

Abstract
Background
Inflammation is known to contribute to the pathogenesis of vascular diseases in which arterial wall extracellular matrix (ECM) homeostasis is disrupted. Tumor necrosis factor-α (TNF-α), a pivotal cytokine that regulates ECM metabolism by increasing degradation and decreasing production of arterial collagens, is associated with vulnerable plaques and aortic aneurysms.
Methods and Results
In the current study, we showed that, when administered in doses of 1 to 100 ng/mL, TNF-α dose-dependently downregulated the expression of prolyl-4-hydroxylase αI [P4Hα (I)]—the rate-limiting subunit for the P4H enzyme essential for procollagen hydroxylation, secretion, and deposition in primary human aortic smooth muscle cells (HASMCs). Using a progressive deletion cloning approach, we characterized the TNF-α–responsive element (TaRE) in the human P4Hα (I) promoter and found that a negative regulatory region at the position of −32 to +18bp is responsible for ≈80% of TNF-α–mediated suppression. Using oligonucleotide-based transcription factor pull-down method in which proteins were resolved in 1-D gel electrophoresis and identified using LC-MS/MS, we identified the NonO protein binds this region. When NonO expression silenced with specific siRNA, we found that 70% of the TNF-α–mediated P4Hα suppression was abolished, which appeared to be mediated by the ASK1-JNK pathway.
Conclusions
Our findings define a novel molecular pathway for inflammation associated extracellular matrix dysregulation, which may account for atherosclerotic plaque rupture and aortic aneurysm formation. Further understanding of this pathway may facilitate development of novel therapeutics for vascular diseases.
Keywords: inflammation, collagen, P4Hα1TNF-α, NonO, JNK, ASK1
Inflammation plays a pivotal role in the pathogenesis of cardiovascular diseases, including cardiomyopathy, heart failure, atherosclerotic plaque rupture, and aortic aneurysm.1,2 Many pathologic changes in the cardiovascular system can be triggered by inflammation, which contributes to the disease process. Metabolic imbalance in the extracellular matrix (ECM) in the myocardium and arterial wall represents one of the key structural changes that mark the development and progression of most cardiovascular diseases. ECM components, especially collagen—the main constituent of the fibrous cap in atheroma—determines plaque stability and vulnerability to rupture.3 Furthermore, dysregulated ECM metabolism in the aortic wall, such as inadequate collagen degradation or elastin disruption, leads to aortic aneurysm and rupture.4 Cytokines, including TNF-α, transforming growth factor (TGF)-β, and various interleukins, are dysregulated in inflammation and may participate in ECM metabolism by increasing ECM degradation through activation of matrix metalloproteinases (MMPs)5,6 and inhibition of collagen synthesis.7,8 TNF-α, which is released by activated macrophages, is one of the most potent cytokines involved in cardiovascular pathogenesis1 and actively regulates ECM metabolism.7
ECM is the structural framework of all tissues including the arterial wall, in which fibrillar proteins (collagen and elastin) and adhesive proteins (eg, laminin and fibronectin) form the structural backbone of the tissue. In the arterial wall, various cells including endothelial cells, smooth muscle cells, and fibroblasts, contribute to ECM metabolism. Collagen is one of the most metabolically active ECM components, with at least 39 subtypes; types I and III are the ones most commonly found in the arterial wall.9 The collagen molecule consists of 3 identical polypeptide chains, called α chains. This molecule has at least 1 triple-helical collagenous domain with repeating (Gly-X-Y) n sequences, ie, a glycine residue at every third amino acid and, frequently, proline and 4-hydroxyproline in the X and Y positions. Collagen biosynthesis involves a number of posttranslational modifications of procollagens and proteolytic conversion to collagens. The intracellular modifications require 5 specific enzymes, including 3 collagen hydroxylases and 2 collagen glycosyltransferases.
Prolyl-4-hydroxylase (P4H) is one of the key intracellular enzymes required for the synthesis of all known types of collagens.10 It catalyzes the formation of hydroxyproline from proline residues located in repeating X-Pro-Gly triplets in the procollagens during posttranslational processing. It is essential for folding the procollagen polypeptide chains into stable triple helical molecules.11 Inhibition of P4H produces unstable collagen associated with collagen decrease.12 P4H is composed of α and β subunits in which α subunit is rate-limiting and essential for collagen maturation and secretion.13 P4H isoenzymes are expressed in most tissues, although certain subtypes may be tissue-specifically expressed14; and are regulated by various cytokines, such as TNF-α, and cigarette smoking.15,16 Previously, we have reported that an E-box–like sequence (CACGGG) located at −135bp of the P4Hα1 promoter is responsible for 80% of the basal transcriptional activity and appears to be upregulated by TGFβ1 and downregulated by cigarette smoking.17
In the current study, we investigated the effects of TNF-α on P4Hα1 expression and the underlying molecular mechanisms of these effects. We have found that TNF-α1 suppresses P4Hα1 expression at the transcriptional level. We have further identified a TNF-α response element (TaRE) located at −32 to +18bp of the P4Hα1 promoter. A transcription factor, NonO (human p54nrb),18 has been shown to bind with the TaRE and determines TNF-α–mediated P4Hα1 suppression. Furthermore, we have found that TNF-α–activated ASK1–JNK pathway appears to be involved in the P4Hα1 suppression.19 Our study offers potentially new therapeutic targets to attenuate inflammation-induced ECM disturbance and consequently cardiovascular diseases.
