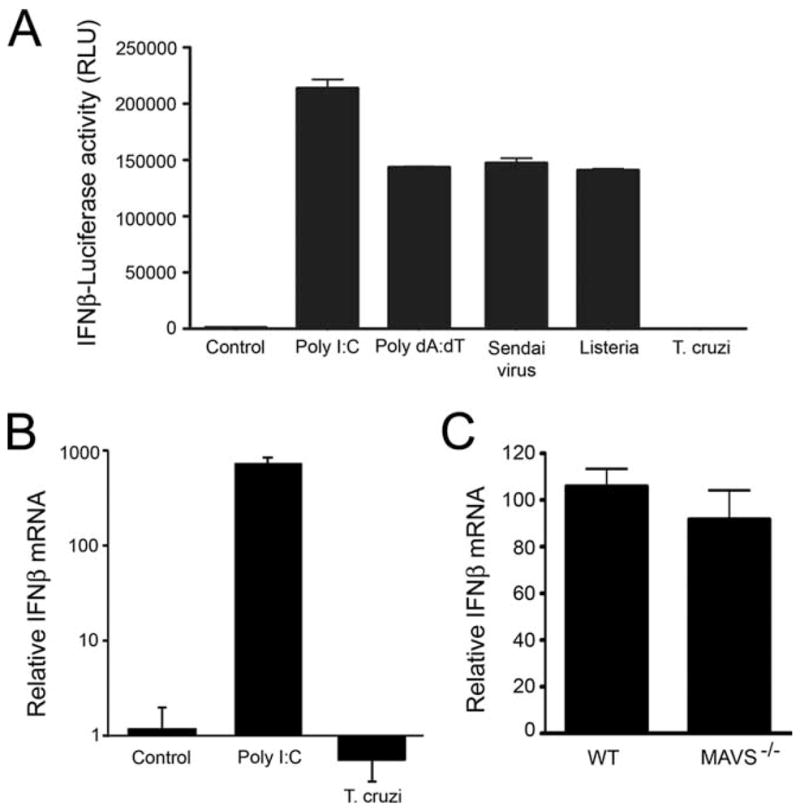
T. cruzi triggers IFN-β expression independently of characterized cytosolic pathways. HEK293-IFN-β-luciferase reporter cells were transfected with dsRNA (poly(I:C)) or dsDNA (poly(dA:dT)) or infected with Sendai virus, L. monocytogenes, or T. cruzi. Luciferase activity (A) or endogenous IFN-β mRNA (B) was measured 24 h after treatment. Relative ifnb transcript abundance was measured by qRT-PCR in WT and MAVS−/− BMDM after 6 h of infection (C). Values reported represent the mean fold induction relative to mock-infected controls ± SD.
Discussion
Pathogen-dependent activation of the type I IFN response in cells is accomplished by engaging one of two general pathways involving recognition by plasma membrane and endosomal TLRs or by cytosolic RNA and DNA-sensing molecules (reviewed in Refs. 4 and 42). In this study, we examined the mechanistic basis for induction of IFN-β in immune and nonimmune cells by the intracellular protozoan parasite T, cruzi. In this study, we show that T. cruzi triggers IFN-β expression in host cells by engaging a novel TLR-independent pathway that requires both TBK1 and IRF3, but does not involve known cytosolic nucleic acid sensors. We demonstrate that the induction of ifnb is stage specific, as the epimastigote stage of the parasite does not induce this response, suggesting that the ligand that triggers the IFN response is differentially expressed within the parasite life cycle. Based on evidence that epimastigotes significantly differ from trypomastigotes with regard to the lipid (43), carbohydrate (44), and protein (45–47) composition of their surface coats, it is not surprising that the host response to these parasite life cycle stages would differ. Additionally, we demonstrate that T. cruzi-mediated induction of IFN-β mRNA results in a functional type I IFN response, leading to expression of the ISGs cxcl10, viperin, and lrg47, in fibroblasts and macrophages. Interestingly, IFN-β- induced lrg47 is known to play a vital role in control of T. cruzi infection in vivo (48). The demonstration here that T. cruzi-induced type I IFNs can also result in the expression of this critical player in the immune response to the parasite suggests a possible mechanism for the type I IFN response to play a protective role in T. cruzi infection.
