

Abstract
Glycogen synthase kinase 3β (GSK3β) is involved in metabolism, neurodegeneration, and cancer. Inhibition of GSK3β activity is the primary mechanism that regulates this widely expressed active kinase. Although the protein kinase Akt inhibits GSK3β by phosphorylation at the N terminus, preventing Akt-mediated phosphorylation does not affect the cell-survival pathway activated through the GSK3β substrate β-catenin. Here, we show that p38 mitogen-activated protein kinase (MAPK) also inactivates GSK3β by direct phosphorylation at its C terminus, and this inactivation can lead to an accumulation of β-catenin. p38 MAPK-mediated phosphorylation of GSK3β occurs primarily in the brain and thymocytes. Activation of β-catenin-mediated signaling through GSK3β inhibition provides a potential mechanism for p38 MAPK-mediated survival in specific tissues.
The p38 mitogen-activated protein kinase (MAPK) is activated through phosphorylation primarily by MAPK kinase 3 (MKK3) and MKK6 in response to cellular stress and cytokines. The p38 MAPK pathway functions in the control of differentiation, the blockade of proliferation, and in the induction of apoptosis (1). It is also activated in response to DNA double-stranded breaks (DSBs) induced by ionizing irradiation or chemotherapeutic drugs, and it participates in the induction of a G2/M cell-cycle checkpoint (2, 3). p38 MAPK can also promote survival (4-6) by unknown mechanisms. During T cell receptor β (TCRβ) rearrangement, V(D)J recombination-mediated DSBs also activate p38 MAPK in immature thymocytes at the double negative 3 (DN3) stage of development (7, 8). The expression of a constitutively active mutant of MKK6 [MKK6(Glu)] in thymocytes of transgenic mice (MKK6 transgenic mice) activates a p53-mediated G2/M phase cell-cycle checkpoint (8). Like recombination-activating gene (Rag) deficiency, persistent activation of p38 MAPK interferes with the differentiation of thymocytes beyond the DN3 stage. However, MKK6 transgenic thymocytes (but not Rag-/- thymocytes) survive and accumulate in vivo (8), suggesting that p38 MAPK may also provide a survival signal. A gene expression profile analysis comparing Rag-/- and MKK6 DN3 thymocytes revealed that the MKK6 DN3 thymocytes expressed more c-myc and lef (fig. S1) [two transcription factors associated with cell survival (9-11)] than did the Rag-/- thymocytes. The increased abundance of c-Myc and Lef proteins in the MKK6 transgenic thymocytes, compared with Rag-/- thymocytes, was confirmed by Western blot analysis (Fig. 1A) (12). Thymocytes from Rag-/- mice crossed with MKK6 transgenic (Rag-/- MKK6) mice contained higher amounts of c-Myc and Lef proteins than did Rag-/- thymocytes, indicating that the activation of p38 MAPK, but not the pre-TCR signals, contributes to the enhanced expression of these transcription factors (Fig. 1B). The c-myc and lef genes are targets of the β-catenin signaling pathway in certain contexts (13, 14). Nuclear accumulation of β-catenin was detected in MKK6 thymocytes, but not in Rag-/- thymocytes (Fig. 1C). Expression of constitutively active MKK6 in 293T cells was also sufficient to increase the amount of β-catenin protein (Fig. 1D), but this had no effect on β-catenin mRNA (fig. S2).
Fig. 1.