Materials and Methods
Plasmid Construction
To define promoter regions that are responsive to TNF-α–mediated P4Hα1 suppression, we generated P4Hα1 promoter-pGL3 constructs with serial deletions of the P4Hα1 promoter. The pGL3-basic vector (Cat#: E1751, Promega, Madison, Wi) was used for the plasmid construction. We generated plasmids containing P4Hα1 promoter regions from −580, −480, −417, −320, −271, −184, −145, −97, −32, and +18 to +76bp between the multiple cloning sites KpnI and HindIII for the purpose of directional cloning. The details of the primers and sequence confirmation of these plasmids have been described previously.17 To define the nucleotides within the region −32 to +18bp in the P4Hα1 promoter responsible for the binding of TNF-α responsive protein NonO, we constructed P4Hα1 promoter pGL3 reporter with serial deletions of −32 to −15bp, −32 to −3bp. The primers of the region −15pGL3 vector were sense: GGTACCACGGGCTCCCTCTGCTGCCCAG, antisense: GCTAGCACCACCACAGCGGGAAGGAATGG. The primers of the region −3pGL3 vector were sense: GGTACCGCTGCCCAGTCGCGCCGCCAG, antisense: GCTAGCACCACCACAGCGGGAAGGAATGG. To examine the isoforms of the JNK that are involved in P4Hα1 regulation, we used wild-type JNK1 (Ad-wtJNK1) and dominant negative (Ad-dnJNK1) expression vectors as described previously.20,21
Cell Culture and Transfection
Human aortic smooth muscle cells (HASMCs) were obtained from fresh human aorta (from organ donors) using an explant culture technique in SMC culture medium (Cat#311 to 500 Cell Application, San Diego, Calif) containing 10% fetal bovine serum (FBS). Cells were cultured up to the 4th passage before the experiments were conducted. For dose-dependent effects, HASMCs were treated with 0, 1, 10, and 100 ng/mL human recombinant TNF-α (Cat#T6674, Sigma-Aldrich) for 8 hours before cells were harvested for measurements of target gene mRNA levels. For the time-course study, we treated cells for 4, 8, 24, and 48 hours.
To search for the TaRE in the P4Hα promoter, recombinant P4Hα-pGL3 plasmids were transfected to HASMCs with Lipo-fectamine 2000 (Invitrogen Corp) in 6-well plates as described previously.17 Twenty-four hours after the transfection, HASMCs were treated with 100 ng/mL TNF-α. At the end of the 8-hour treatment, cells were harvested for mRNA measurement. After the TaRE was defined to the region of −32 to +18bp, we further investigated the signaling pathways that are responsible for the P4Hα1 suppressing effect. Twenty-four hours after the cells were transfected with P4Hα-32-pGL3 plasmid (or P4Hα-271-pGL3 plasmid as a parallel control), they were treated with 50 μmol/L JNK inhibitor SP600125 (Sigma) or 200 ng/mL thioredoxin (Sigma)—an ASK1 inhibitor21–23—and then with TNF-α. After 8-hour treatment, cells were harvested for mRNA measurement.
To explore the effects of transcription factors in TNF-α–mediated P4Hα1 suppression, we used NonO, hnRNP-K, BUB3, and ILF2 specific siRNAs (100 nmol/L) to inhibit the expression of these proteins in cells transfected with P4Hα-271-pGL3 plasmid and then, 24 hours later, treated with TNF-α. The promoter activity of P4Hα1 was assessed by measuring mRNA levels of the reporter luciferase gene. The siRNA duplex was chemically synthesized by Integrated DNA Technologies. The following siRNA sequences for NonO gene were used: sense, 5′rArArUrCrArUrArCrUrCrCrArArGrGrArArGrCrArUrUrU3′; antisense, 5′rArArArUrGrCrUrUrCrCrUrUrGrGrArGrUrArUrGrArUrUr3′. The siRNA duplex sequences for the hnRNP-K gene were the following: sense, 5′rArArGrCrArGrUrArUrUrCrUrGrGrArArArGrUrUrUrUrU3′; antisense, 5′rArArArArArCrUrUrUrCrCrArGrArArUrArCrUrGrCrUrU3′; for the BUB3 gene: sense 5′rArArUrGrGrArArUrUrGrGrCrArCrArGrUrCrGrCrUrUrU3′; antisense, 5′rArArArGrCrGrArCrUrGrUrGrCrCrArArUrUrCrCrArUrU3′; for the ILF2 gene: sense 5′rArArCrArGrArGrArArArGrGrArUrArGrArUrGrCrCrUrGrUrU3′; antisense, 5′rUrUrGrUrCrUrCrUrUrUrCrCrUrArUrCrUrArCrGrGrArCrArA3′.
Quantitative Real-Time RT-PCR
We used quantitative real-time RT-PCR for the mRNA measurements of the target genes. Primers were designed with Beacon Designer 2.0 software and chemically synthesized (Integrated DNA Technologies). The primers for P4Hα1 mRNA were: sense, 5′CATGACCCTGAGACTGGAAA3′; antisense: 5′GCCAGGCACTCTTAGATACT3′. The primers for housekeeping gene human α-actin were: sense, 5′CTGGAACGGTGAAGGTGACA3′; antisense, 5′GGGACTTCCTGTAACAATGCA3′. The primers for the reporter luciferase gene were: sense, 5′TCAAAGAGGCGAACTGTGTG3′; antisense, 5′GTGTTCGTCTTCGTCCCAGT3′.
Total RNA was extracted from the cells with RNA Trizol (Invitrogen) according to the manufacturer’s protocol. The extracted RNA was dissolved in a final volume of 25 μL RNase free water, and concentrations of the total RNA were tested using a spectrophotometer. One microgram of mRNA was used for reverse transcription in a final volume of 20 μL. The reverse transcription from mRNA to cDNA was carried out using a iScript cDNA synthesis kit (Bio-Rad) containing a mixture of oligo(dT) and random primers. One microliter of cDNA in this 20 μL volume was used for real-time PCR with an SYBR Green I Supermix kit (Bio-Rad). The PCR was performed in duplicate using an iCycler iQ real-time PCR detection system; the program ran for 40 cycles at 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds. The mRNA levels were estimated from the value of the threshold cycle (Ct) of the real-time PCR adjusted by that of β-actin through the formula 2ΔCt (ΔCt=β-actin Ct - gene of interest Ct).
Electrophoretic Mobility Shift Assay and Antibody-based Supershift Assay
To discover transcription factors that are responsible for TNF-α–mediated P4Hα1 suppression, we first carried out electrophoretic mobility shift assay (EMSA) to learn whether the TaRE in the P4Hα1 promoter was binding with any transcription factors. Nuclear proteins were isolated from cultured HASMCs treated with or without TNF-α using the Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology Inc). Protein concentrations were determined by the Bradford method (Sigma). EMSA was performed with a nonradioisotope method (Gel-Shift kit, Panomics Inc) that uses a biotin-labeled probe according to the manufacturer’s instructions. Briefly, the human P4Hα1 promoter DNA fragment sense strand oligonucleotides (5′-GGTTATAAAAGGGCTAACGGGCTCCCTCTGCTGCCCAGTCGCGCCGCCAGCGGGCTG-3′, from −32 to +24bp) were annealed with their antisense strands and labeled with biotin at their 5′-ends. The binding reaction was performed with 10 ng 30-fmol biotin-labeled probes incubated with 5 μg nuclear extract in a binding buffer containing 1 μg poly (dI-dC) in a final volume of 20 μL at room temperature for 15 minutes. The DNA-protein complex was resolved through 6% native polyacrylamide gel at 80 V (5 V/cm gel), followed by electro-blotting onto positively charged nylon membrane and UV cross-linking. After being blocked in the blocking buffer, the blot was incubated with streptavidin-labeled antibody conjugated with HRP for 15 minutes at room temperature. The blot was then washed, and chemiluminescent detection was performed with CSPD as the substrate in the ECL kit (Pierce) before being exposed to X-ray film.