Results from this study demonstrate that, comparable to many IFN-β triggering signals, TBK1/IRF3 signaling is required for T. cruzi-induced ifnb expression. We further demonstrate that the intracellular signaling pathways involved in ifnb induction by T. cruzi are TLR independent. Infection of cells deficient for expression of the TLR adaptor proteins TRIF and/or MyD88 showed no impairment of ifnb expression in response to T. cruzi. We also showed that TLR3 and TLR4, which signal through TRIF to activate ifnb expression (33), are dispensable for T. cruzi-elicited ifnb expression. These data provide strong evidence that T. cruzi triggers type I IFN expression in a TLR-independent manner. These findings contrast with results of a previous study that reported a role for TRIF in T. cruzi-triggered expression of ifnb in BMDM and dendritic cells (28). The reason for this difference is unclear but gross differences in experimental design do not appear to account for this. Both studies used BMDM isolated from mice on a C57BL/6 background; control treatments demonstrate that both TRIF−/− and MyD88−/− cells are no longer sensitive to poly(I:C) or the TLR2 agonist, Pam2Csk4, respectively, and both studies clearly show MyD88-dependent induction of proinflammatory cytokines in BMDM in response to T. cruzi. Because different parasite strains were used in the two studies, Y strain here and Tulahuen strain in the previous study (28), a plausible explanation may relate to differences in the T. cruzi strains used in the two studies. An enormous amount of genetic diversity exists between the >100 T. cruzi strains (49) and future studies should address the possibility that different strains of the parasite might differentially activate innate immune signaling pathways. If so, these differences may impact the nature of the host response and pathophysiology of Chagas’ disease. Irrespective of potential differences, our data provide a compelling argument that T. cruzi Y strain can induce IFN-β expression by engaging a TLR-independent pathway.
Similar to Listeria, T. cruzi invades cells and transiently resides within a vacuole before escaping into the host cell cytosol (25). Rupture of the Listeria vacuole is critically dependent upon expression of a pore-forming molecule, listeriolysin O (50). T. cruzi also secretes a pore-forming molecule into the vacuole, optimally active at low pH (TcTOX; Ref. 26) which is thought to be involved in vacuole membrane disruption facilitating parasite delivery to the cytoplasm. As shown in this study, T. cruzi resides within a Lamp-1-positive vacuole for ≥8 h after entry before the gradual process of egress begins. The signaling events initiated by T. cruzi to trigger IFN-β expression clearly precede cytosolic localization of the parasite because parasite-induced phosphorylation of IRF3 and ifnb expression are detectable by 2 h after infection. These observations suggest that signals leading to ifnb expression in response to T. cruzi are likely to be initiated from a plasma membrane receptor or from within the parasite vacuole. The rapid and robust response of cells to heat-killed trypomastigotes, which are not internalized, argues in favor of an extracellular signal. Alternatively, destabilization of the parasite membrane as a result of heat killing may also result in shedding of membrane vesicles that are internalized by the endocytic pathway more rapidly or in higher quantity than the route of ligand delivery that occurs during the invasion process. Although both live and heat-killed trypomastigotes trigger a robust IFN-β response in a TLR-independent manner, we cannot rule out the possibility that the initiating signals differ in these two conditions. If endocytic trafficking is involved in the mechanism of IFN-β induction by T. cruzi, the signaling events leading to ifnb expression are not dependent on maintenance of a pH gradient. Attempts to block the induction of ifnb by blocking the vacuolar H+-ATPase with bafilomycin A failed to change the early kinetics of ifnb expression in T. cruzi-infected cells, while causing a marked delay in vacuole egress for the first 8 h. Similarly, bafilomycin A pretreatment of cells failed to block the IFN response to heat-killed trypomastigotes (data not shown). Thus, the events leading to ifnb expression following T. cruzi invasion appear to be pH independent and not triggered by vacuole egress as reported previously for Listeria (2). Furthermore, as evidenced from the differential ability to trigger ifnb expression in HEK293 cells, the pathways engaged by T. cruzi and Listeria to trigger IFN-β expression are clearly distinct.
Cytosolic surveillance mechanisms play an important role in TLR-independent pathogen recognition and induction of IFN-β (2, 51). Infection of cells with certain viruses can trigger activation of RIG-I-like receptors that recognize dsRNA in the cytoplasm (5). In addition, DNA-sensing pathways may play a role in generating a response to certain intracellular bacteria or viruses (13); however, definitive cytosolic DNA receptors have yet to be identified. A cytosolic DNA-sensing pathway involving Z DNA-binding protein 1 (ZBP-1), also known as DAI was initially reported to act as a cytosolic DNA receptor upstream of ifnb expression (15, 16); however, it was recently demonstrated that DAI is not responsible for cytosolic DNA-dependent ifnb signaling after examining the IFN response in DAI null cells (14).