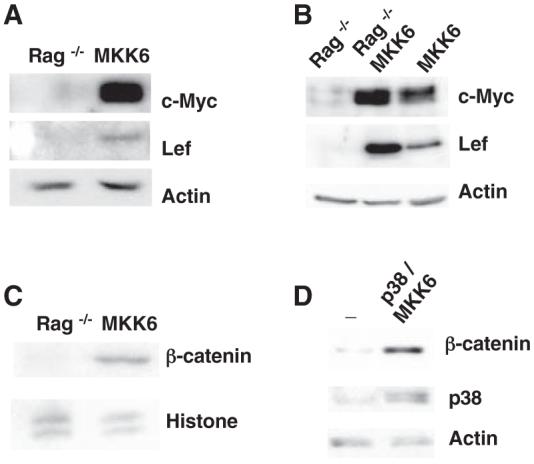
Regulation of the β-catenin pathway by p38 MAPK. (A) Western blot showing c-Myc and Lef in whole-cell extracts from Rag-/- thymocytes (Rag-/-) and MKK6 thymocytes (MKK6). Actin was examined as a control. (B) Western blot showing c-Myc and Lef in thymocytes from Rag-/-, MKK6, and Rag-/-/MKK6 mice. (C) Western blot showing β-catenin in nuclear extracts from Rag-/- and MKK6 thymocytes. (D) Western blot showing β-catenin and p38 MAPK in whole-cell extracts from 293T cells transfected with GSK3β (-) or GSK3β with p38 MAPK and MKK6 (p38/MKK6).
Phosphorylation of β-catenin by glycogen synthase kinase 3β (GSK3β) targets β-catenin for ubiquitination and subsequent degradation (15, 16). The best-characterized mechanism for the inactivation of GSK3β is through phosphorylation of its N terminus at Ser9 by Akt (17). No increase was observed in the amount of phospho-Ser9 GSK3β in MKK6 thymocytes compared with that in Rag-/- thymocytes (Fig. 2A). Similarly, no increase in phospho-Ser9 was observed in 293T cells transfected with constitutively active MKK6 (Fig. 2B). Phosphorylation of Ser9 was impaired by Wortmanin, an inhibitor of the PI3K-Akt pathway, but it was not affected by the pharmacological inhibitor of p38 MAPK SB203580 (fig. S3). Thus, p38 MAPK does not appear to regulate the Akt-mediated phosphorylation of GSK3β on Ser9. p38 MAPK immunoprecipitated from MKK6 thymocytes or MKK6-transfected 293T cells phosphorylated recombinant catalytically inactive GSK3β in vitro, and this phosphorylation was blocked by the p38 MAPK inhibitor (Fig. 2C). No Akt was detected in p38 MAPK immunoprecipitates, and no p38 MAPK was detected in Akt immunoprecipitates (fig. S4), ruling out the presence of residual AKT associated with p38 MAPK. The depletion of Akt before immunoprecipitating p38 MAPK did not affect the phosphorylation of GSK3β (Fig. 2D). A purified recombinant activated p38 MAPK also phosphorylated GSK3β in vitro, and this phosphorylation was blocked by SB203580 (Fig. 2E). Coimmunoprecipitation analysis showed that p38 MAPK was present in GSK3β immunoprecipitates from MKK6 thymocytes (Fig. 2F) and 293T cells (fig. S5). Thus, p38 MAPK physically associates with and phosphorylates GSK3β at a Ser9-independent residue. Although GSK3α and GSK3β are thought to be similarly regulated and can compensate for each other for some functions (18, 19), GSK3α was not phosphorylated by recombinant p38 MAPK in vitro (Fig. 2G).
Fig. 2.
