

Abstract
The ligand-activated nuclear receptor PXR is known to play a role in the regulated expression of drug metabolizing enzymes and transporters. Recent studies suggest a potential clinically relevant role of PXR in breast cancer. However, the relevant pathway or target genes of PXR in breast cancer biology and progression have not yet been fully clarified.
In this study, we show that mRNA expression of OATP1A2, a transporter capable of mediating the cellular uptake of estrogen metabolites, is nearly 10-fold greater in breast cancer compared to adjacent healthy breast tissues. Immunohistochemistry revealed exclusive expression of OATP1A2 in breast cancer tissue. Interestingly, treatment of breast cancer cells in vitro with the PXR agonist rifampin induced OATP1A2 expression in a time- and concentration-dependent manner. Consistent with a role as a hormone uptake transporter, induction of OATP1A2 was associated with increased uptake of estrone 3-sulfate. The rifampin response was abrogated after si-RNA targeting of PXR. We then identified a PXR response element in the human OATP1A2 promoter, located approximately 5.7 kb upstream of the transcription initiation site. The specificity of PXR-OATP1A2 promoter interaction was confirmed using chromatin immunoprecipitation. Importantly we utilized a novel potent and specific antagonist of PXR (A-792611) to demonstrate the reversal of the rifampin effect on the cellular uptake of E1S.
These data provide important new insights into the interplay between a xenobiotic nuclear receptor PXR and OATP1A2 that could contribute to the pathogenesis of breast cancer and may also prove to be heretofore unrecognized targets for breast cancer treatment.
Keywords: OATP1A2, Estrone 3-Sulfate uptake, breast cancer, PXR, A-792611
Introduction
The pregnane X receptor (PXR, SXR, NR1I2) is a member of the nuclear receptor family of ligand-activated transcription factors. This intracellular receptor has been shown to function as a xenobiotic sensor importantly involved in drug metabolism and transport. Indeed, this nuclear receptor is a major regulator of cytochrome P450 (CYP) enzymes including CYP3A4, CYP2C8, CYP2C9 and CYP2B6 (1–5) and drug efflux transporters such as multidrug resistance protein 1 (MDR1, ABCB1) and multidrug resistance associated protein 2 (MRP2, ABCC2) (6–8). The mechanistic basis of many drug-drug interactions in vivo can now be explained by the activation of this nuclear receptor and the resultant induction of hepatic and intestinal enzymes and transporters. Not surprisingly, PXR expression has been noted in several tissues which are exposed to high levels of xenobiotics such as the liver, intestine, lung and kidney (9, 10). Interestingly, PXR expression has also been detected in normal and neoplastic human breast tissue (11). Recently a study by Miki and colleagues noted expression of PXR and the drug uptake transporter organic anion transporter polypeptide 1A2 (OATP1A2) in human breast cancer tissue. Importantly, OATP1A2 and PXR expression was proposed as a histopathological marker of dedifferentiation and progression of breast cancer (12).
OATP1A2 is a member of the OATP superfamily of transporters that mediate the cellular uptake of endo- and xenobiotics. OATP1A2 has been shown to be expressed in hepatic cholangiocytes, the capillary endothelia forming the blood brain barrier (13), and on the apical domain of intestinal enterocytes (14). OATP1A2-expression in normal non-malignant breast tissue has been noted to be very low especially in comparison to other members of the OATP family such as OATP-B (OATPB2B1), OATP-E (OATP4A1) and OATP-D (OATP3A1) (15), although OATP1A2 expression in lactating mammary epithelium cells (MECs) is significantly greater compared to non-lactating MECs, suggesting regulated physiological function of this transporter in breast tissue (16). Of potential significance to breast cancer, OATP1A2 is known to mediate the cellular uptake of hormone conjugates (17). Despite the known ability of this transporter to mediate the cellular uptake of biologically active hormone conjugates such as estrone 3-sulfate (E1S) and estradiol 17β-glucuronide (E2G), the possible role of regulated OATP1A2 expression to hormone associated progression of breast cancer has yet not been clarified. It should be noted that exposure of breast cancer cells to E1S has been shown in vitro to result in increased cellular proliferation and that this effect could be modulated by concomitant treatment with bromosulphthalein, a known non-specific OATP inhibitor (18, 19). However, an association of such effect to the uptake transporter OATP1A2 had not been reported. Note that E1S level is significantly greater in malignant tissue, resulting in increased levels of biological active 17β-estradiol (20). Mechanistically, it is plausible that OATP1A2 plays a pivotal role in regulating proliferation and tumor promotion of breast tissue by enhancing the uptake of estradiol metabolites, thereby increasing intracellular levels of such hormones that activate the estrogen receptor. Importantly, molecular mechanisms that determine OATP1A2 overexpression in breast cancer have not been defined.
In this study, we examined OATP1A2 and PXR expression in neoplastic and adjacent non-neoplastic breast tissue. In addition, expression of OATP1A2 and PXR was assessed in a panel of breast cancer tissues of different tumor stages. To assess whether a direct link between PXR activation and human OATP1A2 expression exists, cell based reporter assays and chromatin immunoprecipitation experiment were performed to identify a PXR response element in the OATP1A2 gene. In addition, using a breast cancer cell model, we were able to show OATP1A2 induction results in greater E1S cellular uptake and enhanced estrogen receptor activation. Importantly, using a newly identified specific and potent PXR antagonist, we demonstrated that inhibition of PXR activity attenuates the proliferative effects of estrogen.
Material & Methods
Chemicals
Rifampin, tamoxifen, estrone 3-sulfate (E1S) and p-nitrophenolphosphate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Tritium labeled E1S was obtained from Perkin Elmer Life Sciences (Waltham, MA, USA). The PXR-inhibitor A-792611 was provided by Dr. J. Waring and D. Kempf (Abbott Labs, Chicago, IL, USA).
Cell culture
MCF-7, T47-D, HeLa and HepG2 cells were purchased from ATCC (Manassas, VA, USA). MCF-7 cells were grown in EMEM supplemented with 10% fetal bovine serum, non-essential amino acids, L-glutamine and penicillin/streptomycin. T47-D and HepG2 were maintained in RPMI and DMEM, respectively. Media and supplements were purchased from Cambrex (Wakersville, CA, USA). Cell culture was performed at 37ºC with 5% CO2 in a humidified atmosphere.