For the antibody-based supershift assay, antibodies against transcription factors, including NonO (0.2 μg, Cat#A300-587A, Lot#A300-587A-1, Bethyl, Montgomery, Tex), hnRNP-K (2 μg, Cat#: R8903, Sigma), AP2 (0.5 μg, Cat#: KAP-TF100, Nventa, Victoria, BC), and v-Myb/c-Myb (1 μg, Cat#: GTX10935 GeneTex) were added to a mixture of biotin-labeled oligonucleotide probes and nuclear proteins that had been reacted for 30 minutes. After an additional 30 minutes of incubation, the protein-DNA mixture was electrophoresized in 6% polyacrylamide gel for 1 hour. The rest of the process was the same as that used in EMSA.
1-D Electrophoresis and Transcription Factor Identification
In searching for the transcription factors that are responsible for TNF-α–induced P4Hα1 suppression, we first used the web-based TFSEARCH program (http://www.cbrc.jp/research/db/TFSEARCH.html, Parallel Application TRC Laboratory, RWCP, Japan) and subjected the −32 to +18bp region to the search. We then tested the most likely transcription factors by using the antibody-based super-shift assay to confirm their presence. To uncover novel proteins that were not revealed by TFSEARCH, we also designed an oligonucleotide-based pull-down technique to identify novel binding proteins that were not revealed by TFSEARCH. In doing this, we biotin-labeled the double-stranded oligonucleotide for DNA fragments containing the TaRE (−32 to +18bp). We then mixed 10 μL (10 ng/μL) labeled probe with 60 μL nuclear proteins (5 μg/μL) for 30 minutes at room temperature. The nuclear proteins were isolated from cultured HASMCs treated with or without TNF-α for 8 hours. The reaction mixture was then mixed with 60 μL streptavidinagarose beads (Sigma) overnight at 4°C. After centrifugation at 10 000g for 5 minutes at 4°C, proteins in binding with the probe were cleaved from the beads in 100 μL sodium dodecyl sulfate (SDS) full lysis buffer (100 mmol/L NaCl, 500 mmol/L Tris, pH=8, 10% SDS wt/vol). The protein mixture was then run in SDS-PAGE gel for 2 hours (120V). After gel was stained in Coomassie blue, clear bands were excised from the gel and sent for LC-MS/MS analysis. The profiles of the peptide masses obtained by LC-MS/MS were matched with the profile of known proteins in the database (http://www.expasy.ch) by the Protein Chemistry Core Facility, Columbia University (New York, NY).
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation (ChIP) assay was performed with the histone H3 ChIP assay kit according to the manufacturer’s protocol (Upstate). In brief, approximately 3×107 cells were used per ChIP assay. After HASMCs were treated with or without TNF-α for 8 hours, cells were cross-linked with 1% formaldehyde at 37°C for 10 minutes and rinsed twice with ice-cold phosphate-buffered saline (PBS). Cells were harvested by brief centrifugation. Cell pellets were resuspended in SDS-lysis buffer (50 mmol/L Tris-HCl, pH 8.1, 10 mmol/L EDTA, 1% SDS, protease inhibitors). Sonication was performed on ice using a Sonifier II 450 (Brason) with a 3-mm tip set at duty cycle 20 and output level 2 to achieve chromatin fragments of 200 to 1000 bp in size. This was followed by centrifugation of the cell pellets at 15 000g for 10 minutes at 4°C. Supernatants were collected and diluted 10-fold in ChIP dilution buffer (a 20-μL aliquot was removed to serve as an input sample), followed by preimmunoprecipitation clearing with 80 μL of a mixture of salmon sperm DNA, Protein A, and Protein G at 4°C with rotation for 30 minutes. Immunoprecipitation was carried out with 2 μg of anti–acetyl-histone H3 antibody at 4°C overnight with rotation. After immunoprecipitation, 60 μL of a mixture of salmon sperm DNA/Protein A/Protein G was added and incubated at 4°C with rotation for 30 minutes and followed by brief centrifuge. The precipitates were washed twice (5 minutes each at 4°C) with low-salt buffer, once with high-salt buffer, and once with LiCl buffer. Then, the precipitates were washed again with TE buffer twice for 5 minutes each. The immune complexes were extracted 3 times with 200 μL of elution buffer. The elutes and the input were heated at 65°C for at least 4 hours to reverse cross-link by addition of 20 μL 5 mol/L NaCl. After proteinase K treatment, DNA was extracted with phenol/chloroform solution and precipitated with ethanol with the aid of yeast tRNA. The recovered DNA was resuspended in 30 μL H2O, and 4 μL was used for PCR. The PCR products were analyzed on 1.5% agarose gel. The primers for PCR were 5′-CTCCCTGGCGCTGCCATCGCG-3′ spanning from −60 to −39bp and 5′-CACCTGGAAAGTGGGACGAGAGG-3′ spanning from +83 to +104bp. To carry out the transcription factor–specific ChIP assay, we also used anti-human NonO or hnRNPk antibodies instead of anti-H3 histone antibody for the immunoprecipitation process.
Western Blot
After the HASMCs underwent these various treatments, their nuclear proteins were extracted, separated with 10% SDS-PAGE, and transferred to nitrocellulose membranes. The membrane was blocked in 5% nonfat milk in TBS-T (50 mmol/Tris, pH 7.5, 150 mmol/L NaCl, 0.05% Tween-20) for 1 hour at room temperature. After incubation with the primary antibodies, including mouse anti-human NonO and hnRNP-K antibodies, in TBS-T containing 1% milk for 2 hours at room temperature, the membrane was washed in TBS-T 4×10 minutes and incubated with secondary horse anti-mouse IgG-HRP conjugate (Cat# 7076, Lot# 15, Cell Signaling) for 1 hour at room temperature. After 3 washes in TBS-T, the membrane was visualized with ECL plus reagents (Amersham Biosciences). For collagen analysis, the cellular protein extracts were subjected to Western blot assay using mouse anti-human type I (Cat# C 2456, Lot# 075K4868, Sigma) and type III (Cat# C 7805, Lot#076K4756, Sigma) collagens as the primary antibodies.