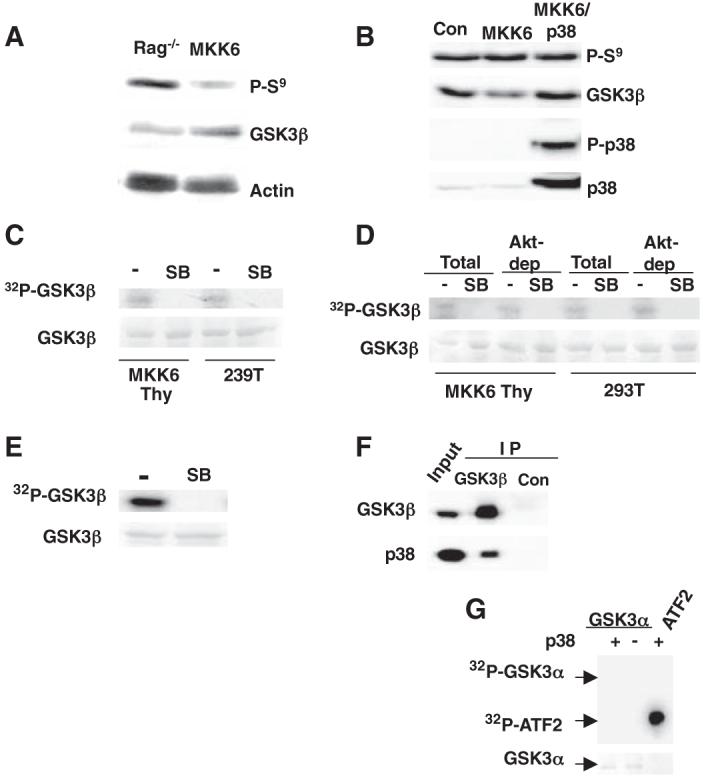
Direct phosphorylation of GSK3β by p38 MAPK. (A) Western blot showing phospho-Ser9 GSK3β (P-S9) and total GSK3β in Rag-/- and MKK6 thymocytes. (B) Western blot showing P-Ser9 GSK3β, GSK3β, phospho-p38 MAPK (P-p38), and p38 MAPK in 293T cells transfected with an empty vector (Con), MKK6 alone, or MKK6 and p38 MAPK (MKK6/p38). (C) In vitro p38 MAPK assay with inactive recombinant GSK3β as the substrate and p38 MAPK immunoprecipitated from MKK6-thymocytes (MKK6 Thy) or MKK6-transfected 293T cells (293T). In vitro reactions were incubated in the presence (SB) or absence (-) of the specific p38 MAPK inhibitor SB203580. Total GSK3β was visualized by PonceauS staining, and phosphorylated GSK3β was detected by autoradiography. (D) In vitro p38 MAPK kinase assay as described in (C), using total or Akt-depleted extracts (Akt-dep) from MKK6 thymocytes (MKK6 Thy) or MKK6-transfected 293T cells (293T). (E) In vitro kinase assay as described in (C), with recombinant active p38 MAPK kinase. (F) Western blot showing GSK3β and p38 MAPK in the GSK3β and p21 (Con) immunoprecipitates (IP) and whole-cell extracts from MKK6 thymocytes (Input). (G) In vitro kinase assay for recombinant active p38 MAPK kinase using catalytically inactive GSK3α as a substrate. Phosphorylation of activating transcription factor 2 (ATF2) was examined as a positive control.
The MAPK extracellular signal-regulated protein kinase (ERK) phosphorylates Thr43 of GSK3β (20) but does not affect GSK3β activity. Although SerPro or ThrPro motifs recognized by ERK are also recognized by other MAPK groups, p38 MAPK was still able to partially phosphorylate a GSK3β-T43A mutant (Fig. 3A), suggesting the existence of additional phosphorylation sites in GSK3β. Mass spectrometric analysis of recombinant GSK3β phosphorylated in vitro by p38 MAPK revealed two GSK3β phospho-peptides containing phosphorylation within a consensus SerPro or ThrPro motif, a phospho-peptide containing Thr43, and a C-terminal peptide (384 to 403) containing the ThrPro motif at Thr390 (corresponding to Ser389 in mouse GSK3β) (figs. S6 and S7). To confirm Thr390 as a target of p38 MAPK in GSK3β, catalytically inactive GSK3β-T390A and GSK3β-T43A/T390A mutants were used as substrates for p38 MAPK in vitro. Phosphorylation of the GSK3β-T390A mutant by p38 MAPK was partially reduced but not abrogated (Fig. 3B), but phosphorylation of the GSK3β-T43A/T390A mutant was abrogated, indicating that these two residues are probably the targets for p38 MAPK in GSK3β. The T43A mutation (but not the T390A mutation) abrogated phosphorylation of GSK3β by ERK (Fig. 3C). Thus, Thr390 of GSK3β appears to be specifically phosphorylated by p38 MAPK.