Tissue samples
Malignant and adjacent non-malignant breast tissue samples were obtained from Vanderbilt Tissue Procurement Core. Paraffin embedded tissue slides were prepared by the Department of Pathology at Vanderbilt University, Nashville, TN, USA. Tissue for RNA isolation was snap frozen in liquid nitrogen and stored at −80ºC. In addition, a commercially available TissueScan Real-Time Breast Cancer Disease Panel (Origene, Rockville, MD, USA) of breast cancer tissue of different stages was used for quantification of gene expression.
RNA isolation from tissue samples and cultured cells
For RNA extraction, the QiaMINI RNA easy Kit (Qiagen, Valencia, CA, USA) was used. Frozen breast tissue was mechanically homogenized in RLT-Buffer supplemented with β-mercaptoethanol. Subsequently, the thawed homogenate was centrifuged through a QiaShredder column and the eluate was loaded on purification columns. The following RNA isolation was performed as described by the manufacturer. Briefly, phenol-chlorophorm extraction was performed to isolate RNA from in vitro experiments using Trizol (Invitrogen, Carlsbad, CA, USA). The integrity and content of the RNA was determined using the Agilent Bioanalyzer (Agilent, Santa Clara, CA, USA). RNA samples were stored at −80ºC.
Real-Time PCR
Total RNA was reverse transcribed in a 50 μl reaction volume containing 1500 ng of RNA with the TaqMan Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) using random hexamers as described by the manufacturer. The amount of OATP1A2, PXR and 18S were measured by SYBRgreen quantitative real-time PCR with an ABI Prism 7700 sequence detection system (Applied Biosystems). The sequences of primers used for establishing the amount of the transporter, the nuclear receptor and the endogenous control were as follows: OATP1A2-for 5′-AGACCAACGCAGGATCCAT-3′, OATP1A2-rev 5′-GAGTTTCACCCATTCCACGTACA-3′; PXR-for 5′-CAGCGGAAGAAAAGTGAAC-3′, PXR-rev 5′-CACAGATCTTTCCGGACCTG-3′; 18S-for 5′-GTAACCCGTTGAACCCATT-3;′ 18S-rev 5′-CCATCCAATCGGTAGTAGCG-3′. CAR-for 5′-CCAGCTCATCTGTTCATCCA-3′ and CAR-rev 5′-GGTAACTCCAGGTCGGTCAG-3′ The quantitative PCR was carried out in 25 μl reaction volume containing 300 nM of each primer, 1x SYBRgreen PCR Master Mix and 40 ng of the reverse transcribed cDNA. The amount of the transporter and the nuclear receptor were normalized to 18S-rRNA.
Immunofluorescence
Protein localization of OATP1A2 in human breast neoplastic and normal tissue was investigated by immunofluorescence microscopy. For OATP1A2 detection, a polyclonal antibody raised in rabbit was used (13). Paraffin-embedded tissue sections were prepared by standard methods. The tissue sections were deparaffinized in xylol. Afterwards the tissue sections were incubated in ethanol of declining concentration from 100% to 50% for rehydratation and then rinsed in distilled water. For heat induced epitope retrieval, the tissue sections were boiled in citrate buffer (10 mM, pH 6.0). After washing twice in ice-cold PBS the slides were blocked with 5%-FBS-PBS. Thereafter the slides were incubated with diluted anti-OATP1A2-antibody (1:50) in a humidified atmosphere at 4ºC overnight. After several washing steps with PBS the sections were incubated with the fluorescent-labeled secondary antibody (Invitrogen). After washing the slides with PBS, the tissue was mounted in anti-fading mounting medium containing DAPI (Vector Laboratories, Burlingame, CA, USA). Images were obtained by fluorescence microscopy. As negative control, the primary antibody was omitted.
Western Blot Analysis
To determine the effect on OATP1A2 protein expression, T47-D cells were cultured in 10 cm-dishes. After treatment for 24 h, the cells were harvested in 5 mM Tris-HCl (pH 7.4). After freeze-shock in liquid nitrogen the cell suspension was homogenized using a Dounce Potter. Membrane fraction was collected by centrifugation at 100.000 × g at 4ºC, 50 μg protein were separated by SDS-PAGE and electrotransfered to NuPAGE nitrocellulose membrane Western Blotting system (Invitrogen). OATP1A2 in HeLa cells overexpressed using a vtf7-virus as previously described was used as positive control (13). 15 μg of the total cell lysate of transfected cells were used. To ehance utility of the OATP1A2-antibody described above (1:1000), for Western blot analysis, the antibody was further purified using the Melon Gel IgG Purification Kit (Thermo-Fisher). An HRP-labeled anti-rabbit-antibody (1:2000) (BioRad, Hercules, CA) was used as the secondary. The immobilized secondary antibody was detected using the ECL Plus Western Blotting Detection System (GE Healthcare, Baie d’Urfe, Quebec, Canada) and KODAK ImageStation 4000MM (Mandel, Guelph, ON, Canada). To normalize sample loading, the blot was reprobed with a monoclonal anti-actin antibody (Sigma-Aldrich).
In silico scan for PXR response elements
A 10kb-fragment upstream of the transcription start of the OATP1A2 gene was screened for potential PXR binding sites using the NUBIscan algorithm (http://www.nubiscan.unibas.ch).