Statistical Analyses
All quantitative data are presented as mean±SD. Between-group differences were compared with independent Student t tests; among-group differences for 2 or more conditions were compared with one-way ANOVA in which Bonferroni post hoc test was applied for the between group comparisons. Two-tailed P<0.05 was regarded as statistically significant. We used the SPSS v12.0 for Windows (SPSS Inc) to carry out the statistical analyses.
Results
TNF-α Suppresses P4Hα1 Expression
In the time course study, we treated HASMCs with TNF-α 100 ng/mL for 4, 8, 24, and 48 hours. At the end of each of these time points, cells were harvested and P4Hα1 mRNA levels were measured. We found that P4Hα1 mRNA levels were significantly decreased up to 8 hours after treatment (Figure 1A), by which time P4Hα1 mRNA had been reduced to 50% of its baseline level before levels were recovered somewhat. To examine whether the recovered P4Hα1 expression at 24 and 48 hours was attributable to diminished TNF-α activity or impeded cellular response, we replenished the medium with fresh TNF-α every 8 hours during the culture. We showed that a plateau was reached by 8 hours by which cells appeared to be refractile to additional TNF-α exposure (Figure 1B). To evaluate the residual TNF-α activity after 8 hours incubation with HASMCs, we transferred the TNF-α–containing culture medium at 8 hours to fresh HASMCs and measured the P4Hα1 mRNA levels at 4 and 8 hours. P4Hα1 mRNA level was reduced only 19% after 4 hours treatment with the “used” TNF-α containing medium. Comparing with the 57% reduction using the fresh TNF-α within the same time period, our experiment suggested that approximately 33% active TNF-α remained in the culture medium 8 hours after incubating with the cells. In examining dose-response effects, we observed a linear reduction in P4Hα1 mRNA levels in HASMCs treated with 1 to 100 ng/mL TNF-α for 8 hours in culture (Figure 1C). In corresponding with the repressed P4Hα1 expression, the levels of type I and III collagen from the TNF-α–treated cells were also significantly reduced (Figure 1D). In all of the experiments described below, treating HASMCs with 100 ng/mL TNF-α for 8 hours was used as the standard treatment condition to reduce the expression of P4Hα1 mRNA.
Figure 1.

TNF-α suppresses P4Hα1 mRNA expression in HASMCs. Quantitative realtime RT-PCR was used to measure the mRNA levels. A, Levels of P4Hα1 mRNA were measured from HASMCs treated with 100 ng/mL TNF-α for 0, 4, 8, 24, and 48 hours. B, HASMCs were treated with 100 ng/mL TNF-α at 0 hours and replenished with fresh 100 ng/mL TNF-α at every 8 hours till 48 hours. Levels of P4Hα1 mRNA were measured at 0, 4, 8, 24, and 48 hours. C, HASMCs were treated with 0, 1, 10, and 100 ng/mL TNF-α for 8 hours. ANOVA was used to compare the among-group differences in both the time course and dose-dependent effects in TNF-α-mediated P4Hα1 inhibition. **P<0.01 indicates the comparison between the treated cells and the control cells in the Bonferroni post hoc between group analyses. D, Western blot of type I and III collagen from HASMCs treated with 100 ng/mL TNF-α for 8 hours. The total cellular proteins were extracted and measured using Bradford method. Equal amount of proteins were then loaded to 8% SDS-PAGE gel for electrophoresis. ECL chemiluminescent method was used for the type I and III collagen detection using anti-human type I and III antibodies.
Identification of TNF-α Regulatory Element (TaRE) in the P4Hα1 5′-Flanking Sequence
To clarify the promoter region responsible for TNF-α–induced P4Hα1 suppression, we transfected HASMCs with several pGL3 reporter constructs containing the progressively deleted 5′-flanking regions of the P4Hα1 gene and measured mRNA levels of the luciferase gene after treatment with TNF-α. Compared with no treatment, TNF-α dramatically reduced promoter activity in all P4Hα1 promoter vectors except when the region of −32bp to +18bp was deleted in the promoter region (Figure 2), which abolished more than 80% of the TNF-α–induced inhibition. These results suggest that TaRE is located in the −32 to +18bp region of the P4Hα1 gene.
Figure 2.

Characterization of P4Hα1 promoter responsible for TNF-α–mediated suppression. We transfected HASMCs with 9 serially deleted P4Hα1 promoter-pGL3 constructs: pGL3–580, pGL3–480, pGL3–417, pGL3–320, pGL3–271, pGL3–184, pGL3–97, pGL3–32, and pGL3-+18. PGL-3 basic vector was used as a control. The transfected cells were treated with or without 100 ng/mL TNF-α for 8 hours. Quantitative real-time RT-PCR was used to measure the levels of luciferase mRNA. **P<0.01 is result of the Student t test between cells treated with and without TNF-α.
AP2, v-Myb, and c-Myb as Potential Transcription Factors According to TFSEARCH
As the initial strategy, we used a web-based bioinformatic tool (TFSEARCH) to identify sequence similarities within the TaRE region with known transcription factors. By subjecting the DNA sequence (−32 to +18bp) to the TFSEARCH, we found potential sequence matches for AP-2 (Score 94), v-Myb (Score of 91.4), and c-Myb (90.7). The consensus binding sequences for AP-2, v-Myb, and c-Myb were 5′-GCCCCAGGC-3′, 5′-GCTAACGG-3′, and 5′-GCTAACGGG-3′, respectively, which match partial sequences in the −32 to +18bp region: 5′-GGTTATAAAAGGGCTAACGGGCTCCCTCTGCTGCCCAGTCGCGCCGCCAGCGGGCTG-3′. Using specific antibodies against these 3 transcription factors in the supershift assays, however, we revealed no shift in the DNA-protein bands (data not shown). To further confirm the involvement of these transcription factors in TaRE, we mutated these 3 potential binding regions in the P4Hα1 to 32-pGL3 vectors (AP-2: 5′-GCCCCAGGC-3′ to 5′-GCCTTAGTC-3′, c-Myb/v-Myb: 5′-GCTAACGG-3′ to 5′-GCTGGAGG-3′, respectively). We then transfected these mutant P4Hα1 to 32-pGL3 vectors to HASMCs and treated them with TNF-α for 8 hours. The transcription efficiency in these TNF-α–treated cells was not different from that in the untreated cells (data not shown). Our results suggest that these transcription factors are unlikely to be the binding proteins responsible for the TNF-α–mediated P4Hα1 suppression.