Considering our finding that T. cruzi triggers IFN-β in a TLR-independent manner, we postulated that one of the known cytosolic recognition pathways is activated by internalized parasites. The cytosolic RNA signaling pathways acting through RIG-I and MDA5 were ruled out following infection of MAVS-deficient macrophages which mounted a normal IFN response to T. cruzi. Although DNA isolated from different organisms can trigger expression of type I IFNs when delivered to the cytoplasm of mammalian cells (11, 13), there is no clear evidence to support the idea that DNA is the principal ligand recognized by the host during the infection process. A recent report demonstrating that T. cruzi genomic DNA is able to induce a potent proinflammatory response when delivered to the endosomes of TLR9-expressing cells (52) suggests that parasite DNA in the vacuole could readily access the host cell cytoplasm and trigger IFN-β expression through a DNA-sensing mechanism. The ability to couple the proinflammatory cytokine response with IFN-β expression via TLR-dependent and -independent mechanisms in infected host cells is an attractive idea that warrants further investigation. Although we were unable to directly assess the role of dsDNA-sensing mechanisms in the parasite-mediated IFN response, HEK293 IFN-β-luc reporter cells, which lack TLRs but remain responsive to many cytosolic triggers of ifnb transcription, including dsDNA and dsRNA, were completely unresponsive to parasite infection. These findings strongly suggest that T. cruzi does not engage known cytosolic surveillance pathways. In the future, HEK293 cells could be used as a potential tool for genetic complementation approaches toward identification of the components of the signaling pathway that are engaged by T. cruzi to trigger type I IFN expression.
Findings reported here indicate the existence of a novel TLR-independent pathway that is distinct from the cytosolic dsRNA or DNA recognition pathways upstream of IFN-β expression that have been previously described in the literature. In addition to the novel pathway through which T. cruzi induces IFN-β described in this study, a recent report by Ishii et al. (14) also describes a novel TBK1-dependent pathway for type I IFN induction. Although these novel TBK1-dependent pathways have not been characterized at the molecular level, together these findings highlight the concept that the full range of pathogen and cytosolic recognition receptors has yet to be identified. A more complete understanding of the novel pathway through which T. cruzi triggers IFN-β in immune and nonimmune cells will provide valuable insight into the complexity of the signaling networks that trigger IFN-β expression and regulation of the innate immune response to intracellular pathogens.
Acknowledgments
We thank K. Krumholz and C. Wells for maintenance of parasite cultures as well as L. Waggoner and N. Goutagny for help with obtaining mouse femurs. We thank J. Kagan and R. Medzhitov for providing MyD88−/−, MyD88−/−/TIRAP−/−, and TLR2−/−/TLR4−/− MEF and J. Hiscott for providing the anti-phosphoSer396 IRF3 Ab.
Footnotes
This work was supported by National Institutes of Health Grant AI047960 (to B.A.B.) and AI067497 (to K.A.F.).
