

Abstract
To potentiate the response of acute myelogenous leukemia (AML) cells to TNF-Related Apoptosis-Inducing Ligand (TRAIL) cytotoxicity, we have examined the efficacy of a combination with perifosine, a novel phosphatidylinositol 3-kinase (PI3K)/Akt signaling inhibitor. The rationale for using such a combination is that perifosine was recently described to increase TRAIL-R2 receptor expression and decrease the cellular FLICE-Inhibitory Protein (cFLIP) in human lung cancer cell lines. Perifosine and TRAIL both induced cell death by apoptosis in the THP-1 AML cell line, which is characterized by constitutive PI3K/Akt activation, but lacks functional p53. Perifosine, at concentrations below IC50, dephosphorylated Akt and increased TRAIL-R2 levels, as demonstrated by western blot, RT-PCR, and flow cytometric analysis. Perifosine also decreased the long isoform of cFLIP (cFLIP-L) and the X-linked Inhibitor of Apoptosis Protein (XIAP) expression. Perifosine and TRAIL synergized to activate caspase-8 and induce apoptosis, which was blocked by a caspase- 8 selective inhibitor. Upregulation of TRAIL-R2 expression was dependent on a protein kinase Cα/c-Jun-NH2-kinase 2/c-Jun signaling pathway activated by perifosine through reactive oxygen species production. Perifosine synergized with TRAIL also in primary AML cells displaying constitutive activation of the Akt pathway, by inducing apoptosis, Akt dephosphorylation, TRAIL-R2 upregulation, cFLIP-L and XIAP downregulation, and c-Jun phosphorylation. The combined treatment negatively affected the clonogenic activity of CD34+ cells from AML patients. In contrast, CD34+ cells from healthy donors were resistant to perifosine and TRAIL treatment. Our findings suggest that the combination perifosine and TRAIL might offer a novel therapeutic strategy for AML.
Keywords: Akt signaling, apoptosis, caspase-8, TRAIL, combination therapy
INTRODUCTION
The TNF family member TNF-Related Apoptosis-Inducing Ligand (TRAIL) was originally reported to induce apoptosis in many tumor cells but not in normal cells both in vitro and in vivo and thus represents a promising anticancer cytokine (1). TRAIL is expressed as a type-II TNF transmembrane protein, however its extracellular domain can be proteolytically cleaved from the cell surface and acts as a soluble cytokine. TRAIL transmits the death signal via TRAIL-R1 and TRAIL-R2 (also referred to as DR4 and DR5, respectively) receptors, which, upon TRAIL binding, are oligomerized at the cell surface, thereby enabling the recruitment of the adaptor molecule Fas- Associated Death Domain (FADD) and assembly of the Death-Inducing Signaling Complex (DISC) (2). Two other TRAIL receptors, TRAIL-R3 and TRAIL-R4 (also referred to as DcR1 and DcR2) contain no or only a truncated death domain and do not induce apoptosis upon TRAIL binding. TRAIL-R3 and –R4 act, therefore, as decoy receptors (3). It has been suggested that preferential expression of decoy receptors on normal cells is one of the mechanisms underlying the proapoptotic action of TRAIL on neoplastic but not healthy cells (4). Upon binding of TRAIL to –R1 and –R2 receptors, the extrinsic apoptosis pathway is activated (3). In recent years, TRAIL has stimulated hope for its therapeutic potential as an anti-neoplastic agent in different types of tumors, including hematological malignancies such as acute myelogenous leukemia (AML) (5). The in vitro cytotoxic response of AML cell lines to recombinant TRAIL varies from good to moderate (6, 7), however, a number of in vitro studies have convincingly demonstrated that AML primary cells are resistant to the proapoptotic activity of TRAIL used as a single agent (e.g. 8). TRAIL sensitivity of AML blasts could be increased by cotreatment with cytotoxic drugs such as daunorubicin (9) or histone deacetylase inhibitors (10). A recent report has highlighted that TRAIL sensitivity of human lung cancer cell lines could be considerably increased by cotreatment with the novel Akt inhibitor, perifosine (11). The phosphatidylinositol (PI3K)/Akt signaling pathway is activated in many AML patients (12–14) and markedly influences AML sensitivity to various drugs, including TRAIL (6). Therefore, small molecules which inhibit this pathway are currently being developed for clinical use, including perifosine (15). Perifosine is a phospholipid analogue which has shown promising preclinical activity and is currently undergoing phase I/II clinical evaluation, also for AML treatment. Serum concentrations up to 20 μM perifosine, have been reached during clinical evaluation (16, 17). We have recently demonstrated the cytotoxic activity of perifosine, alone or in combination with chemotherapeutic drugs, in AML cells (18). Therefore, it was investigated whether perifosine could increase AML cell sensitivity to recombinant TRAIL. Here, we demonstrate in THP-1 AML cells that perifosine increased TRAIL-R2 expression and decreased levels of the long isoform of the cellular FLICE-Inhibitory Protein (cFLIP-L) and X-linked Inhibitor of Apoptosis Protein (XIAP) at concentrations below the IC50. When perifosine was combined with TRAIL, there was a synergistic induction of apoptosis and increased levels of caspase-8 activation. Similar results were obtained using AML blasts with constitutively active PI3K/Akt pathway. Perifosine and TRAIL combined treatment also decreased the clonogenic activity of CD34+ cells from AML patients with active Akt, while it had no effect on CD34+ cells from healthy donors. Therefore, our findings suggest that perifosine, in combination with TRAIL, may represent an effective approach for treatment of AML patients.
MATERIALS AND METHODS
Chemicals and antibodies
Perifosine was provided by AEterna Zentaris GmbH, Frankfurt, Germany. For cell viability determination, Cell Viability Kit I (3-[4, 5-diMethylThiazol-2-yl]-2, 5-diphenylTetrazolium bromide, or MTT) was purchased from Roche Applied Science, Penzberg, Germany. Propidium Iodide (PI, DNA-Prep kit) was from Beckman Coulter Immunology, Miami, FL, USA. The Annexin V-FITC (Fluorescein IsoThioCyanate) staining kit was from Tau Technologies BV, Kattendijke, The Netherlands, while carboxyfluorescein FLICA (Fluorescent-Labeled Inhibitor of CAspases) Apoptosis Detection kit for caspase activity assay was from AbD Serotec, Oxford, UK. Recombinant human TRAIL, the c-Jun NH2-terminal kinase (JNK) inhibitor SP600125, and the p38 MAP kinase inhibitor SB203580, were from EMD Biosciences, La Jolla, CA, USA. The Protein Kinase C (PKC) inhibitor Gö6976, Phorbol 12-Myristate 13-Acetate (PMA), the Reactive Oxygen Species (ROS) scavenger N-Acetyl-L-Cysteine (NAC), and DiChlorodiHydroFuorescein DiAcetate (DCHF-DA) were from Sigma-Aldrich, St. Louis, MO, USA. Antibodies to the following proteins were employed for western blot analysis: Akt, Ser 473 p-Akt, X-linked Inhibitor of Apoptosis Protein (XIAP), FADD, PKCα, caspase-8, Ser 638/641 p-PKCα/β, c-Jun, Ser 63 p-c-Jun, Thr183/Tyr185 p-JNK 1/2, and β-tubulin were from Cell Signaling, Danvers, MA, USA; PKCβ2 was from Santa Cruz Biotechnology, Santa Cruz, CA, USA; TRAIL receptors -R1, -R2, -R3, -R4, and cFLIP-S/L (which recognizes both the short and the long isoform of the protein) were from ProSci Inc., Poway, CA, USA. Horseradish peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Cell Signaling Technology. For flow cytometric analysis: AlexaFluor 488-conjugated anti-Ser 473 p-Akt was from Cell Signaling; mouse antihuman TRAIL receptors antibodies conjugated to PhycoErythrin (PE) were from R&D Systems, Minneapolis, MN, USA; mouse anti-human CD33 FITC-conjugated antibody was from BD Biosciences Pharmingen, San Diego, CA, USA. Controls were performed with normal rabbit IgG conjugated to AlexaFluor 488 or normal mouse IgG conjugated to either PE or FITC (Upstate, Charlottesville, VA, USA).