Plasmids
CYP3A4-XREM-Luc plasmid containing the proximal promoter (−362/+53) and distal XREM (−7836/−7208) inserted in pGL3 basic (Promega, Madison, WI, USA) was used as positive control for PXR response (21). OATP1A2-regulatory regions containing the basal promoter (−440/+50) and distal fragments that contained putative PXR response elements (PXRREs), namely the −1130 to −1960, the −4400 to −3100, the −6600 to −5750 and the −6600 to 5120 segments of the SLCO1A2 gene were prepared by PCR using the following primer: -450-OATP1A2-for 5′-GGGTGTGCGCTCGAGGTCTTACATCTTAGTTTG-′3, +50-OATP1A2-rev 5′-GGGTGTGCGAGATCTAGAAAATCATGGTGTTAG-′3, -1960-OATP1A2-for 5′-GGGTGTGCGCCCGGGACTGTGTGGTCCTGGGAG-′3, -1130-OATP1A2-rev 5′-GGGTGTGCGCTCGAGTAGAAGGAAATGCAATTT-′3, -4400-OATP1A2-for 5′-GGGTGTGCGACGCGTACTAGCTATCATCATCAC-′3, -3100-OATP1A2-rev 5′-GGGTGTGCGCCCGGGTACCTACCCTATCACTTT-′3, -6600-OATP1A2-for 5′-GGGTGTGCGGAGCTCTAGTCAAGAACACAGCAT-′3, -5120-OATP1A2-rev 5′-GGGTGTGCGACGCGTTCGCTATTCATATTTAACA-′3, -5750-OATP1A2-rev 5′-GGGTGTGCGACGCGTCAATACTTGTCTGTCTCG-′3. The distal fragments of the SLCO1A2 gene were subcloned into pGL3-basic immediately upstream of the basal promoter. MutOATP1A2-5120-Luc plasmids, containing mutated PXR (DR4, DR3) response elements, were prepared by site-directed mutagenesis (QuickChange, Stratagene, La Jolla, CA, USA).
PXR reporter gene assays
T47-D or HepG2 cells were plated in 24-well plates. After 24 hrs cells were transfected with 250 ng of the reporter vector (pGL3 basic variants), 25 ng of pRL-CMV (Promega) to normalize transfection efficiency and 250 ng of human PXR expression plasmid (pEF-hPXR) or vector control in 200 μl Opti-MEM (Invitrogen) using Lipofectin (Invitrogen). Cells were incubated for 16 hrs with the transfection mixture then treated for 24 hrs with the tested agents. The reporter enzyme activities were assayed with a dual luciferase reporter assay system as described by the manufacturer (Promega). Luminescence was quantified using a plate reader (Fluoroskan Ascent FL, Thermo-Fisher). Luciferase activities in the presence of transfected PXR were expressed as a percent of cells transfected with blank vector.
Estrogen receptor reporter gene assays
T47-D cells were plated in 24-wells in RPMI supplemented with 5% charcoal stripped FBS. After 24 hrs cells were transfected with 250 ng of a commercially available vector containing a secreted alkaline phosphatase gene (SEAP), whose expression is controlled by an estrogen responsive element (ERE) (Clontech, Mountain View, CA, USA). After pretreatment with rifampin or DMSO in RPMI without phenol-red supplemented with L-glutamine and 1% charcoal stripped FBS (Invitrogen), the cells were exposed to E1S (10 nM). After treatment, activity of alkaline phosphatase was determined by combining30 μl of the sample with 150 μl of 5 mM p-nitrophenolphosphate prepared in 80 mM Tris-HCl, pH 9.6 for 60 min at 37ºC. The reaction was stopped by addition of 20 μl 0.1 M NaOH. The amount of reduced p-nitrophenolphosphate was determined by UV-spectroscopy at 405 nm using a plate reader (Multiskan Spectrum, Thermo-Fisher).
Cellular modulation of PXR expression with siRNA
T47-D cells were suspended to a final concentration of 1 × 105 cells/mL in growth medium and incubated at 37ºC. The lipid-based transfection agent siPort™NeoFX™ (Ambion, Austin, TX, USA) was used for transfection of PXR si-RNA (siRNA ID # 138584 NR1I2, Ambion) as described by the manufacturer. The transfection was incubated for 12 h at 37ºC at 5% CO2. Afterwards, cells were treated with rifampin (1μM) or DMSO for 24 hrs.
Estrone 3-sulfate uptake experiments
T47-D cells were grown in 12-wells and pretreated with rifampin or DMSO. Cells were incubated with tritium-labeled E1S (1 μM containing 400.000dpm/well). After 10 min incubation at 37ºC the cells were washed twice with ice cold Opti-MEM and lysed using 1%-SDS. The cellular uptake of E1S was determined using Ultima Gold ® scintillation liquid and a Liquid Scintillation Counter (Tri-Carb 2900TR, Perkin Elmer Life Sciences).
AlamarBlue™ cell proliferation Assay
T47-D or MCF-7 cells were pre-treated with rifampin or DMSO and then exposed to E1S. In order to determine cell viability and proliferation, the alamarBlue™ assay reagent (Biosource, Camarillo, CA, USA) was used as described by the manufacturer. The production of the reduced product was monitored by spectrophotometrically at 570 nm and 600 nm.
Chromatin Immunoprecipitation (ChIP) Assay
For DNA-crosslinking and chromatin immunoprecipitation, the EZ ChIP Assay (Millipore, Billerica, MA, USA) was used as described by the manufacturer. Briefly, T47D-cells were cultured in 10 cm-dishes, treated 48 hrs with DMSO or rifampin (1 M). After DNA crosslinking, the cells were lysed. DNA was then sheared by sonication (Virsonic 100, Virtis, Gardiner, NY, USA). The shearing efficacy was analyzed by agarose gel electrophoresis to ensure that the average size of DNA was 1500 kb. Thereafter, the sheared and crosslinked chromatin was incubated with 5 μg of each antibody overnight at 4ºC. Two polyclonal anti-PXR sera were used (A-20 and N-16, Santa Cruz Biotechnology, Santa Cruz, CA, USA). An anti-acetyl-histone (H3) antibody was used as control.
The antibody/antigen/chromatin complex was gathered with protein G agarose and centrifugation. After several washing steps, the antibody/chromatin complex was eluted and bound DNA was released by incubation at 65º C overnight after adding 8 μl of 5 M NaCl, treated with RNase A and Proteinase K and purified. Binding of human PXR to the SLCO1A2 gene was confirmed by PCR amplification of a 210 bp fragment of genomic DNA encompassing the putative PXR (DR4-1) binding site. The following primers were used in the PCR: ChIP for 5′-CCACCTTTCTTTCTCCATCTT-3′, ChIP rev 5′-TCTAATTTAAGCCACATTTC-3′. PCR was carried out in 50 μl volume using the AmpliTaqGold PCR system (Applied Biosystems). The annealing temperature was 54 ºC, followed by elongation at 72 ºC for 30 sec. The PCR was performed at 32 cycles. For the acetyl Histone H3 the PCR was performed as described by the manufacturer using the AmpliTaq Gold PCR system. PCR products were separated on 4% agarose gels.