Direct Binding of NonO and hnRNP-K to the P4Hα1 Promoter
Using 1-D electrophoresis and LC-MS/MS methods, we found that several nuclear proteins—ILF2, BUB3, SPFH2, NonO, and hnRNP-K—bound to the TaRE. Among these proteins, NonO (human p54nrb, 55 kDa) and hnRNP-K (65 kDa) appeared to be biologically relevant to gene regulation. We confirmed the binding of these 2 proteins with the TaRE in the supershift assay (data not shown) for both NonO and hnRNP-K antibodies in the reaction with nuclear proteins extracted from untreated HASMCs. The density of the supershift band with the anti-NonO antibody was significantly increased when nuclear proteins extracted from TNF-α–treated HASMCs were used in the binding reaction. The binding patterns between the NonO protein from HASMCs treated with or without TNF-α and the TaRE were also confirmed by the ChIP assay, in which the anti-NonO antibody was used instead of anti-H3 histone antibody (Figure 3). Only TNF-α–treated HASMCs showed a NonO-TaRE interaction in the ChIP assay. These results show that, whereas both NonO and hnRNP-K bound to the TaRE of the P4Hα1 in the in vitro supershift EMSA, only NonO appears to have responded to TNF-α treatment and bound to the TaRE of P4Hα1 in the native condition. Using Western blot analysis, we found that TNF-α did not alter the NonO expression nor the JNK inhibitor (Figure 4A). The finding indicates that the increased NonO-TaRE binding in TNF-α–treated HASMCs may be caused by other mechanisms such as post-translational modification, which increases the binding affinity.
Figure 3.
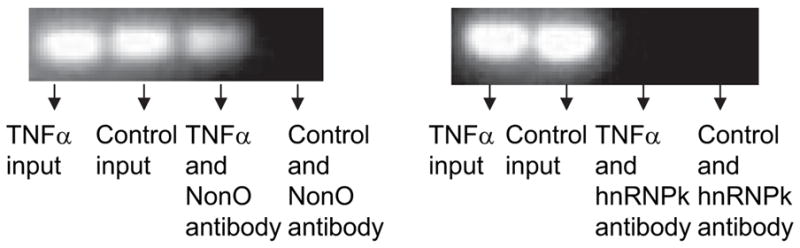
ChIP assay for the NonO or hnRNP-K proteins and P4Hα1 promoter interaction. HASMC nuclei were extracted by using the Nuclear and Cytoplasmic Extraction Reagents kit (Pierce). HASMCs were treated with or without 100 ng/mL TNF-α for 8 hours before nuclear extraction. The ChIP assay was then performed. Anti-human NonO or hnRNP-K antibodies were used in immunoprecipitation. The recovered protein-bound DNA was amplified by PCR with primers that cover the −60 to +104bp region of the P4Hα1 promoter. The PCR products were subjected to electrophoresis in 1.5% agarose gel. NonO-P4Hα1 promoter binding was only found in the TNF-α–treated cells. However, hnRNP-K did not appear to directly bind with the P4Hα1 promoter in the ChIP assay.
Figure 4.

Western blot of the NonO expression in HASMCs. A, Expression of the NonO protein in HASMCs treated by 100 ng/mL TNF-α for 8 hours with or without JNK inhibitor. B, HASMCs were treated with NonO specific siRNA with or without TNF-α treatment. After the treatment, total cellular proteins were extracted and measured using Bradford method. Equal amount of proteins were electrophoresized in 8% SDS-PAGE gel. The NonO band was detected with anti-human NonO antibody and anti-rabbit IgG-HRP conjugate using ECL chemiluminescent detection method.
NonO Responsible for TNF-α–Mediated P4Hα1 Suppression Through TaRE
To investigate the role of NonO in TNF-α–mediated P4Hα1 suppression, we used NonO-specific siRNA to suppress NonO expression before TNF-α treatment. The NonO siRNA was cotransfected with the P4Hα1 to 32-pGL3 vectors to the HASMCs. Twenty-four hours after the transfection, HASMCs were treated with TNF-α for 8 hours. We confirmed the siRNA-induced NonO silencing by measuring NonO protein levels using Western blot analysis and found an 80% reduction in the NonO protein levels (Figure 4B). NonO silencing abolished more than 60% of TNF-α–mediated P4Hα1 suppression (Figure 5). NonO silencing alone did not change P4Hα1 promoter activity significantly (Figure 5A). In contrast, hnRNP-K silencing did not seem to affect TNF-α–mediated P4Hα1 suppression (Figure 5A). The rescuing effect of the NonO silencing on TNF-α–mediated P4Hα1 suppression was also reflected in the P4Hα1 mRNA levels of the cultured HASMCs (Figure 5B). We also tested the possible involvements of BUB3 and ILF2 in the TNF-α–induced P4Hα1 repression using the gene specific siRNA. The results showed no effects by the BUB3 siRNA and ILF2 on TNF-α–induced P4Hα1 suppression (data not shown). To define the NonO binding sequence on P4Hα1 promoter, we transfected 2 P4Hα-pGL3 vectors into HASMCs. As demonstrated by the mRNA levels of the luciferase gene, deletion of the −32 to 15bp region abolished TNF-α–mediated P4Hα1 suppression. The result suggests that the binding site for the NonO protein is located within the sequence of GTTATA-AAAGGGCTA from −32 to −15bp in the P4Hα1 promoter.
Figure 5.

Effects of NonO and hnRNP-K on TNF-α–mediated P4Hα1 promoter suppression. A, Expression of NonO or hnRNP-K siRNA was silenced by transfection with NonO- and hnRNP-K–specific siRNAs at the same time that pGL3-P4Hα1 promoter vectors were transfected to HASMCs. Twenty-four hours after the transfection, cells were treated with or without 100 ng/mL TNF-αfor 8 hours. Luciferase mRNA levels measured by real-time RT-PCR were used to evaluate the effects. B, Expression of NonO or hnRNP-K was silenced by specific siRNA in HASMCs with no simultaneous pGL3 transfection. Twenty-four hours after the siRNA transfection, HASMCs were treated with or without 100 ng/mL TNF-αfor 8 hours. The levels of P4Hα1 mRNA were measured and compared among different conditions. ANOVA was used for comparisons among groups, and Bonferroni post hoc analysis was used to test between-group differences. **P<0.01 for TNF-αtreatment vs all other conditions except cells treated with TNF-α+hnRNPk siRNA. There was no statistical difference between controls and cells treated with NonO siRNA alone, TNF-α+NonO siRNA, and hnRNPK siRNA alone.