Cell culture, patients, and clonogenic assays
THP-1 human acute monocytic leukemia cells were grown as previously reported (18). Samples were obtained from patients at presentation of AML, before chemotherapy treatment. Informed consent was obtained from all patients prior to obtaining the samples, according to Institutional guidelines. Bone marrow or peripheral blood mononuclear cells were isolated by Ficoll-Paque (Amersham Biosciences, Uppsala, Sweden) density-gradient centrifugation. Percentage of blasts in the samples ranged between 75% and 91% and was checked by flow cytometry staining, depending on the phenotype of the leukemia (usually CD13, CD33, CD34, CD45, alone or in combination). Blasts cells were cultured in RPMI 1640 medium supplemented with 20% FCS. CD34+ progenitor cells from AML patients or from cord blood were isolated using immunomagnetic cell separation (Miltenyi Biotec, Bergisch Gladbach, Germany) and cultured as reported previously (14).
Cell viability analysis by MTT assay
An MTT assay was employed to analyze cell growth and viability, as reported elsewhere (19)
Flow cytometric detection of apoptosis and ROS generation
Whole cell lysate preparation, cell fractionation, western blot, and densitometric analysis of blots
This was performed as described (14). For analysis of THP-1 cells, 40 μg protein per lane was loaded, whereas for AML blasts, 80 μg protein per lane was loaded. Densitometric analysis was as reported by Nyakern et al. (21). For each blot, the band with the highest intensity was normalized to 1, while other bands were expressed as fraction. Values from densitometric scanning are indicated above each protein band. All the blots shown are representative of at least three separate experiments.
Immunoprecipitation
Cells were lysed in 50 mM Tris, pH 8.0, 50 mM KCl, 10 mM EDTA, 1% Nonidet P-40, Protease Inhibitor Cocktail, 2 mM Na3VO4. Immunoprecipitation was performed overnight using polyclonal antibodies to either PKCα or β2, according to standard procedures. Antibodies were captured using Protein A/G-agarose and immunoprecipitates washed with lysis buffer.
Caspase activity assay
Flow cytometric assays were performed to determine caspase activity, using the FLICA Apoptosis Detection kit according to the manufacturer’s instructions, as reported elsewhere (18).
Flow cytometric detection of Ser 473 p-Akt, TRAIL receptors, and CD33
P-Akt detection was performed essentially as previously reported (13). Anti-TRAIL, -TRAIL receptors, and -CD33 antibodies (final concentration: 10 μg/mL) were employed on fresh, unfixed cells (5 × 105), according to the manufacturer’s procedure. Then, cells were washed three times with PBS, fixed with 0.5% paraformaldehyde in PBS, washed again three times with PBS, and immunostained for p-Akt. At least 5000 events were analyzed for each sample in all flow cytometric analyses. All the flow cytometric data are representative of three different experiments.
RT-PCR analysis for TRAIL-RI and R2 mRNA
This was performed exactly according to Zhang et al. (22). PCR-amplified products were electrophoresed on a 1.5% agarose gel containing 0.5 μg/ml ethidium bromide and were visualized under UV light.
Combined drug effects analysis
To characterize the interactions between TRAIL and perifosine, the combination effect and a potential synergy was evaluated from quantitative analysis of dose-effect relationships described by Chou and Talalay (23). The CalcuSyn software was employed (Biosoft, Cambridge, UK) to calculate combination index (CI).
Transient protein downregulation by short interfering RNA (siRNA)
Scrambled (sc-44230) and specific siRNAs to either PKCα (sc-36243) or c-Jun (sc-29223) were from Santa Cruz Biotechnology. Transfection of THP-1 cells was performed using the Amaxa system (Amaxa, Cologne, Germany) following their specifications (24). Briefly, 106 cells in 100 μL of medium was mixed with 3 μg of siRNA and transferred to an Amaxa-certified cuvette. For transfection, we used the program V-01. Transfection efficiency was between 75% and 85% (data not shown), as checked by flow cytometry, using a fluorescein-labeled non-targeted siRNA control (Cell Signaling). Cells were examined for gene downregulation and other properties 48 h after transfection.
Statistical evaluation
The data are presented as mean values from 3 separate experiments ± sd. Data were statistically analyzed by a Dunnet test after one-way analysis of variance (ANOVA) at a level of significance of p < 0.05 vs. control samples.
RESULTS
Effect of TRAIL and perifosine on THP-1 cell survival
Exposure for 24 hr to increasing concentrations of recombinant TRAIL or perifosine, induced a dose-dependent decrease in cell survival, as evaluated by MTT assays (Figure 1A). At the highest tested concentration of TRAIL (800 ng/ml) or perifosine (16 μM), survival was 60% or 32%, respectively. To establish whether a combined treatment consisting of TRAIL and perifosine was synergistic, THP-1 cells were cultured with serial concentrations of TRAIL (range: 12.5–800 ng/ml) and perifosine (range: 0.25–16.0 μM) at a constant ratio, for 24 hr and data were analyzed by the method of Chou and Talalay (23). The combined treatment was much more cytotoxic than either of the single treatments. All the combinations gave an effect which ranged from synergistic (CI<0.6) to highly synergistic (CI<0.3) (Figure 1B). To determine whether decreased cell survival was related to apoptosis, an Annexin V-FITC/PI analysis was performed. When samples were analyzed by flow cytometry, it became evident that the combined TRAIL and perifosine treatment induced apoptotic cell death of THP-1 cells, whereas when the single drugs were used alone, much lower effects were observed (Figure 1C). Western blot analysis demonstrated a marked decrease in Ser 473 p-Akt phosphorylation already at 0.5 μM perifosine. Akt dephosphorylation was complete at 1.0 μM, while total Akt levels remained unchanged (Figure 1D).
Figure 1. Perifosine synergistically enhances TRAIL-induced cell death in THP-1 cells.