Statistical Analysis
Determination of statistical differences between group parameters was determined using Student’s t-test and Mann-Whitney U-test (GraphPad Software, Inc., San Diego, CA). A p value of <0.05 was taken to be the minimum level of statistical significance.
Results
OATP1A2 and PXR expression in Malignant and Normal Human Breast Tissue
OATP1A2 and PXR expression were examined using real-time PCR. Both transcripts were detectible in all malignant (n=4) and non-neoplastic (n=4) tissue samples. Breast cancer tissue showed an 8-fold higher expression of OATP1A2 compared to the adjacent normal tissue in all samples tested (fig. 1A). Similarly, the level of PXR expression was nearly 8-fold higher in malignant tissue compared to the adjacent normal tissue of each subject (fig. 1C). Immunofluorescence microscopy revealed restricted expression of OATP1A2 in human malignant breast tissue using a specific antibody against OATP1A2. Distinct immunofluorescent signals were not observed in normal human breast tissue and in tumor or normal tissue when the primary antibody was omitted (fig. 1B).
Figure 1. Expression of OATP1A2 and PXR comparing non-neoplastic and neoplastic breast tissue.
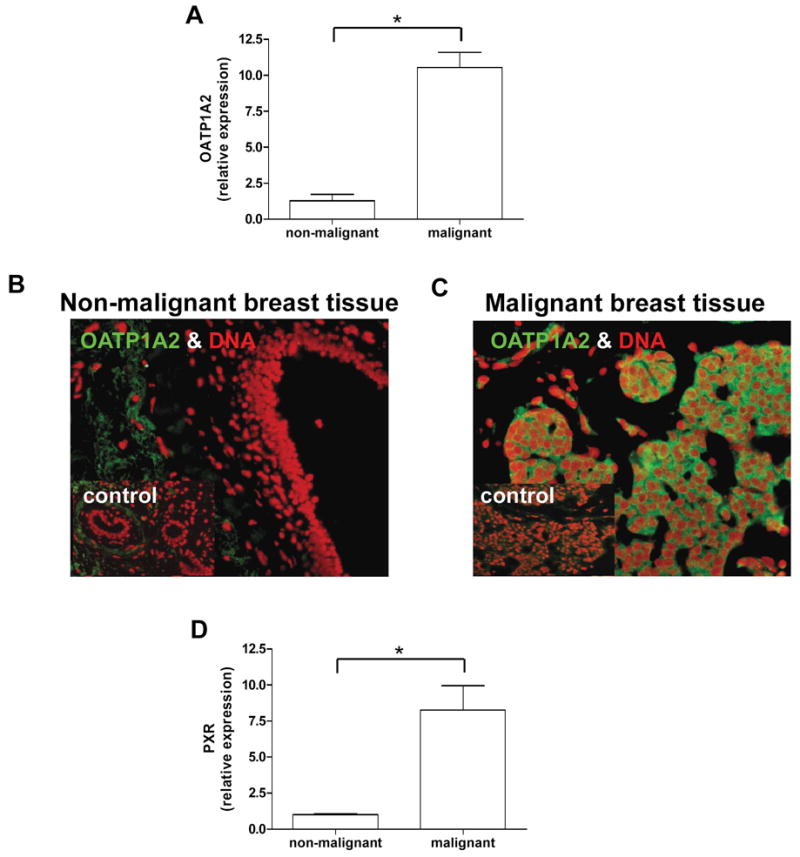
OATP1A2 (1A) and PXR (1C) mRNA-expression was assessed in neoplastic and adjacent non-neoplastic breast tissue samples of 4 individuals using real-time PCR revealing significant higher expression of both genes in the cancerous tissue. Immunofluorescent staining of the uptake transporter OATP1A2 (green) in human malignant and non-malignant breast tissue showed an intense and restricted expression in malignant cells (right panel 1B). No distinct staining pattern was detected in non-malignant breast tissue (left panel 1B). DNA-chromatin was counterstained with DAPI (red). As control the primary antibody was omitted (insert 1B). Data are expressed as mean ± SD, * p<0.05, t-test.
Detection of OATP1A2 mRNA in Breast Cancer Tissue cDNA Panel
OATP1A2 expression was also assessed in a panel of different stages of breast cancer by real-time PCR. The number of OATP1A2 transcripts was greater in tumor stage I and IIA compared to stage 0 (containing samples of patients with carcinoma in situ or fibrocystic changes) (OATP1A2 expression relative to stage 0, mean ± SD; stage 0: 1.23 ± 0.898 n=4; stage I: 3.03 ± 3.37 n=11; stage IIA: 5.10 ± 3.95, n=15; stage IIB: 1.07 ± 0.28, n=4; stage IIIA: 0.55 ± 0.14, n=4; stage IIIC: 0.88 ± 0.41, n=2). It is noteworthy, that OATP1A2 was undetectible in some samples of higher stages. In 3 of 7 samples of stage IIB, in 4 of 8 samples of stage IIIA, and in 1 of 3 samples of stage IIIC, OATP1A2-expression did not reach the minimum level of detection. The absence of OATP1A2 expression was not related to Estrogen Receptor (ER), Progesterone Receptor or Her2 status reported by the company. PXR-mRNA was detectible in all samples and the tumor stage-dependant expression pattern was similar to OATP1A2 (PXR expression relative to stage 0, mean ± SD; stage 0: 1.05 ± 0.35, n=4; stage I: 2.57 ± 1.44, n =11; stage IIA: 4.37 ± 2.03, n=15; stage IIB: 1.82 ± 0.52, n=7; stage IIIA: 1.88 ± 2.35, n=7; stage IIIC: 3.29 ± 1.53 n= 3).
PXR regulates OATP1A2 expression and estrogen-mediated cell proliferation
To examine the interaction between PXR and OATP1A2, T47-D breast cancer cells were treated with the known PXR activator rifampin. Treatment of T47-D cells were associated with a time-dependant induction of OATP1A2. Increased transporter mRNA levels were evident by 6 hrs after initiation of rifampin treatment and reached maximum levels by 8 hrs. OATP1A2 expression upon PXR activation was concentration dependent (fig. 2A). Another ER-positive breast cancer cell line (MCF-7 cells), was used to determine if PXR expression levels differ compared to T47-D cells resulting in changes in rifampin response. We noted no difference in PXR expression (relative PXR-expression ± SEM; MCF-7 cells: 1.01 ± 0.34; T47-D cells: 1.09 ± 0.57). However, the effect of rifampin on OATP1A2 expression was more pronounced in T47-D cells after treatment with rifampin for 8 hours (relative OATP1A2 expression ± SEM; MCF-7 cells 8.02 ± 0.93; T47-D cells 13.15 ± 2.87).