Abbreviations used in this paper: IRF, IFN regulatory factor; DAI, DNA-dependent activator of IRF; TBK1, TANK-binding kinase 1; IFNAR, type I IFN receptor; MEF, mouse embryonic fibroblast; WT, wild type; Pen-Strep, penicillin-streptomycin; BMDM, bone marrow-derived macrophage; qRT-PCR, quantitative RT-PCR.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Bogdan C, Mattner J, Schleicher U. The role of type I interferons in non-viral infections. Immunol Rev. 2004;202:33–48. doi: 10.1111/j.0105-2896.2004.00207.x. [DOI] [PubMed] [Google Scholar]
- 2.O’Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci USA. 2002;99:13861–13866. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noppert SJ, Fitzgerald KA, Hertzog PJ. The role of type I interferons in TLR responses. Immunol Cell Biol. 2007;85:446–457. doi: 10.1038/sj.icb.7100099. [DOI] [PubMed] [Google Scholar]
- 4.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 6.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-β promoter. Proc Natl Acad Sci USA. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 9.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 11.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 12.Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med. 2005;202:1333–1339. doi: 10.1084/jem.20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, Akira S. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- 15.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi O, Akira S. Signaling pathways activated by microorganisms. Curr Opin Cell Biol. 2007;19:185–191. doi: 10.1016/j.ceb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Soulat D, Bauch A, Stockinger S, Superti-Furga G, Decker T. Cytoplasmic Listeria monocytogenes stimulates IFN-β synthesis without requiring the adapter protein MAVS. FEBS Lett. 2006;580:2341–2346. doi: 10.1016/j.febslet.2006.03.057. [DOI] [PubMed] [Google Scholar]
- 18.Stockinger S, Reutterer B, Schaljo B, Schellack C, Brunner S, Materna T, Yamamoto M, Akira S, Taniguchi T, Murray PJ, et al. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol. 2004;173:7416–7425. doi: 10.4049/jimmunol.173.12.7416. [DOI] [PubMed] [Google Scholar]
- 19.Barnes B, Lubyova B, Pitha PM. On the role of IRF in host defense. J Interferon Cytokine Res. 2002;22:59–71. doi: 10.1089/107999002753452665. [DOI] [PubMed] [Google Scholar]
- 20.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 21.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 22.Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol Cell. 1998;1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 23.Malmgaard L, Salazar-Mather TP, Lewis CA, Biron CA. Promotion of α/β interferon induction during in vivo viral infection through α/β interferon receptor/STAT1 system-dependent and -independent pathways. J Virol. 2002;76:4520–4525. doi: 10.1128/JVI.76.9.4520-4525.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burleigh BA. Host cell signaling and Trypanosoma cruzi invasion: do all roads lead to lysosomes? Sci STKE. 2005;2005:pe36. doi: 10.1126/stke.2932005pe36. [DOI] [PubMed] [Google Scholar]
- 25.Ley V, Robbins ES, Nussenzweig V, Andrews NW. The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the pH of acidic compartments. J Exp Med. 1990;171:401–413. doi: 10.1084/jem.171.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews NW, Abrams CK, Slatin SL, Griffiths G. A T. cruzi-secreted protein immunologically related to the complement component C9: evidence for membrane pore-forming activity at low pH. Cell. 1990;61:1277–1287. doi: 10.1016/0092-8674(90)90692-8. [DOI] [PubMed] [Google Scholar]
- 27.de Avalos SV, I, Blader J, Fisher M, Boothroyd JC, Burleigh BA. Immediate/early response to Trypanosoma cruzi infection involves minimal modulation of host cell transcription. J Biol Chem. 2002;277:639–644. doi: 10.1074/jbc.M109037200. [DOI] [PubMed] [Google Scholar]
- 28.Koga R, Hamano S, Kuwata H, Atarashi K, Ogawa M, Hisaeda H, Yamamoto M, Akira S, Himeno K, Matsumoto M, Takeda K. TLR-dependent induction of IFN-β mediates host defense against Trypanosoma cruzi. J Immunol. 2006;177:7059–7066. doi: 10.4049/jimmunol.177.10.7059. [DOI] [PubMed] [Google Scholar]
- 29.Costa VMA, Torres KCL, Mendonca RZ, Gresser I, Gollob KJ, Abrahamsohn IA. Type I IFNs stimulate nitric oxide production and resistance to Trypanosoma cruzi infection. J Immunol. 2006;177:3193–3200. doi: 10.4049/jimmunol.177.5.3193. [DOI] [PubMed] [Google Scholar]