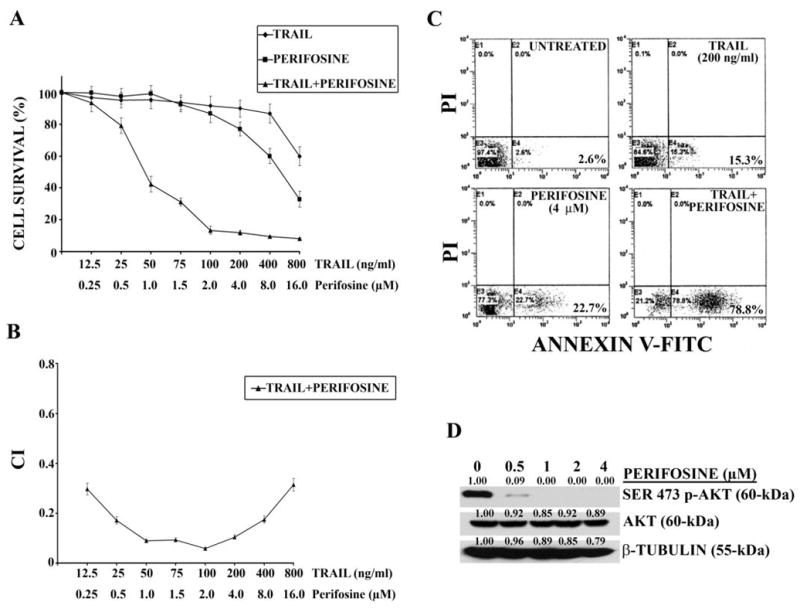
A: cells were treated for 24 hr with either single agent alone or in combination at the indicated concentrations. Cell viability was then analyzed by MTT assays. B: CI as calculated from experiments reported in A. C: Annexin V-FITC/PI staining analysis of THP-1 cells treated with either perifosine or TRAIL alone and with two drugs together for 24 hr. The numbers in the lower right quadrants correspond to the percentage of cells which are Annexin V-positive and PI-negative (early apoptotic cells). D: western blot analysis demonstrating Ser 473 p-Akt and total Akt levels in THP-1 cells treated with increasing concentrations of perifosine for 16 hr. β-tubulin served as loading control.
Perifosine increases TRAIL-R2 expression in THP-1 and downregulates cFLIP-L and XIAP levels
Given that in human lung carcinoma cells perifosine upregulates TRAIL-R2 expression (11), it was investigated whether this was also true for THP-1 cells. The expression of TRAIL receptors in untreated THP-1 cells was examined by flow cytometry. Under basal conditions, it was observed that these cells expressed TRAIL-R1,-R2, and -R4, but no –R3 (data not shown), in agreement with others (25). Western blot analysis demonstrated that perifosine increased the levels of TRAIL-R2 in a dose dependent manner. The amount of the other TRAIL receptors expressed by THP-1 cells was almost unchanged (Figure 2A). A dose-dependent decrease in c-FLIP-L and XIAP expression was also observed in THP-1 cells treated with perifosine, whereas FADD levels were not affected. Increased expression of TRAIL-R2 protein by western blot was corroborated by RT-PCR analysis, which showed an increase in TRAIL-R2, but not TRAIL-R1, mRNA (Figure 2A). Also flow cytometric analysis highlighted selective enhanced TRAIL-R2 expression in response to perifosine treatment (Figure 2B). Moreover, this technique demonstrated no changes in surface TRAIL expression by perifosine (data not shown)
Figure 2. Perifosine decreases cFLIP-L and XIAP expression, and downregulates TRAIL-R2 expression in THP-1 cells.
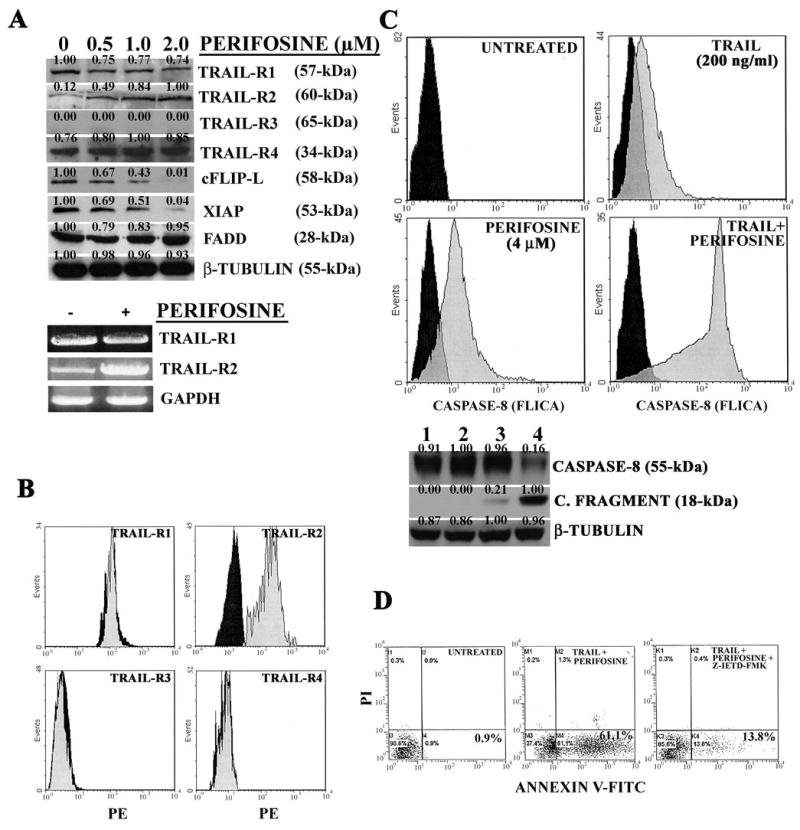
A: western blot and RT-PCR analysis performed on THP-1 cells treated for 16 hr with perifosine. In case of RT-PCR analysis for TRAIL-R1 and –R2 mRNA, perifosine concentration was 0.5 μM for 16 hr. GAPDH served as a control. B: flow cytometric analysis demonstrating surface expression of TRAIL receptors in untreated cells (black shaded histograms) and cells treated with 2.0 μM perifosine (grey shaded histograms) for 16 hr. C: FLICA and western blot analysis of THP-1 cells treated with either perifosine or TRAIL alone and with two drugs together for 24 hr. For FLICA analysis: black shaded histograms correspond to untreated cells, while grey shaded histograms represent drug-treated cells. For western blot analysis: Lane 1: untreated cells; Lane 2: TRAIL-treated cells; Lane 3: perifosine-treated cells; lane 4: TRAIL and perifosine treated cells. For western blot analysis, TRAIL and perifosine concentrations were as for FLICA analysis. Drug treatment was for 24 hr. D: Annexin V-FITC/PI staining analysis of THP-1 cells treated with the two drugs together for 24 hr. Samples had been pre-treated for 1 hr with the caspase-8 inhibitor, Z-IETD-FMK (20 μM). Drug concentrations were as in Panel C. The numbers in the lower right quadrants correspond to the percentage of cells which are Annexin V-positive and PI-negative (early apoptotic cells).