Figure 2. Association of PXR with OATP1A2 expression and activity in T47-D cells.
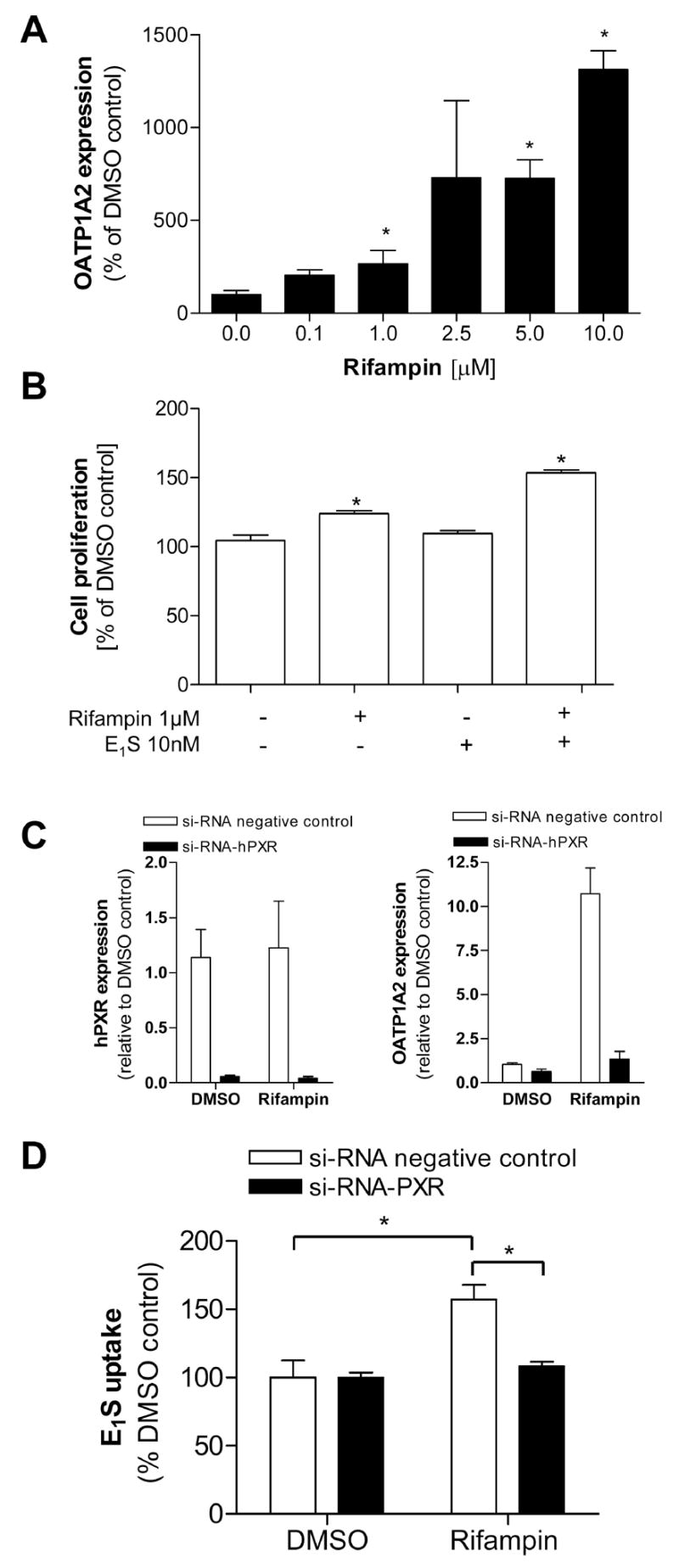
T47-D cells were treated with different concentrations of rifampin for 8h and the OATP1A2 expression was determined showing a concentration dependant effect on transporter mRNA-expression (2A). To assess the effect of the transporter associated uptake of estrone 3-sulfate (E1S) on the cell viability of T47-D cells, those breast cancer cells were pretreated with rifampin 1 μM for 24 hrs, respectively and then exposed to estrone 3-sulfate (10nM). After 24 hrs incubation the cell proliferation was determined using the alamar blue™ assay reagent (2B). T47-D cells were transfected with si-RNA PXR to validate the interplay between PXR activation and OATP1A2 expression and function. The efficacy of the cellular knock down of PXR was assessed by real-time-PCR in cells transfected with siRNA-PXR (black) or siRNA-negative control (white) (2C). The level of OATP1A2 and PXR mRNA expression in cells transfected with the PXR-siRNA was determined by real-time RT-PCR after 8 hrs incubation with rifampin (1μM) (2C). To assess the effect of the cellular knock-down of PXR on the rifampin modulated OATP1A2 function, cells were transfected with the si-RNA and treated with rifampin, subsequently the cellular accumulation of 3[H]-E1S was determined (2D). Data are expressed as mean ± SD; n=3; * p<0.05, t-test.
One of the known effects of the OATP1A2 substrate, E1S, is the enhancement of cell proliferation in vitro. Cell proliferation was assessed after pretreatment of T47-D cells with rifampin (1 μM) or DMSO and subsequent exposure to E1S for 24 hours (10 nM) using alamarBlue™. As shown in figure 2B, T47-D cells treated with the PXR-activator exhibited a significantly higher ability to metabolize the alamarBlue™ reagent compared to cells exposed to DMSO prior to the E1S treatment. Similar results were obtained in MCF-7 cells (cell viability %-DMSO control; mean ± SEM; pre-treatment with DMSO followed by E1S: 120.31 ± 6.38; pre-treatment with rifampin followed by DMSO 94.47 ± 11.61 pre-treatment with rifampin followed by E1S 155.9 ± 25.46) (data not shown).