Fig. 3.

Inhibition of GSK3β by p38 MAPK is mediated by phosphorylation at Thr390. (A) In vitro kinase assays for recombinant p38 MAPK with catalytically inactive GSK3β and GSK3β-T43A mutantas substrates. (B) In vitro kinase assay for recombinant p38 MAPK with kinase-inactive GSK3β (WT), GSK3β-T43A, GSK3β-T390A, and GSK3β-T43A/T390A mutants as substrates. (C) In vitro kinase assay for recombinant active ERK with catalytically inactive GSK3β, GSK3β-T43A, GSK3β-T390A, and GSK3β-T43A/T390A mutants as substrates. (D) In vitro kinase assay for active GSK3β, GSK3β-T43A, and GSK3β-T390A mutants before (Con) or after incubation with activated Akt or activated p38 MAPK. GSK3β activity relative to the activity without Akt or p38 MAPK (Con) is shown. Error bars represent SD (n =3 replicates). (E) In vitro GSK3β kinase reactions alone (-) or in the presence of unphosphorylated-Thr390 (Thr390), phospho-Ser9 (P-Ser9), or phospho-Thr390 (P-Thr390) peptides, as described in (D). Error bars represent SD. (F) GSK3β in vitro kinase assays as in (D), using various concentrations of phospho-Thr390, phospho-Ser9, and unphosphorylated-Thr390 peptides. Each point is the average of two measurements. GSM, modified glycogen synthase peptide.
We examined the activity of wild-type (WT) GSK3β and GSK3β-T43A and GSK3β-T390A mutants before or after incubation with p38 MAPK or Akt. p38 MAPK inhibited both WT GSK3β and the GSK3β-T43A mutant (Fig. 3D), but not the GSK3β-T390A mutant (Fig. 3D). Akt inhibited WT GSK3β and the two mutants (Fig. 3D). p38 MAPK did not affect the activity of GSK3α (fig. S8), in which the Thr390 residue from GSK3β is not conserved. Together, these results demonstrate that p38 MAPK-mediated phosphorylation of GSK3β at Thr390 (but not Thr43) is sufficient to inhibit GSK3β activity. A peptide derived from the N terminus of GSK3β containing phospho-Ser9 specifically inhibits GSK3β in vitro (21, 22). A phospho-Thr390 peptide also inhibited GSK3β activity, whereas the unphosphorylated-Thr390 peptide did not (Fig. 3E). The phospho-Thr390 peptide inhibited GSK3β activity as efficiently as the phospho-Ser9 peptide (Fig. 3F). Thus, phosphorylation at Thr390 by p38 MAPK may cause an inhibition of GSK3β comparable to the phosphorylation of Ser9 by Akt.
To demonstrate the phosphorylation of this residue in intact cells and in vivo, we generated a specific antibody (Ab) to a mouse phospho-Ser389 GSK3β peptide. A band corresponding to GSK3β was detected with this Ab in WT and GSK3α-\- embryonic stem (ES) cells, but not in the GSK3β-/- ES cells by Western blot analysis (Fig. 4A). This specific band was also present in GSK3β-/- ES cells transfected with a WT GSK3β, but not with a GSK3β-S389A mutant (Fig. 4B). Phospho-Ser389 GSK3β was detected in mouse GSK3β-transfected 293T cells, but only if active MKK6 was present (Fig. 4C). The presence of the phospho-Ser389 GSK3β in these cells correlated with an increased amount of β-catenin (Fig. 4C), which is indicative of an inhibition of GSK3β activity. Ser389-phosphorylation was also detected in WT GSK3β, but not the GSK3β-S389A mutant after in vitro incubation with activated p38 MAPK (fig. S9). Phosphatase treatment of GSK3β previously incubated with activated p38 MAPK abrogated its recognition by the phospho-Ser389 Ab (fig. S9). Together, these results show the specificity of this Ab for phospho-S389 GSK3β and the phosphorylation of GSK3β at S389 by p38 MAPK in vitro.