Perifosine and TRAIL combined treatment results in enhanced caspase-8 activation
The combined treatment was associated with increased activation of caspase-8, as demonstrated by FLICA assay (Figure 2C). Western blot analysis corroborated flow cytometric findings, demonstrating a dramatic decrease in procaspase-8 levels and the appearance of the p18 cleaved fragment of caspase-8 in cells treated with perifosine and TRAIL combination (Figure 2C). In contrast, no p18 fragment was detected in cells treated with TRAIL alone, while a very faint band was visible in samples exposed to perifosine alone. The relevance of caspase-8 activation for perifosine-induced apoptosis was demonstrated by experiments in which cells were preincubated with a selective caspase-8 inhibitor (Z-IETD-FMK) prior to being treated with the combined drugs. This resulted in a much lower percentage of apoptotic cells (Figure 2D).
Upregulation of TRAIL-R2 requires PKC activity
A recent report has highlighted that TRAIL-R2 upregulation in non-small cell lung cancer cells required PKC activity (26). Therefore, it was investigated if this was true also in THP-1 cells. Preliminary experiments indicated that THP-1 cells expressed only two of the conventional PKC isoforms, α and β2, whereas β1 and γ were not expressed (data not shown). When cells were treated with perifosine and Gö6976, a well established inhibitor of PKC conventional isoforms (27), the increase in TRAIL-R2 expression was lower. No changes in the expression of other TRAIL receptors (-R1 and –R4) were observed in response to Gö6976 and perifosine treatment, whereas cFLIP-L and XIAP levels did not significantly change in cells treated with Gö6976 and perifosine when compared to perifosine alone (Figure 3A). A time-dependent increase in TRAIL-R2, but not TRAIL-R1, expression levels was detected when THP-1 cells were treated with PMA, an activator of PKC conventional isoforms (28) (Figure 3B). Downregulation of PKCα levels by specific siRNA, but not treatment with scrambled siRNA, resulted in a much lower induction of TRAIL-R2 by perifosine (Figure 3C). MTT assays demonstrated a lower cytotoxic effect of the perifosine and TRAIL combined treatment in cells with downregulated PKCα expression, but not in those treated with scrambled siRNA (Figure 3D). Efficacy of PKCα downregulation by specific siRNA was evaluated by western blot analysis (Figure 3D). Overall, these findings indicated that upregulation of TRAIL-R2 by perifosine was dependent on PKCα and was required to maximally potentiate the proapoptotic effect of TRAIL.
Figure 3. Increased TRAIL-R2 expression in THP-1 cells is dependent on PKCα.

A: western blot analysis of THP-1 cell extracts. Lane 1: untreated cells; Lane 2: cells treated with perifosine (2 μM); Lane 3: cells treated with perifosine (2 μM) + Gö6976 (0.5 μM). Treatments were for 16 hr. B: western blot analysis for TRAIL-R1 and -R2 expression in THP-1 cells treated with PMA (100 ng/mL) for increasing periods of time. C: western blot analysis for TRAIL-R2 expression levels in cells incubated with perifosine (2 μM for 16 h). Cells treated for 48 hr with PKCα specific siRNA or scrambled siRNA. D: results from MTT assays in cells treated with TRAIL and perifosine for 24 hr at the indicated concentrations. The western blot analysis for PKCα levels in THP-1 cell extracts is also shown. Lane 1: untreated cells; Lane 2: cells treated with siRNA specific for PKCα; Lane 3: cells treated with scrambled siRNA. Cells were analyzed 48 hr after transfection. β-tubulin served as loading control.
Uperegulation of TRAIL-R2 by perifosine is dependent on a ROS/PKCα/JNK1/c-Jun pathway
The mechanism of PKCα activation by perifosine was next investigated. It has been shown that PKCα could be activated (phosphorylated) by ROS (29, 30) which also induced its membrane binding. Moreover, perifosine caused ROS production in U937 AML cells (31). Therefore, it was investigated if perifosine caused ROS production also in THP-1 cells. ROS generation was analyzed by flow cytometry after labeling of cells with the ROS selective probe, DCFH-DA. Perifosine (2 μM) caused an increase in ROS levels, which was blocked by the ROS scavenger, NAC (Supplementary Figure 1A). Cell fractionation experiments demonstrated that in response to perifosine treatment, the amount of membrane-bound PKCα increased, whereas cytosolic PKCα decreased (Figure 4A). These changes in PKCα subcellular localization were largely prevented by NAC, suggesting they were dependent on ROS production. Moreover, perifosine treatment resulted in increased Thr 638/641 p-PKCα levels but not PKCβ2 isoform, as demonstrated by immunoprecipitation experiments with antibodies selective for total (unphosphorylated and phosphorylated) PKCα and β2 isoforms, followed by western blot with an antibody which recognizes both PKCα and β1/2 phosphorylated on Thr 638/641 (Figure 4A).
Figure 4. Perifosine induces PKCα and c-Jun activation in THP-1 cells.
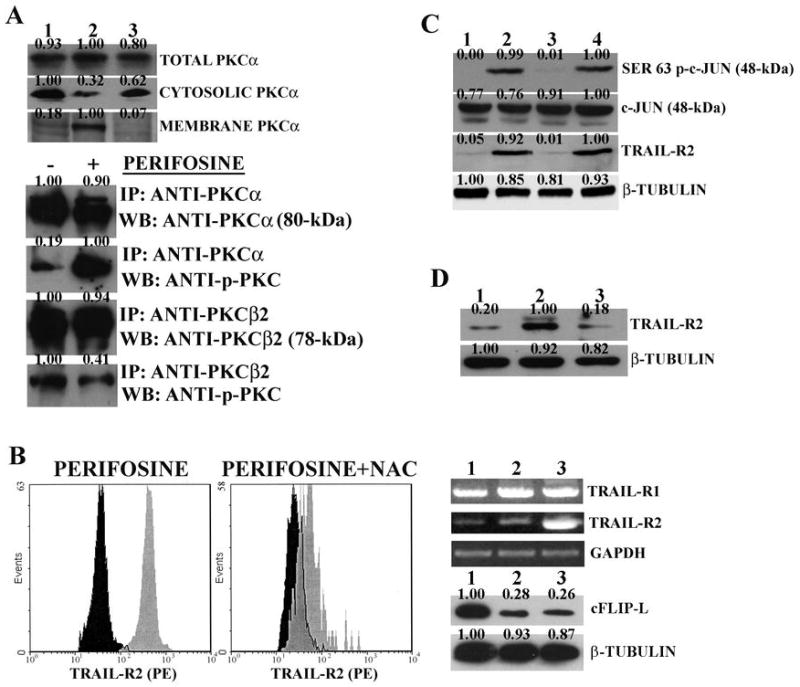
A: western blot analysis for PKCα expression in different subcellular fractions. Lane 1: untreated cells; Lane 2: cells treated with perifosine (2 μM for 16 hr); Lane 3: cells treated with perifosine (2 μM for 16 hr) + NAC (15 mM). Immunoprecipitation of cell extracts from untreated and perifosinetreated (2 μM for 16 hr) samples. IP: antibody used for immunoprecipitation; WB: antibody used for probing the blots. B: flow cytometric analysis RT-PCR, and western blot. Flow cytometric analysis shows surface expression of TRAIL-R2 receptor in untreated cells (black shaded histograms) and cells treated with 2.0 μM perifosine for 16 h (grey shaded histograms) with or without NAC (15 mM). In case of RT-PCR analysis for TRAIL-R1 and –R2 mRNA, perifosine and NAC concentrations were as for flow cytometry. Lane 1: untreated cells; Lane 2: NAC +perifosine; Lane 3: perifosine alone. In case of western blot analysis for cFLIP-L expression, lanes were as for RT-PCR analysis. C: western blot analysis for Ser 63 p-c-Jun, c-Jun, and TRAIL-R2 levels in cells treated with perifosine (2 μM for 16 hr) in the presence or in the absence of JNK 1/2 or p38 MAP kinase inhibitors. Lane 1: untreated cells. Lane 2: perifosine only; Lane 3: perifosine + SP600125 (10 μM); Lane 4: perifosine + SB203580 (5 μM). D: western blot analysis for TRAIL-R2 in cells with downregulated c-Jun levels. Lane 1: untreated cells; Lane 2: cells treated with perifosine (2 μM for 16 hr); Lane 3: perifosine-treated cells with c-Jun downregulated by siRNA specific for c- Jun. β-tubulin served as loading control.