Effect of PXR-siRNA and OATP1A2 mediated E1S uptake in T47-D cells
si-RNA mediated cellular knock-down of PXR was used to validate the involvement of PXR in regulating OATP1A2 expression and the resultant increase in cellular uptake of E1S. In si-RNA treated T47-D cells a marked reduction of PXR level (fig. 2C) was associated with the loss of rifampin-induced OATP1A2 expression (fig. 2C) and rifampin-stimulated E1S uptake.
Cells transfected with control si-RNA and treated with rifampin (10 μM) exhibited an 1.5-fold induction of E1S-uptake compared to cells treated 24 hrs with DMSO (E1S-uptake %-DMSO control; mean ± SEM; DMSO treated 100.00 ± 12.45; rifampin treated 157.18 ± 10.73), whereas cells transfected with PXR-siRNA and treated with rifampin do not exhibit an increase in E1S uptake (E1S-uptake %-DMSO control; mean ± SEM, DMSO treated 99.99 ± 3.59; rifampin treated 108.06 ± 3.31) (fig 2D).
Identification of functional PXR response elements in the SLCO1A2
We next aimed to determine the molecular mechanisms involved in PXR-mediated regulated OATP1A2 expression. Hence a 10kb-fragment of the SLCO1A2 gene was scanned using the NUBIscan algorithm (18) to reveal potential PXR response elements (PXRREs). Included in the search were known PXR responsive DNA motifs of tandem hexameric repeats with various spacing and orientation such as DR4, DR3, ER6 and ER8. To confirm whether the predicted binding sites were functional, fragments of the SLCO1A2 gene containing potential PXRREs were subcloned into a luciferase vector containing a basal promoter fragment of SLCO1A2 (−440bp to +50bp)- and assessed in cell based reporter gene assays.
The promoter constructs were tested by co-transfecting PXR into T47-D cells. Addition of the known PXR-activator rifampin (10 μM) resulted in PXR-dependent enhanced luciferase activation of the construct containing the −5120 bp to −6600 bp fragment of the SLCO1A2 gene. Rifampin-stimulated, PXR-dependent reporter activity was not observed in other fused reporter constructs tested (fig. 3A). However, a slight but statistically significant activation of the basal promoter construct was seen in rifampin treated cells. These results are in agreement with previous findings testing the PXR dependent activation of CYP3A4 showing that activation of the basal promoter is significantly increased by a distal enhancer module containing the actual PXRRE of CYP3A4 (22, 23). We observed similar findings performing the experiment using hepatocellular carcinoma (HepG2) cells or using the PXR-activator tamoxifen (10 μM) (data not shown).
Figure 3. Luciferase reporter gene assays of OATP1A2 promoter fragments.

Luciferase reporter gene assay of subcloned OATP1A2 promoter fragments revealed increased activity of luciferase in T47-D cells transfected with the −5120 to −6600 OATP1A2 promoter fragment after treatment with rifampin (10μM) (3A). In silico analysis of the responsive fragment revealed several potential DNA motifs (insert). Subsequent mutation of those motifs revealed a loss of transactivation mediated by rifampin (3B) in HepG2 transfected with a construct containing a mutated DR4 motif. To validate the link between OATP1A2 transactivation by PXR a chromatin immunoprecipitation was performed using different anti-PXR antibodies (A-20 and N-16 supplied by Santa Cruz). The PCR detecting a DNA fragment which includes the identified DR4 motif suggests that this DNA motif is involved in the PXR mediated induction of OATP1A2 (3C). Data are expressed as mean ± SD; n=3, * p<0.05, t-test.
Since another reporter construct containing overlapping sequences from −5790bp to −6600bp was unresponsive to PXR activation, we focused on possible PXRREs located between −5120 to −5790. Within this region, NUBIscan analysis exposed five potential PXR binding sites (fig 3 insert). These putative PXRREs were systematically mutated using a site directed mutagenesis approach and tested in reporter assays. Interestingly, mutation of the DR4-1 DNA motif (localized at −5786 to −5801) resulted in the complete loss of rifampin-stimulated, PXR-dependent, luciferase reporter activity in HepG2 cells (fig. 3B) and T47-D cells (data not shown). These findings suggest that this DR4-element in the SLCO1A2 promoter is transactivated by agonist-bound PXR.
Chromatin immunoprecipitation (ChIP) using anti-PXR antibodies
To further confirm the role of the DR4-1 element in the inductive regulation of OATP1A2 expression, we used antibody-mediated immunoprecipitation of PXR bound to chromatin. T47-D cells were treated with rifampin (1 μM) or DMSO and ChIP analysis was performed on formaldehyde cross-linked DNA with subsequent immunoprecipitation with two different PXR antibodies (A-20 and N-16). PCR amplification of a 210 bp fragment of the OATP1A2 gene surrounding the putative DR4-1 PXRRE from immunoprecipiated DNA revealed specific signals in cells treated with rifampin but not in vehicle treated cells using both antibodies (fig. 3C). These results demonstrate rifampin activated PXR binds to the OATP1A2 promoter and strongly suggests that the DR4-1 motif is a key element responsible for OATP1A2 transactivation by PXR.
Pharmacologic modulation of OATP1A2 expression and function by PXR antagonism
Recently, we have identified a novel potent and selective PXR antagonist A-792611 (24). As expected, addition of A-792611 abolished the PXR activation of the OATP1A2 reporter construct (fig. 4A). Importantly, the inductive effect of rifampin on OATP1A2 expression could be abolished by treatment of T47-D cells with A-792611 (fig. 4B). Similar results were obtained when OATP1A2 expression was assessed using Western Blot analysis. T47-D cells treated with rifampin exhibited significant higher OATP1A2 protein levels compared to those cells treated with rifampin and A-792611 (fig. 4C). Co-treatment with A-792611 abolished the increased cellular accumulation of E1S associated with rifampin treatment (fig. 5A).
Figure 4. Inhibition of the PXR effects on OATP1A2 using a specific PXR inhibitor.

Cell based reporter gene assays in HepG2 cells using the reporter vector containing the −5120 to −6600 fragment of the OATP1A2 gene was performed to determine the ability of A-792611 (10 μM) to inhibit the rifampin (10 μM) associated PXR mediated transactivation (4A). T47-D cells were treated with rifampin (10 μM) in the presence of different concentrations of A-792611 to determine the modulation of OATP1A2 expression using real-time RT-PCR (4B). Western Blot analysis was performed to evaluate the effect of rifampin (1μM) in the presence and absence of the PXR inhibitor A-792611 (1μM) on OATP1A2 expression in T47-D cells (4C). Cell lysates of HeLa cells transfected with OATP1A2-pEF6 or pEF6-vector only were used as positive and negative controls. Actin expression was assessed in the same blot to visualize equivalent sample loading for the cell lysates (4C).