Fig. 4.

Phosphorylation of endogenous GSK3β by p38 MAPK. (A) Western blot showing the presence of endogenous phospho-Ser389 GSK3β (P-Ser389) in WT, GSK3α-/-, and GSK3β-/- ES cells. Total GSK3α, GSK3β, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were examined as controls. (B) Western blot showing P-Ser389 in GSK3β-/- ES cells transfected with WT GSK3β or GSK3β-S389A mutant alone or with p38 MAPK and MKK6 (p38/MKK6). (C) Western blot showing P-Ser389, Flag-tagged mouse GSK3β, and β-catenin in nontransfected 293T cells (-) or cells transfected with mouse GSK3β alone or in combination with p38 MAPK and MKK6 (p38/MKK6). (D) P-Ser389, total GSK3β, and β-catenin in 293T cells transfected with GSK3β, p38, and MKK6 in the absence (-) or presence of SB203580. (E) P-Ser389 and total GSK3β in MEF or total GSK3α and GSK3β in ES cells nontreated (-) or treated with SB203580. (F) P-Ser389, P-Ser9, total GSK3β, P-p38, total p38, and β-catenin in WT and MKK3-/-MKK6-/- (3/6-/-) MEF. (G) The tissue distribution of P-Ser389, P-Ser9, and total GSK3β. Quantification of the levels of P-Ser389 relative to P-Ser9 in each tissue is also shown (lower panel). Thy, thymocytes; Spl, spleen cells. (H) P-Ser389 and total GSK3β in thymocytes and brain from WT mice treated in vivo with vehicle (-) or SB203580 (SB). (I) P-S389 and total GSK3β in thymocytes from Rag-/- and MKK6 transgenic mice.
To determine whether activation of p38 MAPK was required for phosphorylation of GSK3β at Ser389 in intact cells, we treated mouse GSK3β-transfected 293T cells with SB203580. The inhibition of p38 MAPK abrogated the phosphorylation of Ser389 (Fig. 4D). Similarly, treatment with SB203580 inhibited phosphorylation of endogenous GSK3β at Ser389 in WT mouse embryonic fibroblasts (MEFs) and ES cells (Fig. 4E). We also examined phospho-Ser389 abundance in MEFs deficient for the major upstream activators of p38 MAPK, MKK3, and MKK6 (23). Phospho-Ser389 was barely detectable in MKK3-/-MKK6-/- MEFs (Fig.4F). In contrast, the amounts of phospho-Ser9 were comparable in WT and MKK3-/-MKK6-/- MEFs (Fig. 4F). Thus, activation of p38 MAPK appears to be required for the phosphorylation of GSK3β at Ser389. Inhibition of p38 MAPK by either SB203580 (Fig. 4D) or the absence of MKK3 and MKK6 (Fig. 4F) also decreased the amount of β-catenin, consistent with the possibility that p38 MAPK activation is required for repressing GSK3β activity.
We also examined phospho-Ser389 in different mouse tissues. A high amount of phospho-S389 was detected in the brain, and lesser amounts were detected in thymocytes and spleen cells (Fig. 4G). Phospho-Ser389 was not detected in the kidney (Fig. 4G), liver, or heart (fig. S10). Phosphorylation of GSK3β at Ser9 was detected in practically all of the examined tissues (Fig. 4G). Analysis of the relative abundance of phospho-S389 and phospho-S9 showed a predominance of the former in the brain and thymocytes (Fig. 4G), which correlated with the selective high activation of p38 MAPK in these tissues (fig. S11). Inhibition of p38 MAPK by treating animals with SB203580 reduced the levels of phospho-Ser389 GSK3β in both thymocytes and the brain (Fig. 4H). Analysis of phospho-Ser389 in MKK6 and Rag-/- thymocytes showed that phospho-S389 GSK3β was present selectively in MKK6 thymocytes (Fig. 4I). Together, these results support our proposal that GSK3β is phosphorylated at S389 in vivo by p38 MAPK and that this alternative regulatory mechanism of GSK3β is tissue-specific.