Flow cytometric analysis also demonstrated that the perifosine-induced increase in TRAIL-R2 expression was markedly reduced if cells were treated with NAC in addition to perifosine, while RT-PCR documented that NAC was indeed capable of almost completely blocking the perifosine- dependent increase in TRAIL-R2 mRNA level. However, NAC did not affect the perifosine-evoked decrease in cFLIP-L levels (Figure 4B). Recent findings have highlighted that TRAIL-R2 gene expression could be under the control of a JNK/c-Jun pathway (32) and that in some cell models PKCα could be upstream of JNK (33, 34). Perifosine (2 μM) upregulated p-JNK 2 (54-kDa) but not p-JNK 1 (46-kDa) in THP-1 cells, and this event could be blocked by NAC (Supplementary Figure 1B). Perifosine treatment resulted in c-Jun phosphorylation on Ser 63, and this phosphorylation was inhibited by SP600125 (a JNK 1/2 selective inhibitor) but not by SB203580 (a p38 MAP kinase selective inhibitor). Moreover, SP600125, but not SB203580, suppressed the perifosine-dependent increase in TRAIL-R2 expression (Figure 4C). Finally, when c-Jun levels were downregulated by siRNA specific for c-Jun (Supplementary Figure 1C), the perifosine-evoked increase in TRAIL-R2 expression was reduced significantly (Figure 4D). Taken together, these findings strongly suggested that perifosine could upregulate TRAIL-R2 expression through a ROS→PKCα→JNK 2→c-Jun signaling pathway.
Synergistic cytotoxic effecs of TRAIL/perifosine combined treatment on AML blasts with activated Akt
The efficacy of the perifosine and TRAIL combined treatment was then analyzed on samples obtained from patients with AML. Samples from 12 patients were studied (Table 1). Since levels of caspase-8 could influence TRAIL sensitivity of AML blasts (35), we analyzed samples with comparable expression of caspase-8, as evaluated by western blot analysis (data not shown). Activation of PI3K/Akt signaling was studied by western blot and/or flow cytometric analysis. Seven patients were positive for Ser 473 p-Akt (Table 1). The combination of perifosine and TRAIL was much more effective than either drug alone, as demonstrated by MTT assays (Figure 5A, patient M4#1). Cytotoxicity was due to apoptotic cell death, as documented by Annexin V-FITC/PI staining (Figure 5B), and was characterized by increased TRAIL-R2 expression which was detected in CD33+ AML cells by flow cytometric analysis of samples double stained for TRAIL-R2 and CD33 (Figure 5C). Combined treatment also resulted in higher levels of caspase-8 activation than single treatment, as demonstrated by FLICA analysis (data not shown). Western blot analysis demonstrated Ser 473 p-Akt downregulation by perifosine, in patient blasts with Akt activation, as well as a decrease of both cFLIP-L and XIAP, and unpregulation of Ser 63 p-c-Jun (Figure 5D). FADD levels were not affected by perifosine. In contrast, in patients with no activated Akt, we did not detect increased apoptosis in response to the combined treatment (Supplementary Figure 6A, patient M4#2). In samples from these patients, there was neither increased TRAIL-R2 expression, nor decreased cFLIP-L and XIAP expression. Moreover, c-Jun phosphorylation on Ser 63 was not increased after treatment with perifosine (Supplementary Figure 6B, C). FADD levels did not change in a significant manner. In Table 1, we summarize the results obtained by treating AML samples, showing the effects of perifosine on TRAIL-R2 expression and the average CI of the combined treatment.
TABLE 1. Patient classification and response to combined treatment.
Patients were classified according to the French-American-British (FAB) classification. The levels of Ser 473 p-Akt and TRAIL-R2 were evaluated by flow cytometry and/or western blot. P: perifosine. T: TRAIL. ED: Effective Dose. A CI < 0.9 was considered synergistic, whereas a CI between 0.9 and 1.1. was considered additive. Results are from 3 different experiments ± sd.
| FAB TYPE | Ser 473 p-Akt | TRAIL-R2 UPREG | P+T SYNERGISM AVERAGE CI (ED50+ED75+ED90) |
|---|---|---|---|
| M1 # 1 | ++ | ++++ | 0.23 ± 0.03 |
| M1 # 2 | + | ++ | 0.51 ± 0.05 |
| M2 # 1 | − | − | 0.95 ± 0.10 |
| M2 # 2 | +++ | ++++ | 0.26 ± 0.04 |
| M2 # 3 | +++ | ++++ | 0.15 ± 0.02 |
| M2 # 4 | − | − | 1.08 ± 0.15 |
| M2 # 5 | + | +/− | 0.84 ± 0.09 |
| M2 # 6 | − | − | 0.99 ± 0.12 |
| M2 # 7 | ++ | +++ | 0.39 ± 0.05 |
| M4 # 1 | +++ | ++++ | 0.18 ± 0.02 |
Figure 5. Perifosine and TRAIL combination is synergistic in AML primary cells with activated Akt.

A: cells from patient M4#1 were treated for 48 hr with either single agent alone or in combination at the indicated concentrations. Cell viability was analyzed by MTT assays. B: Annexin V-FITC/PI staining analysis of cells from patient M4#1 treated with either perifosine or TRAIL alone and with two drugs together for 48 hr. The numbers in the lower right quadrants correspond to the percentage of cells which are Annexin V-positive and PI-negative (early apoptotic cells). C: flow cytometric analysis showing surface expression of TRAIL receptors and CD33 in cells from patient M4#1, either untreated or treated with 4.0 μM perifosine for 24 hr. Anti-TRAIL receptors antibodies were conjugated to PE, while anti-CD33 antibody was FITC-conjugated. D: western blot analysis for Ser 473 p-Akt, cFLIP-L, XIAP, FADD, and Ser 63 p-c-Jun in extracts from AML patient primary cells. Perifosine treatment was for 24 hr at 4.0 μM concentration. β-tubulin served as loading control.