Figure 5. Estrogenic effects modulated by PXR activation and determination of the potential involvement of CAR.

Estrone 3-sulfate uptake (1 μM containing 400,000 dpm/well) was measured in T47-D cells pretreated 24h with rifampin (1 μM) in presence or absence of the PXR inhibitor A-792611 (1 μM) to assess the effect on transport activity in this cell model (5A). Finally, T47-D cells were transfected with a vector containing an ERE driven alkaline phosphatase activity (black bars). After pretreatment of the cells with rifampin (1 μM) in presence or absence of the novel specific PXR inhibitor A-792611 (1 μM) for 24h the cells were exposed to E1S subsequently, activity of alkaline phosphatase was measured in comparison to the vector control (white bars) (5B). A cell based reporter gene assay was performed to determine the effect of constitutive and CITCO (10μM) activated expression of the nuclear receptor CAR on the responsive OATP1A2 reporter fragment compared to the fragment harboring the mutated DR4 DNA motif (5C). In addition, T47-D breast cancer cells were treated with CITCO (1 μM) and rifampin (1 μM) and OATP1A2 expression was determined after 24 hrs incubation (5D). Data are expressed as mean ± SD; * p<0.05, t-test.
Stimulated estrogen receptor signaling with OATP1A2 induction is attenuated by PXR antagonism
To determine whether OATP1A2-mediated E1S-transport is associated with increased estrogenic effects, ER-positive T47-D cells were transfected with an alkaline phosphatase reporter construct driven by an estrogen responsive element (ERE) and then treated with rifampin. When the cells were exposed to E1S and alkaline phosphatase activity was determined, ER activation was significantly higher after pretreatment with the PXR-activator rifampin. This increase of rifampin-stimulated ER activity could be reduced by co-treatment with the PXR inhibitor A-792611 (fig. 5B).
Determination of responsiveness of the OATP1A2 promoter fragment to constitutive and activated CAR
To determine whether the identified PXRRE (DR4-1) in the OATP1A2 promoter fragment is activated by CAR, a nuclear receptor which shares the same DNA motif as PXR (25) we performed a reporter gene assay on the responsive OATP1A2 promoter element. In fact, expression of CAR in HepG2 cells resulted in a significant induction of luciferase activity, which increased with treatment of the known CAR-ligand CITCO (10μM). This effect was not seen using the OATP1A2 promoter fragment harboring themutation in the DR4-1 motif (fig. 5C).
Expression of CAR in non-malignant and malignant breast tissue
Assessing CAR mRNA-expression by performing real-time PCR in human breast cancer tissue revealed significantly higher expression of CAR in non-malignant compared to malignant breast tissue (CAR expression relative to non-malignant tissue mean ± SD, non-malignant tissue 1.01 ± 0.72, n=4; malignant tissue 0.21 ± 0.74, n=4, t-test p<0.05, data not shown). However in T47-D and MCF-7 cells detection of CAR did not reach the minimum level of detection, and this is in agreement with analysis of OATP1A2 expression in T47-D cells after 24 hrs treatment with CITCO (1μM), which failed to result in induction of OATP1A2 expression (fig. 5D).
Discussion
The mammary gland is widely recognized as an estrogen target tissue. Estrogens are thought to act through the intracellular estrogen receptors to direct normal lobular development, regulate epithelial cell growth and increase the expression of steroid hormone metabolizing enzymes (26, 27). In addition to those physiological processes, the relationship between estrogens and the development and progression of breast cancer has been widely studied. Starting with the findings from George Beatson in 1896 reporting improved outcome from breast cancer in a postmenopausal woman after removal of her ovaries, subsequent studies have established a clear link between estrogens and the pathogenesis of breast cancer (28). Several studies have focused on plasma levels of estradiol and breast cancer risk. However, such studies have been inconsistent in linking breast cancer risk with increased plasma levels of estradiol (29, 30). Interestingly, endogenous E1S levels have been linked with a greater risk of breast cancer (29). E1S is a major circulating estrogen metabolite with a long half live in humans (~9 hrs), is found at high plasma levels in women (280 pg/mL), and is thought to be one of the major precursors of active estrogen in postmenopausal women (31). Due to the hydrophilic nature of this compound, it is likely that an uptake transporter is needed to facilitate the transmembrane entry into tissues such as breast.
In this report, we show a tumor specific overexpression of the uptake transporter OATP1A2 in breast tissue. Importantly, we demonstrate a PXR-mediated induction of this transporter can markedly enhance the extent of E1S uptake in breast cancer cells, and significantly increase estrogen receptor mediated gene transcription. Regulation of gene expression by PXR has been studied for a few Oatps/OATPs. For example, activation of Pxr in mouse causes hepatic up-regulation of Oatp1a4 (Oatp2) (32). In rats, Oatp2 (Oatp1a4) gene expression is induced by phenobarbital and pregnenolone-16α-carbonitrile (PCN), well-known activators of CAR and PXR (33).
In breast tissues and breast cancer cell lines including T47-D and MCF-7 cells, E1S is efficiently converted to bioactive estrone due to high steroid sulfatases (STS) activity (34, 35). Moreover, in breast cancer there is a compelling association of tumor size and risk of recurrence with STS expression (36). In addition pharmacological targeting of STS is considered to be one of the potential strategies of breast cancer treatment. In fact, therapeutic reduction of STS activity resulted in a significant reduction in tumor growth when assessed using an animal model (37). Estrone is assumed to be converted to highly biological active 17β-estradiol by 17β-Hydroxysteroid Dehydrogenase type 1 (HSD1). Expressed level of this enzyme between non-malignant and malignant tissue revealed not to differ (38). However, recent findings suggest that gene duplications of HSD1 may result in increased risk of breast cancer recurrence among ER-positive breast cancer cases (39).