- 30.Une C, Andersson J, Orn A. Role of IFN-α/β and IL-12 in the activation of natural killer cells and interferon-γ production during experimental infection with Trypanosoma cruzi. Clin Exp Immunol. 2003;134:195–201. doi: 10.1046/j.1365-2249.2003.02294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 34.Tardieux I, Nathanson MH, Andrews NW. Role in host cell invasion of Trypanosoma cruzi-induced cytosolic-free Ca2+ transients. J Exp Med. 1994;179:1017–1022. doi: 10.1084/jem.179.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nogueira N, Cohn Z. Trypanosoma cruzi: mechanism of entry and intracellular fate in mammalian cells. J Exp Med. 1976;143:1402–1420. doi: 10.1084/jem.143.6.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tardieux I, Webster P, Ravesloot J, Boron W, Lunn JA, Heuser JE, Andrews NW. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- 37.Woolsey AM, Sunwoo L, Petersen CA, Brachmann SM, Cantley LC, Burleigh BA. Novel PI3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J Cell Sci. 2003;116:3611–3622. doi: 10.1242/jcs.00666. [DOI] [PubMed] [Google Scholar]
- 38.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem. 2004;279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- 39.Servant MJ, Grandvaux N, tenOever BR, Duguay D, Lin R, Hiscott J. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem. 2003;278:9441–9447. doi: 10.1074/jbc.M209851200. [DOI] [PubMed] [Google Scholar]
- 40.Campos MA, Closel M, Valente EP, Cardoso JE, Akira S, Alvarez-Leite JI, Ropert C, Gazzinelli RT. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J Immunol. 2004;172:1711–1718. doi: 10.4049/jimmunol.172.3.1711. [DOI] [PubMed] [Google Scholar]
- 41.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signaling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 42.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 43.Esteves MG, Gonzales-Perdomo M, Alviano CS, Angluster J, Goldenberg S. Changes in fatty acid composition associated with differentiation of Trypanosoma cruzi. FEMS Microb Lett. 1989;50:31–34. doi: 10.1016/0378-1097(89)90453-9. [DOI] [PubMed] [Google Scholar]
- 44.de Andrade AF, Esteves MJ, Angluster J, Gonzales-Perdomo M, Goldenberg S. Changes in cell-surface carbohydrates of Trypanosoma cruzi during metacyclogenesis under chemically defined conditions. J Gen Microbiol. 1991;137:2845–2849. doi: 10.1099/00221287-137-12-2845. [DOI] [PubMed] [Google Scholar]
- 45.Schenkman S, Eichinger D, Pereira ME, Nussenzweig V. Structural and functional properties of Trypanosoma trans-sialidase. Annu Rev Microbiol. 1994;48:499–523. doi: 10.1146/annurev.mi.48.100194.002435. [DOI] [PubMed] [Google Scholar]
- 46.Tomas AM, Kelly JM. Stage-regulated expression of cruzipain, the major cysteine protease of Trypanosoma cruzi is independent of the level of RNA1. Mol Biochem Parasitol. 1996;76:91–103. doi: 10.1016/0166-6851(95)02545-6. [DOI] [PubMed] [Google Scholar]
- 47.Avila AR, Dallagiovanna B, Yamada-Ogatta SF, Monteiro-Goes V, Fragoso SP, Krieger MA, Goldenberg S. Stage-specific gene expression during Trypanosoma cruzi metacyclogenesis. Genet Mol Res. 2003;2:159–168. [PubMed] [Google Scholar]
- 48.Santiago HC, Feng CG, Bafica A, Roffe E, Arantes RM, Cheever A, Taylor G, Vieira LQ, Aliberti J, Gazzinelli RT, Sher A. Mice deficient in LRG-47 display enhanced susceptibility to Trypanosoma cruzi infection associated with defective hemopoiesis and intracellular control of parasite growth. J Immunol. 2005;175:8165–8172. doi: 10.4049/jimmunol.175.12.8165. [DOI] [PubMed] [Google Scholar]
- 49.de Freitas JM, Augusto-Pinto L, Pimenta JR, Bastos-Rodrigues L, Goncalves VF, Teixeira SM, Chiari E, Junqueira AC, Fernandes O, Macedo AM, et al. Ancestral genomes, sex, and the population structure of Trypanosoma cruzi. PLoS Pathog. 2006;2:e24. doi: 10.1371/journal.ppat.0020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Portnoy DA, Jones S. The cell biology of Listeria monocytogenes infection (escape from a vacuole) Ann NY Acad Sci. 1994;730:15–25. doi: 10.1111/j.1749-6632.1994.tb44235.x. [DOI] [PubMed] [Google Scholar]
- 51.Jefferies CA, Fitzgerald KA. Interferon gene regulation: not all roads lead to Tolls. Trends Mol Med. 2005;11:403–411. doi: 10.1016/j.molmed.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 52.Bartholomeu DC, Ropert C, Melo MB, Parroche P, Junqueira CF, Teixeira SM, Sirois C, Kasperkovitz P, Knetter CF, Lien E, et al. Recruitment and endolysosomal activation of TLR9 in dendritic cells infected with Trypanosoma cruzi. J Immunol. 2008;181:1333–1344. doi: 10.4049/jimmunol.181.2.1333. [DOI] [PubMed] [Google Scholar]