Perifosine/TRAIL combined treatment negatively affects clonogenic activity of CD34+ cells from AML patients
Finally, we investigated the cytotoxic effect of perifosine/TRAIL combined treatment on CD34+ cell clonogenic activity from cord blood and AML patients. As expected, neither TRAIL nor perifosine alone influenced clonogenic activity of CD34+ cells from healthy donors, and the same was true of the combined treatment (Supplementary Figure 6D). In contrast, in leukemic CD34+ cells TRAIL moderately affected the clonogenic activity, and perifosine alone exhibited a statistically significant inhibitory effect in some samples (see for example, patient M2#7). However, we consistently observed in all patient samples with activated Akt a strong inhibitory effect of the perifosine and TRAIL combined treatment on the CD34+ cell clonogenic activity.
DISCUSSION
In this study, we demonstrated that perifosine sensitizes AML cells from both a cell line (THP-1) and patients, to TRAIL-induced apoptosis, at least in part via a decrease in cFLIP-L and XIAP levels, and a PKCα-mediated increase in TRAIL-R2 expression. cFLIP-L, which is structurally similar to caspase-8, can be recruited to the DISC to inhibit the binding and activation of caspase-8 and acts as a powerful repressor of TRAIL-induced death signaling (3). Also XIAP, a potent cellular caspase inhibitor, is an important factor in TRAIL-induced cell death (36).
A recent report from Carter et al. (36) has demonstrated that triptolide, an anticancer agent from a Chinese herb, sensitized AML cells to TRAIL by decreasing XIAP levels and increasing TRAIL-R2 expression through a p53-mediated mechanism. However, perifosine was able to upregulate TRAIL-R2 in THP-1 cells which have a non-functional p53 (37). It is established that TRAIL-R2 levels could be regulated through mechanisms which are p53- dependent or independent (38). Among p53-independent mechanisms, it has been demonstrated that the JNK/c-Jun axis could positively regulate TRAIL-R2 transcription (32). A functional Activator Protein-1 (AP-1) binding site has been demonstrated in the promoter region of TRAIL-R2. JNK, by increasing c-Jun phoshorylation, leads to an increase in AP-1 activity (39). Interestingly, we have recently shown JNK-dependent increased AP-1 activity in T-lymphoblastic acute leukemia cells treated with perifosine (19).
Our findings point to PKCα as an important mediator of TRAIL-R2 upregulation in THP-1 cells, as PKCα downregulation by siRNA resulted in a much lower induction of TRAIL-R2 and a decrease in perifosine-dependent sensitization to TRAIL cytotoxic effect, whereas PMA, a conventional PKC isoform activator, increased TRAIL-R2 expression. Perifosine increased ROS production in THP-1 cells, and ROS could promote PKCα binding to the plasma membrane. Perifosine also induced increased levels of phosphorylation of JNK 2 (but not JNK 1) and c-Jun in THP-1 cells. NAC impaired upregulation of TRAIL-R2 by perifosine. The involvement of JNK 2/c-Jun signaling in TRAIL-R2 increased expression was demonstrated by the fact that a JNK 1/2 selective inhibitor, but not a p38 MAP kinase inhibitor, blocked both c-Jun phosphorylation on Ser 63 induced by perifosine and TRAIL-R2 upregulation. Furthermore, downregulation of c-Jun by siRNA also opposed the increase in TRAIL-R2. Given that in cells with downregulated PKCα, perifosine was unable to increase JNK 2 phosphorylation, we propose a mechanism whereby perifosine generates ROS, which in turn activate a PKCα→JNK→2 c-Jun signaling pathway which leads to increased expression of TRAIL-R2.
XIAP and c-FLIP-L downregulation caused by perifosine in THP-1 cells, could be due to an inhibition of the NF-κB activity which is under the control of the PI3K/Akt axis in AML cells (12). A recent investigation carried out on Waldenstrom macroglobulinemia cells has indeed demonstrated that perifosine targets NF-κB (40). Future studies should address the mechanism(s) which underlie XIAP and cFLIP-L downregulation by perifosine in AML cells. Nevertheless, our unpublished findings, obtained by siRNA technology, have indicated that downregulation of these two proteins was not as critical as that of PKCα for the potentiating effect of perifosine on TRAIL cytotoxicity on THP-1 cells.
Our results demonstrated that a perifosine and TRAIL combination was much more effective than either treatment alone also in primary AML cells. Even though we could not perform an as comprehensive analysis as in THP-1 cells due to the insufficient amount of cells recovered from most of the patients, perifosine dephosphorylated Akt, downregulated XIAP and cFLIP-L expression, and upregulated the levels of TRAIL-R2 and Ser 63 p-c-Jun in some primary AML cells. AML blasts died by apoptosis and the combined treatment was much more effective in activating caspase-8 than either treatment alone. All the patient samples expressed TRAIL-R2 to some extent (not shown), however perifosine increased TRAIL-R2 expression only in samples with activated PI3K/Akt signaling. Accordingly, synergism was only observed in those AML samples which displayed activated Akt. The fact that despite expression of TRAIL-R2 even under basal conditions, AML blasts were not sensitive to TRAIL alone, could be explained by the contemporaneous expression of TRAIL decoy receptors (8). After perifosine treatment, TRAIL-R2 was markedly upregulated in AML blasts, and most likely this could overcome the anti-apoptotic effect played by high levels of decoy receptors. However, it should not be ruled out that other proteins, which are critically important for TRAIL sensitivity (35, 41), are downregulated by perifosine in AML primary cells.
At present it is unclear why perifosine increased ROS generation only in primary AML cells with upregulated PI3K/Akt signaling. Nevertheless, a recent report has highlighted that 7-ketochetosterol, which is incoporated in lipid rafts of THP-1 cells (42), was able to increase ROS production by upregulating the levels of NAD(P)H oxidase (NOX-4) in THP-1 cells. Interestingly, this was accompanied by a downregulation of Akt (43). It might be that perifosine, by disrupting PI3K/Akt signaling at the lipid rafts, positively affects NOX-4 gene expression. Therefore, it would be interesting to investigate if NOX-4 gene expression is under the control of the PI3K/Akt axis in AML cells.
Our results point to the fact that a combination consisting of TRAIL and perifosine had no effect on the clonogenic activity of CD34+ cells from healthy donors, whereas it was markedly cytotoxic for CD34+ cells isolated from leukemic patients. Previous results have demonstrated that TRAIL was not cytotoxic for normal CD34+ cells (44, 45) reflecting the lack of TRAIL receptors expressed in these cells (46). In contrast, TRAIL displayed pro-apoptotic activity in CD34+ cells from AML patients (45), and we have confirmed these findings. Therefore, CD34+ AML cells express TRAIL receptors. Future investigations should be aimed to investigating whether leukemic stem cells also express TRAIL receptors and whether they could be targeted by the combination of TRAIL and perifosine.