Involvement of the ASK1–JNK Pathway
Whereas several pathways can be triggered by TNF-α, c-Jun N-terminal kinase (JNK) appears to be one of the key downstream molecules responsible for the transcriptional effect of TNF-α. Several pathways can lead to JNK activation. Among them, apoptosis signal-regulating kinase 1 (ASK1) may be more relevant to stress and inflammation-related stimulation. We therefore investigated the relevance of the ASK1-JNK pathway to TNF-α–mediated P4Hα1 suppression. JNK inhibitor (SP600125, Sigma) and ASK1 inhibitor (Thioredoxin, Sigma) were used to suppress the signaling pathway. After HASMCs were transfected with P4Hα1 to 32-pGL3 or P4Hα1 to 271-pGL3 vectors, cells were treated with JNK or ASK1 inhibitor for 1 hour before TNF-α treatment for 8 hours. We then measured luciferase mRNA levels. We found that JNK inhibitor recovered almost 100% of the TNF-α–mediated P4Hα1 suppression (Figure 6A). This recovery was also evident in the P4Hα1 mRNA levels of the nontransfected HASMCs (Figure 6B). Furthermore, after treatment with the ASK1 inhibitor, TNF-α–mediated P4Hα1 suppression recovered to a degree similar to that observed in both reporter vector promoter activity (Figure 6C) and P4Hα1 transcription (Figure 6D). These results suggest that the ASK1–JNK pathway is involved in TNF-α–mediated P4Hα1 suppression. To further identify the JNK isoform responsible for the effects, we transfected HASMCs with the wild-type and dominant negative JNK1 overexpression vectors. Whereas wild-type JNK1 induced a 67% reduction in P4Hα1 expression, the dominant negative JNK1 vector resulted in no change in P4Hα1 mRNA levels.
Figure 6.
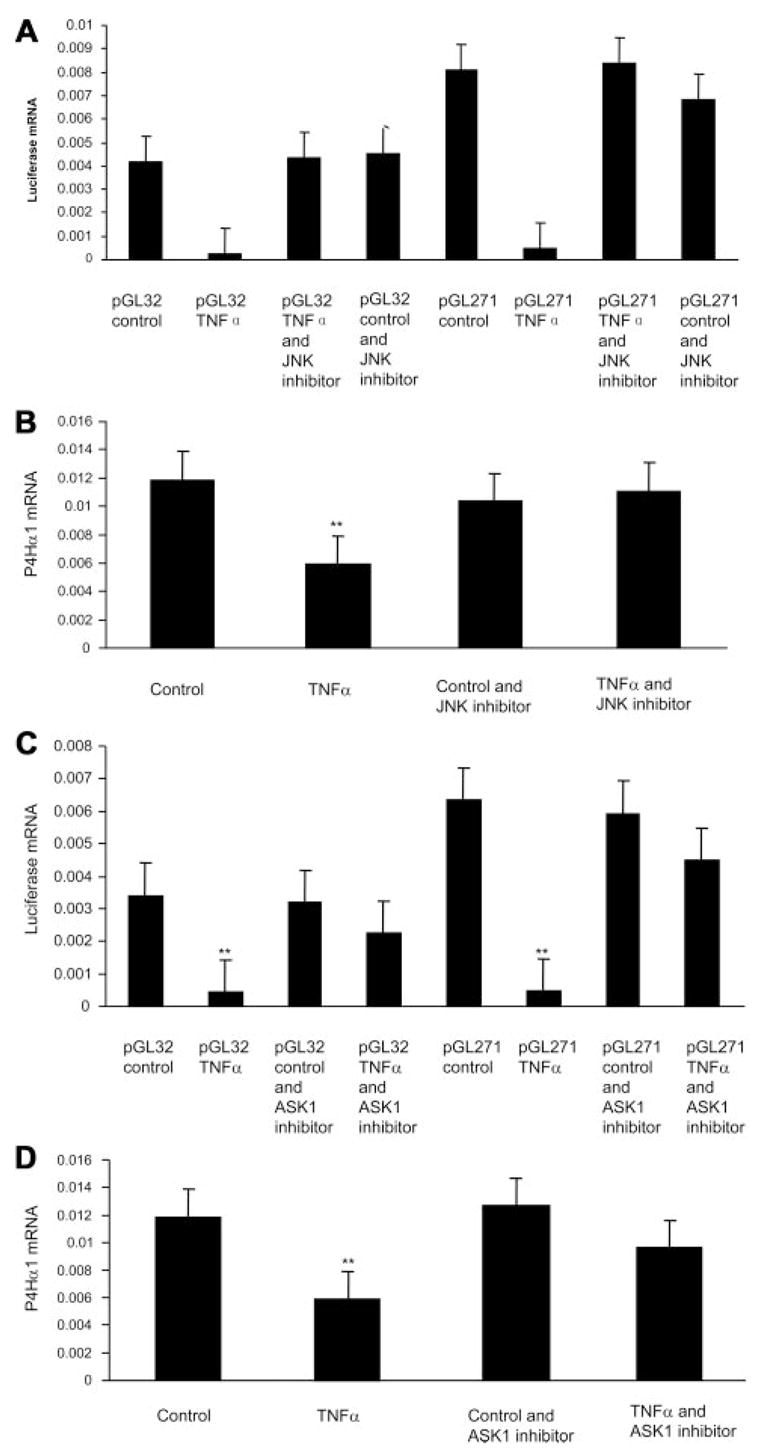
ASK1-JNK pathway is involved in TNF-α–mediated P4Hα1 suppression. Levels of luciferase and P4Hα1 mRNA were measured with quantitative real-time RT-PCR. A, HASMCs were transfected with pGL-32 or pGL-271 (a parallel control) constructs for 24 hours. The JNK inhibitor SP600125 (50 μmol/L) was added 1 hour before cells were treated with or without TNF-α for 8 hours. B, HASMCs were treated with 50 μmol/L JNK inhibitor SP600125 for 1 hour, followed by treatment with or without 100 ng/mL TNF-α for 8 hours. C, HASMCs were transfected with pGL-32or pGL-271 constructs for 24 hours. The ASK1 inhibitor thioredoxin (200 ng/mL) was added 1 hour before cells were treated with or without TNF-α for 8 hours. D, HASMCs were treated with 200 ng/mL ASK1 inhibitor thioredoxin for 1 hour, followed by treatment with or without 100 ng/mL TNF-α for 8 hours. **P<0.01 for pGL-32 (or pGL-271) TNF-α vs all other treatment conditions.