To date, phosphorylation at Ser9 by Akt is the best-characterized mechanism for the inhibition of GSK3β activity. However, knockin mice in which Ser9 was replaced by Ala have only a subtle defect related to insulin regulation of glycogen synthase in their skeletal muscle tissue (24), indicating that alternative mechanisms may be involved in the negative regulation of GSK3β for certain functions. We propose that the phosphorylation of GSK3β at S389 by p38 MAPK may be one such mechanism. Conditions that promote the activation of p38 MAPK promote the accumulation of β-catenin in certain scenarios; thus, the activation of the p38 MAPK pathway could be an alternative mechanism to regulate β-catenin/T cell factor signaling (and, potentially, cell survival) through the inactivation of GSK3β.
Supplementary Material
Acknowledgments
We thank C. Charland for flow cytometry analysis and cell sorting, T. Hunter and the DNA Sequencing Facility for sequencing, and the Univ. of Vermont Protein Core Facility for peptide synthesis. This work was supported by NIH grants R01 AI051454 and National Center for Research Resources (NCRR) grant P20 RR15557 (M.R.), NCRR grants P20 RR021905 and P20 RR16462 (D.E.M.), and Canada Research Chairs and Canadian Institutes of Health Research grant MOP-85057 (B.D.).
References and Notes
- 1.Zarubin T, Han J. Cell Res. 2005;15:11. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 2.Mikhailov A, Shinohara M, Rieder CL. Cell Cycle. 2005;4:57. doi: 10.4161/cc.4.1.1357. [DOI] [PubMed] [Google Scholar]
- 3.She QB, Chen N, Dong Z. J. Biol. Chem. 2000;275:20444. doi: 10.1074/jbc.M001020200. [DOI] [PubMed] [Google Scholar]
- 4.Kurosu T, et al. Apoptosis. 2005;10:1111. doi: 10.1007/s10495-005-3372-z. [DOI] [PubMed] [Google Scholar]
- 5.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. Cancer Cell. 2007;11:175. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dmitrieva NI, Bulavin DV, Fornace AJ, Jr, Burg MB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:184. doi: 10.1073/pnas.231623498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diehl NL, et al. J. Exp. Med. 2000;191:321. doi: 10.1084/jem.191.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pedraza-Alva G, et al. EMBO J. 2006;25:763. doi: 10.1038/sj.emboj.7600972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ioannidis V, Beermann F, Clevers H, Held W. Nat. Immunol. 2001;2:691. doi: 10.1038/90623. [DOI] [PubMed] [Google Scholar]
- 10.Gounari F, et al. Nat. Immunol. 2001;2:863. doi: 10.1038/ni0901-863. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman B, Amanullah A, Shafarenko M, Liebermann DA. Oncogene. 2002;21:3414. doi: 10.1038/sj.onc.1205400. [DOI] [PubMed] [Google Scholar]
- 12.Materials and methods are available as supporting material on Science Online.
- 13.He TC, et al. Science. 1998;281:1509. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 14.Filali M, Cheng N, Abbott D, Leontiev V, Engelhardt JF. J. Biol. Chem. 2002;277:33398. doi: 10.1074/jbc.M107977200. [DOI] [PubMed] [Google Scholar]
- 15.Liu C, et al. Cell. 2002;108:837. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 16.Salahshor S, Woodgett JR. J. Clin. Pathol. 2005;58:225. doi: 10.1136/jcp.2003.009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378:785. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 18.Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Dev. Cell. 2007;12:957. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doble BW, Woodgett JR. J. Cell Sci. 2003;116:1175. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding Q, et al. Mol. Cell. 2005;19:159. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Dajani R, et al. Cell. 2001;105:721. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 22.Frame S, Cohen P. Biochem. J. 2001;359:1. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brancho D, et al. Genes Dev. 2003;17:1969. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McManus EJ, et al. EMBO J. 2005;24:1571. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.