In conclusion, we have demonstrated the in vitro efficacy of a TRAIL and perifosine combination treatment in AML cells. This combination was also synergistic in cells (THP-1) lacking functional p53. Even if p53 deletion and/or inactivating mutations are observed only in approximately 10% of AML patients, p53 levels are frequently low in AML blasts due to overexpression of the negative regulator murine double minute (MDM2) (47). Thus, the use of a drug which could upregulate TRAIL-R2 levels independently of p53 could be extremely useful in leukemia therapy. In this connection, it must be emphasized that the cytotoxic effect of triptolide and TRAIL combination was enhanced by the addition of the MDM2 antagonist, Nutlin-3a (36). In case of triptolide, another drug was required to maximize TRAIL effects which could result in additional toxic side effects if administered to patients.
In summary, the combination of perifosine and TRAIL could represent a novel strategy for treating AML patients by overcoming critical mechanisms of apoptosis resistance.
Supplementary Material
Acknowledgments
This work was supported by grants from: Fondazione CARISBO (to A.M.M.), Progetti Strategici Università di Bologna EF2006 (to A.M.M.), European LeukemiaNet (to G.M.), and a grant from the National Institutes of Health (USA) (RO1CA091025 to J.A.M.).
References
- 1.Kelley SK, Ashkenazi A. Targeting death receptors in cancer with Apo2L/TRAIL. Curr Opin Pharmacol. 2004;4:333–9. doi: 10.1016/j.coph.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14:337–48. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 3.LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 4.Koschny R, Walczak H, Ganten TM. The promise of TRAIL--potential and risks of a novel anticancer therapy. J Mol Med. 2007;85:923–35. doi: 10.1007/s00109-007-0194-1. [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann SH, Steensma DP. On the TRAIL of a new therapy for leukemia. Leukemia. 2005;19:2195–202. doi: 10.1038/sj.leu.2403946. [DOI] [PubMed] [Google Scholar]
- 6.Bortul R, Tazzari PL, Cappellini A, et al. Constitutively active Akt1 protects HL60 leukemia cells from TRAIL-induced apoptosis through a mechanism involving NF-κB activation and cFLIP(L) up-regulation. Leukemia. 2003;17:379–89. doi: 10.1038/sj.leu.2402793. [DOI] [PubMed] [Google Scholar]
- 7.Secchiero P, Zerbinati C, di Iasio MG, et al. Synergistic cytotoxic activity of recombinant TRAIL plus the non-genotoxic activator of the p53 pathway nutlin-3 in acute myeloid leukemia cells. Curr Drug Metab. 2007;8:395–403. doi: 10.2174/138920007780655432. [DOI] [PubMed] [Google Scholar]
- 8.Riccioni R, Pasquini L, Mariani G, et al. TRAIL decoy receptors mediate resistance of acute myeloid leukemia cells to TRAIL. Haematologica. 2005;90:612–24. [PubMed] [Google Scholar]
- 9.Jones DT, Ganeshaguru K, Mitchell WA, et al. Cytotoxic drugs enhance the ex vivo sensitivity of malignant cells from a subset of acute myeloid leukaemia patients to apoptosis induction by tumour necrosis factor receptor-related apoptosis-inducing ligand. Br J Haematol. 2003;121:713–20. doi: 10.1046/j.1365-2141.2003.04340.x. [DOI] [PubMed] [Google Scholar]
- 10.Guo F, Sigua C, Tao J, et al. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. 2004;64:2580–9. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- 11.Elrod HA, Lin YD, Yue P, et al. The alkylphospholipid perifosine induces apoptosis of human lung cancer cells requiring inhibition of Akt and activation of the extrinsic apoptotic pathway. Mol Cancer Ther. 2007;6:2029–38. doi: 10.1158/1535-7163.MCT-07-0004. [DOI] [PubMed] [Google Scholar]
- 12.Martelli AM, Nyakern M, Tabellini G, et al. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20:911–28. doi: 10.1038/sj.leu.2404245. [DOI] [PubMed] [Google Scholar]
- 13.Tazzari PL, Cappellini A, Ricci F, et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia. 2007;21:427–38. doi: 10.1038/sj.leu.2404523. [DOI] [PubMed] [Google Scholar]
- 14.Tazzari PL, Tabellini G, Bortul R, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. 2007;21:886–96. doi: 10.1038/sj.leu.2404643. [DOI] [PubMed] [Google Scholar]
- 15.Sale EM, Sale GJ. Protein kinase B: signalling roles and therapeutic targeting. Cell Mol Life Sci. 2008;65:113–27. doi: 10.1007/s00018-007-7274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vink SR, van Blitterswijk WJ, Schellens JH, Verheij M. Rationale and clinical application of alkylphospholipid analogues in combination with radiotherapy. Cancer Treat Rev. 2007;33:191–202. doi: 10.1016/j.ctrv.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 17.van der Luit AH, Vink SR, Klarenbeek JB, et al. A new class of anticancer alkylphospholipids uses lipid rafts as membrane gateways to induce apoptosis in lymphoma cells. Mol Cancer Ther. 2007;6:2337–45. doi: 10.1158/1535-7163.MCT-07-0202. [DOI] [PubMed] [Google Scholar]
- 18.Papa V, Tazzari PL, Chiarini F, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia. 2008;22:147–60. doi: 10.1038/sj.leu.2404980. [DOI] [PubMed] [Google Scholar]
- 19.Chiarini F, Del Sole M, Mongiorgi S, et al. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008;22:1106–16. doi: 10.1038/leu.2008.79. [DOI] [PubMed] [Google Scholar]
- 20.Ko CH, Shen SC, Chen YC. Hydroxylation at C4′ or C6 is essential for apoptosis-inducing activity of flavanone through activation of the caspase-3 cascade and production of reactive oxygen species. Free Radic Biol Med. 2004;36:897–910. doi: 10.1016/j.freeradbiomed.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 21.Nyakern M, Cappellini A, Mantovani I, Martelli AM. Synergistic induction of apoptosis in human leukemia T cells by the Akt inhibitor perifosine and etoposide through activation of intrinsic and Fas-mediated extrinsic cell death pathways. Mol Cancer Ther. 2006;5:1559–70. doi: 10.1158/1535-7163.MCT-06-0076. [DOI] [PubMed] [Google Scholar]
- 22.Zhang XD, Franco A, Myers K, Gray C, Nguyen T, Hersey P. Relation of TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma. Cancer Res. 1999;59:2747–53. [PubMed] [Google Scholar]