Discussion
Inflammation, one of the most dynamic biological processes, is a natural response to the encountering of endogenous defenses and external invaders. Although regulated inflammation in certain tissues is beneficial for health maintenance, uncontrolled inflammatory processes may be harmful, and participating in the development and progress of most cardiovascular diseases.1 Of the many cytokines, TNF-α is the most active, and it is significantly elevated during inflammation. Our study revealed a novel pathway in which TNF-α suppresses collagen synthesis by specifically inhibiting one of the key enzymes—P4Hα1—in essential collagen post-translational modification, a process that may be implicated in the pathogenesis of vulnerable atherosclerotic plaque and aortic aneurysm. We have described the TaRE in the P4Hα1 promoter and the transcription factor NonO that is responsible for the suppressive effect of TNF-α, which is mediated through ASK1–JNK pathway.
Functional tissue structure, such as the distensibility of an artery, is maintained by a balanced ECM metabolism of production and degradation. Disproportionately low production or overdegradation can both result in weakened tissue structure, which predisposes the tissue to distension or rupture, as in aortic aneurysms and dissection. TNF-α has been reported to activate MMPs,6 which leads to increased degradation of ECM components, including elastin and collagen. On the other hand, TNF-α also downregulates α1(I) collagen and α2(I) collagen expression through a p20C/EBPb and C/EBP mechanism at the transcription level.24 Both actions will tilt the balance toward disrupted ECM and weakened tissue structure. Our study has revealed an additional mechanism that disrupts the collagen maturation and deposition, which inevitably result in a disarrayed ECM structure and weaken the affected tissue. Together with the findings of others, our results suggest that reduced ECM production and increased degradation induced by TNF-α contribute to the formation of vulnerable atherosclerotic plaques and aortic aneurysms and dissection.
It would be therapeutically valuable if interventional strategies can be developed in TNF-α– or inflammation-initiated P4Hα inhibition and collagen reduction. Our study constitutes a step toward this goal because it characterized molecular mechanisms responsible for this relationship. Previously, we have reported that the E-box–like sequences located at −184 to −97bp are responsible for basal human P4Hα1 expression,17 which is different from that in the rat P4Hα1 promoter.16 In the current study, we located a NonO binding sequence at −32 to +18bp region that acts as a TaRE and controls 70% of TNF-α–mediated P4Hα suppression. NonO is a 55-kDa ubiquitously expressed protein originally identified as a non–POU domain-containing octamer-binding protein.25 NonO is highly homologous to the C terminus of PSD18; NonO can bind via its NTH domain to double-stranded DNA with restricted A/T specificity reminiscent of octamer motifs.26 In addition to its own DNA-binding abilities, NonO has the ability to induce the binding of several transcription factors to their response elements and bind directly to Spi-1/PU.1 transcription factors.18,27 It also enhances the binding of some sequence-specific transcription factors (E47, OTF-1, and OTF-2) to their recognition sites.25 It is obvious that the suppressing effect of NonO is not specific to P4Hα or TNF-α signaling because other genes such as CYP17 were also repressed by NonO in a complex with corepressors (Sin3A, HDAC).28
When cells are exposed to TNF-α, several intracellular pathways can be triggered, among which JNK—an important member of the mitogen-activated protein kinase (MAPK) superfamily—appears to be most important for mediating transcription-factor–gene interactions.29 To activate JNK, TNF-α first binds to TNFR1 on the cell surface,30 and activates the downstream cascade.31 Using a JNK-specific inhibitor (SP600125) or the ASK1-specific inhibitor (thioredoxin) to block the AKS1–JNK pathway has successfully prevented the TNF-α–mediated P4Hα1 inhibition in HASMCs. Our study indicates that TNF-α may activate the ASK1–JNK–NonO pathway that directly suppresses P4Hα1 expression, which represents an important class of gene regulators that mediate responses to stimulation. These regulators may be more responsive to manipulation than maintaining the baseline expression and are potentially useful therapeutic targets.
In summary, our study shows that TNF-α can effectively inhibit P4Hα1 expression. This effect represents a previously undiscovered mechanism for the progressive structural destruction that occurs during inflammation and contributes to disease processes such as aortic aneurysm formation. The discovery of TaRE at the P4Hα1 promoter region and the revelation of molecular events mediating the TNF-α suppression, which are distinct from the mechanisms responsible for the basal expression of TNF-α, suggest unique therapeutic targets for modulating inflammation-induced pathologic processes without changing basal TNF-α expression. Disrupting TNF-α–mediated ECM destruction caused by inflammation may reinforce the fibrous cap of the atherosclerotic plaque, rendering it less vulnerable to rupture. Inhibition of the TNF-α activation may strengthen the dissected aorta and make it less likely to dilate. The importance of differentiating among the elements of basal and responsive expressions cannot be overemphasized and should be investigated for all genes involved in environmentally induced pathogenesis.
Acknowledgments
Sources of Funding
This work is supported by an Established Investigator Award (AHA-0440001N) from the American Heart Association to Dr Xing Li Wang, and a National 973 Research Project (No. 2006CB503803) grant awarded to Dr Yun Zhang.
Footnotes
Disclosures
None.