- 23.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 24.Theurl I, Theurl M, Seifert M, et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood. 2008;111:2392–9. doi: 10.1182/blood-2007-05-090019. [DOI] [PubMed] [Google Scholar]
- 25.Phillips TA, Ni J, Pan G, et al. TRAIL (Apo-2L) and TRAIL receptors in human placentas: implications for immune privilege. J Immunol. 1999;162:6053–9. [PubMed] [Google Scholar]
- 26.Chen W, Wang X, Zhuang J, Zhang L, Lin Y. Induction of death receptor 5 and suppression of survivin contribute to sensitization of TRAIL-induced cytotoxicity by quercetin in non-small cell lung cancer cells. Carcinogenesis. 2007;28:2114–21. doi: 10.1093/carcin/bgm133. [DOI] [PubMed] [Google Scholar]
- 27.Mahanivong C, Chen HM, Yee SW, Pan ZK, Dong Z, Huang S. Protein kinase Calpha-CARMA3 signaling axis links Ras to NF-κB for lysophosphatidic acid-induced urokinase plasminogen activator expression in ovarian cancer cells. Oncogene. 2008;27:1273–80. doi: 10.1038/sj.onc.1210746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cuschieri J, Billigren J, Maier RV. Endotoxin tolerance attenuates LPS-induced TLR4 mobilization to lipid rafts: a condition reversed by PKC activation. J Leukoc Biol. 2006;80:1289–97. doi: 10.1189/jlb.0106053. [DOI] [PubMed] [Google Scholar]
- 29.Kitatani K, Idkowiak-Baldys J, Hannun YA. Mechanism of inhibition of sequestration of protein kinase Cα/βII by ceramide. Roles of ceramide-activated protein phosphatases and phosphorylation/dephosphorylation of protein kinase C alpha/betaII on threonine 638/641. J Biol Chem. 2007;282:20647–56. doi: 10.1074/jbc.M609162200. [DOI] [PubMed] [Google Scholar]
- 30.Johann AM, von Knethen A, Lindemann D, Brune B. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-gamma and attenuates the oxidative burst. Cell Death Differ. 2006;13:1533–40. doi: 10.1038/sj.cdd.4401832. [DOI] [PubMed] [Google Scholar]
- 31.Rahmani M, Reese E, Dai Y, et al. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res. 2005;65:2422– 32. doi: 10.1158/0008-5472.CAN-04-2440. [DOI] [PubMed] [Google Scholar]
- 32.Zou W, Liu X, Yue P, et al. c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1, 9-dien-28-oate in human lung cancer cells. Cancer Res. 2004;64:7570–8. doi: 10.1158/0008-5472.CAN-04-1238. [DOI] [PubMed] [Google Scholar]
- 33.Mauro A, Ciccarelli C, De Cesaris P, et al. PKCα-mediated ERK, JNK and p38 activation regulates the myogenic program in human rhabdomyosarcoma cells. J Cell Sci. 2002;115:3587–99. doi: 10.1242/jcs.00037. [DOI] [PubMed] [Google Scholar]
- 34.Chow JM, Shen SC, Wu CY, Chen YC. 12-o-Tetradecanoylphorbol 13-acetate prevents baicalein-induced apoptosis via activation of protein kinase C and JNKs in human leukemia cells. Apoptosis. 2006;11:1999–2011. doi: 10.1007/s10495-006-0085-x. [DOI] [PubMed] [Google Scholar]
- 35.Riccioni R, Senese M, Diverio D, et al. Resistance of acute myeloid leukemic cells to the triterpenoid CDDO-Imidazolide is associated with low caspase-8 and FADD levels. Leuk Res. 2008;32:1244–58. doi: 10.1016/j.leukres.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Carter BZ, Mak DH, Schober WD, et al. Triptolide sensitizes AML cells to TRAIL-induced apoptosis via decrease of XIAP and p53-mediated increase of DR5. Blood. 2008;111:3742–50. doi: 10.1182/blood-2007-05-091504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akashi M, Osawa Y, Koeffler HP, Hachiya M. p21WAF1 expression by an activator of protein kinase C is regulated mainly at the post-transcriptional level in cells lacking p53: important role of RNA stabilization. Biochem J. 1999;337:607–16. [PMC free article] [PubMed] [Google Scholar]
- 38.Kim YH, Jung EM, Lee TJ, et al. Rosiglitazone promotes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Radic Biol Med. 2008;44:1055–68. doi: 10.1016/j.freeradbiomed.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Verde P, Casalino L, Talotta F, Yaniv M, Weitzman JB. Deciphering AP-1 function in tumorigenesis: fra-ternizing on target promoters. Cell Cycle. 2007;6:2633–9. doi: 10.4161/cc.6.21.4850. [DOI] [PubMed] [Google Scholar]
- 40.Leleu X, Eeckhoute J, Jia X, et al. Targeting NF- B in Waldenstrom macroglobulinemia. Blood. 2008;111:5068–77. doi: 10.1182/blood-2007-09-115170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riccioni R, Senese M, Diverio D, et al. M4 and M5 acute myeloid leukaemias display a high sensitivity to Bortezomib-mediated apoptosis. Br J Haematol. 2007;139:194–205. doi: 10.1111/j.1365-2141.2007.06757.x. [DOI] [PubMed] [Google Scholar]
- 42.Berthier A, Lemaire-Ewing S, Prunet C, et al. Involvement of a calcium-dependent dephosphorylation of BAD associated with the localization of Trpc-1 within lipid rafts in 7-ketocholesterol-induced THP-1 cell apoptosis. Cell Death Differ. 2004;11:897–905. doi: 10.1038/sj.cdd.4401434. [DOI] [PubMed] [Google Scholar]
- 43.Palozza P, Serini S, Verdecchia S, et al. Redox regulation of 7-ketocholesterol-induced apoptosis by beta-carotene in human macrophages. Free Radic Biol Med. 2007;42:1579–90. doi: 10.1016/j.freeradbiomed.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 44.Zang DY, Goodwin RG, Loken MR, Bryant E, Deeg HJ. Expression of tumor necrosis factor-related apoptosis-inducing ligand, Apo2L, and its receptors in myelodysplastic syndrome: effects on in vitro hemopoiesis. Blood. 2001;98:3058–65. doi: 10.1182/blood.v98.10.3058. [DOI] [PubMed] [Google Scholar]
- 45.Plasilova M, Zivny J, Jelinek J, et al. TRAIL (Apo2L) suppresses growth of primary human leukemia and myelodysplasia progenitors. Leukemia. 2002;16:67–73. doi: 10.1038/sj.leu.2402338. [DOI] [PubMed] [Google Scholar]
- 46.Secchiero P, Vaccarezza M, Gonelli A, Zauli G. TNF-related apoptosis-inducing ligand (TRAIL): a potential candidate for combined treatment of hematological malignancies. Curr Pharm Des. 2004;10:3673–81. doi: 10.2174/1381612043382747. [DOI] [PubMed] [Google Scholar]
- 47.Kojima K, Konopleva M, Samudio IJ, Ruvolo V, Andreeff M. Mitogen-activated protein kinase kinase inhibition enhances nuclear proapoptotic function of p53 in acute myelogenous leukemia cells. Cancer Res. 2007;67:3210–9. doi: 10.1158/0008-5472.CAN-06-2712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.