References
- 1.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 2.Chamorro A, Hallenbeck J. The harms and benefits of inflammatory and immune responses in vascular disease. Stroke. 2006;37:291–293. doi: 10.1161/01.STR.0000200561.69611.f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libby P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 4.Rahkonen O, Su M, Hakovirta H, Koskivirta I, Hormuzdi SG, Vuorio E, Bornstein P, Penttinen R. Mice with a deletion in the first intron of the Col1a1 gene develop age-dependent aortic dissection and rupture. Circ Res. 2004;94:83–90. doi: 10.1161/01.RES.0000108263.74520.15. [DOI] [PubMed] [Google Scholar]
- 5.Wilson WR, Anderton M, Schwalbe EC, Jones JL, Furness PN, Bell PR, Thompson MM. Matrix metalloproteinase-8 and -9 are increased at the site of abdominal aortic aneurysm rupture. Circulation. 2006;113:438–445. doi: 10.1161/CIRCULATIONAHA.105.551572. [DOI] [PubMed] [Google Scholar]
- 6.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 7.Greenwel P, Tanaka S, Penkov D, Zhang W, Olive M, Moll J, Vinson C, Di Liberto M, Ramirez F. Tumor necrosis factor alpha inhibits type I collagen synthesis through repressive CCAAT/enhancer-binding proteins. Mol Cell Biol. 2000;20:912–918. doi: 10.1128/mcb.20.3.912-918.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verrecchia F, Wagner EF, Mauviel A. Distinct involvement of the Jun-N-terminal kinase and NF-kappaB pathways in the repression of the human COL1A2 gene by TNF-alpha. EMBO Rep. 2002;3:1069–1074. doi: 10.1093/embo-reports/kvf219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plenz GA, Deng MC, Robenek H, Volker W. Vascular collagens: spotlight on the role of type VIII collagen in atherogenesis. Atherosclerosis. 2003;166:1–11. doi: 10.1016/s0021-9150(01)00766-3. [DOI] [PubMed] [Google Scholar]
- 10.Kivirikko KI, Helaakoski T, Tasanen K, Vuori K, Myllyla R, Parkkonen T, Pihlajaniemi T. Molecular biology of prolyl 4-hydroxylase. Ann N Y Acad Sci. 1990;580:132–142. doi: 10.1111/j.1749-6632.1990.tb17925.x. [DOI] [PubMed] [Google Scholar]
- 11.Kivirikko KI, Pihlajaniemi T. Collagen hydroxylases and the protein disulfide isomerase subunit of prolyl 4-hydroxylases. Adv Enzymol Relat Areas Mol Biol. 1998;72:325–398. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 12.Rocnik EF, Chan BM, Pickering JG. Evidence for a role of collagen synthesis in arterial smooth muscle cell migration. J Clin Invest. 1998;101:1889–1898. doi: 10.1172/JCI1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Annunen P, Autio-Harmainen H, Kivirikko KI. The novel type II prolyl 4-hydroxylase is the main enzyme form in chondrocytes and capillary endothelial cells, whereas the type I enzyme predominates in most cells. J Biol Chem. 1998;273:5989–5992. doi: 10.1074/jbc.273.11.5989. [DOI] [PubMed] [Google Scholar]
- 14.Nissi R, Autio-Harmainen H, Marttila P, Sormunen R, Kivirikko KI. Prolyl 4-hydroxylase isoenzymes I and II have different expression patterns in several human tissues. J Histochem Cytochem. 2001;49:1143–1153. doi: 10.1177/002215540104900908. [DOI] [PubMed] [Google Scholar]
- 15.Raveendran M, Senthil D, Utama B, Shen Y, Dudley D, Wang J, Zhang Y, Wang XL. Cigarette suppresses the expression of P4Halpha and vascular collagen production. Biochem Biophys Res Commun. 2004;323:592–598. doi: 10.1016/j.bbrc.2004.08.129. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi S, Dohi N, Takahashi Y, Miura T. Cloning and characterization of the 5′-flanking region of the rat P4Halpha gene encoding the prolyl 4-hydroxylase alpha(I) subunit. Biochim Biophys Acta. 2002;1574:354–358. doi: 10.1016/s0167-4781(01)00360-8. [DOI] [PubMed] [Google Scholar]
- 17.Chen L, Shen YH, Wang X, Wang J, Gan Y, Chen N, Lemaire SA, Coselli JS, Wang XL. Human Prolyl-4-hydroxylase {alpha}(I) Transcription Is Mediated by Upstream Stimulatory Factors. J Biol Chem. 2006;281:10849–10855. doi: 10.1074/jbc.M511237200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shav-Tal Y, Zipori D. PSF and p54(nrb)/NonO–multifunctional nuclear proteins. FEBS Lett. 2002;531:109–114. doi: 10.1016/s0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- 19.He Y, Zhang W, Zhang R, Zhang H, Min W. SOCS1 inhibits tumor necrosis factor-induced activation of ASK1-JNK inflammatory signaling by mediating ASK1 degradation. J Biol Chem. 2006;281:5559–5566. doi: 10.1074/jbc.M512338200. [DOI] [PubMed] [Google Scholar]
- 20.Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, Izumo S. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- 21.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 22.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct. 2001;30:421–455. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- 24.Kahari VM, Chen YQ, Su MW, Ramirez F, Uitto J. Tumor necrosis factor-alpha and interferon-gamma suppress the activation of human type I collagen gene expression by transforming growth factor-beta 1. Evidence for two distinct mechanisms of inhibition at the transcriptional and posttranscriptional levels. J Clin Invest. 1990;86:1489–1495. doi: 10.1172/JCI114866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang YS, Yang MC, Tucker PW, Capra JD. NonO enhances the association of many DNA-binding proteins to their targets. Nucleic Acids Res. 1997;25:2284–2292. doi: 10.1093/nar/25.12.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang YS, Hanke JH, Carayannopoulos L, Craft CM, Capra JD, Tucker PW. NonO, a non-POU-domain-containing, octamer-binding protein, is the mammalian homolog of Drosophila nonAdiss. Mol Cell Biol. 1993;13:5593–5603. doi: 10.1128/mcb.13.9.5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hallier M, Tavitian A, Moreau-Gachelin F. The transcription factor Spi-1/PU. 1 binds RNA and interferes with the RNA-binding protein p54nrb. J Biol Chem. 1996;271:11177–11181. doi: 10.1074/jbc.271.19.11177. [DOI] [PubMed] [Google Scholar]
- 28.Sewer MB, Waterman MR. Adrenocorticotropin/cyclic adenosine 3′,5′-monophosphate-mediated transcription of the human CYP17 gene in the adrenal cortex is dependent on phosphatase activity. Endocrinology. 2002;143:1769–1777. doi: 10.1210/endo.143.5.8820. [DOI] [PubMed] [Google Scholar]
- 29.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 30.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 31.Pantano C, Shrivastava P, McElhinney B, Janssen-Heininger Y. Hydrogen peroxide signaling through tumor necrosis factor receptor 1 leads to selective activation of c-Jun N-terminal kinase. J Biol Chem. 2003;278:44091–44096. doi: 10.1074/jbc.M308487200. [DOI] [PubMed] [Google Scholar]; Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]