Of relevance to breast cancer biology, it has been shown that estrone and 17β-estradiol are moderate but sufficient activators of PXR (40, 41). In addition, several anti-estrogens such as tamoxifen are PXR activators. Our data, which show direct functional interaction of PXR with promoter elements in SLCO1A2 would suggest that a feed forward regulation of OATP1A2 by PXR likely results in greater uptake of E1S. Accordingly, pharmacological inhibition of PXR activation may have potential therapeutic effects by modulating breast cancer progression. Assuming that estrogen metabolites such as E1S are important driving forces in the development of breast cancer, the impact of uptake transporters in governing the accumulation of this steroid precursor in estrogenic target tissues should also be considered as an important pathophysiological determinant. As shown in figure 6, the link between PXR activation and E1S accumulation may be of clinical relevance since estrone and 17β-estradiol are activators of PXR and therefore likely to increase their own cellular uptake in responsive tissues.
Figure 6. Illustration of PXR and OATP1A2 interplay in breast cancer.

The interplay of PXR activation and therefore modulation of OATP1A2 expression and function in breast cancer cells is illustrated. Induction of OATP1A2 is expected to result in increased cellular uptake of hydrophilic estrogen metabolites leading to the increased estrogenic effects. This interplay can be modulated by the specific PXR antagonist A-792611.
It is of interest to note recent reports describing beneficial effects of HMG-CoA reductase inhibitors in terms of breast cancer risk (42–44). Efficacy of statins in breast cancer prevention is assumed to be related to the pleiotropic effects including antiproliferative, proapoptotic, anti-invasive and radiosensitizing properties of statins (45). In fact, it has been shown that lipophilic statins (simvastatin and fluvastatin) inhibit the growth of mammary tumor cells inoculated in neuTg mice. This reduction appeared to be due to statin-associated reduction in tumor proliferation and increased apoptosis (46). It should be noted that OATP1A2 not only transports E1S but also statins (47). Therefore one plausible mechanism for their apparent beneficial effect to breast cancer proliferation might relate to reduced OATP1A2-mediated E1S uptake due to competition for uptake by statins.
It should be noted that PXR has also been linked to anti-apoptotic effects in several cell types including hepatocytes and prostate cancer cells. Recently, constitutive and pharmacological activation of PXR was associated with anti-apoptotic effects in colon cancer cells. Specifically, PXR activation resulted in increased expression of anti-apoptotic genes including BAG3, BIRC2 and MCL-1, thereby counteracting the effects of pro-apoptotic compounds such as deoxycholic acid, adriamycin, staurosporine and dimethylhydracine (48). Accordingly, if such an anti-apoptotic effect is extended to our findings in relation to breast cancer, higher expression of PXR in neoplastic tissue would translate into a higher proliferative activity due to PXR-mediated activation of anti-apoptotic genes.
In summary we have shown that OATP1A2 expression was highly increased in malignant human breast cancer tissues. Expression of the uptake transporter was associated with similarly elevated expression of PXR in malignant tissue suggesting an interplay between the nuclear receptor PXR and OATP1A2. We show direct transactivation of SLCO1A2 by PXR at a distal PXRRE and reduced PXR expression via siRNA technology or PXR function using a novel PXR antagonist significantly reduced the E1S associated estrogen receptor signaling in a breast cancer cell line. In conclusion, the current finding suggest an important role of the xenobiotic nuclear receptor PXR to the regulated expression of a drug/hormone uptake transporter OATP1A2 to the pathophysiology of breast cancer and that these proteins may be novel therapeutic targets for intervention.
Acknowledgments
Financial support: This study was supported by USPHS grants (GM31304, GM54724) and a grant from the Deutsche Forschungsgemeinschaft (ME 3090/1-1).
References
- 1.Kliewer SA, Moore JT, Wade L, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann JM, McKee DD, Watson MA, et al. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferguson SS, Chen Y, LeCluyse EL, Negishi M, Goldstein JA. Human CYP2C8 is transcriptionally regulated by the nuclear receptors constitutive androstane receptor, pregnane X receptor, glucocorticoid receptor, and hepatic nuclear factor 4alpha. Mol Pharmacol. 2005;68:747–57. doi: 10.1124/mol.105.013169. [DOI] [PubMed] [Google Scholar]
- 4.Gerbal-Chaloin S, Pascussi JM, Pichard-Garcia L, et al. Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab Dispos. 2001;29:242–51. [PubMed] [Google Scholar]
- 5.Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol. 2001;60:427–31. [PubMed] [Google Scholar]
- 6.Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–87. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 7.Burk O, Arnold KA, Nussler AK, et al. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol Pharmacol. 2005;67:1954–65. doi: 10.1124/mol.104.009019. [DOI] [PubMed] [Google Scholar]
- 8.Kast HR, Goodwin B, Tarr PT, et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277:2908–15. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, LeCulyse E, Liu L, et al. Rat pregnane X receptor: molecular cloning, tissue distribution, and xenobiotic regulation. Arch Biochem Biophys. 1999;368:14–22. doi: 10.1006/abbi.1999.1307. [DOI] [PubMed] [Google Scholar]
- 10.Miki Y, Suzuki T, Tazawa C, Blumberg B, Sasano H. Steroid and xenobiotic receptor (SXR), cytochrome P450 3A4 and multidrug resistance gene 1 in human adult and fetal tissues. Mol Cell Endocrinol. 2005;231:75–85. doi: 10.1016/j.mce.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Dotzlaw H, Leygue E, Watson P, Murphy LC. The human orphan receptor PXR messenger RNA is expressed in both normal and neoplastic breast tissue. Cancer Res. 1999;5:2103–07. [PubMed] [Google Scholar]
- 12.Miki Y, Suzuki T, Kitada K, et al. Expression of the steroid and xenobiotic receptor and its possible target gene, organic anion transporting polypeptide-A, in human breast carcinoma. Cancer Res. 2006;66:535–42. doi: 10.1158/0008-5472.CAN-05-1070. [DOI] [PubMed] [Google Scholar]
- 13.Lee W, Glaeser H, Smith LH, et al. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J Biol Chem. 2005;280:9610–17. doi: 10.1074/jbc.M411092200. [DOI] [PubMed] [Google Scholar]
- 14.Glaeser H, Bailey DG, Dresser GK, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81:362–70. doi: 10.1038/sj.clpt.6100056. [DOI] [PubMed] [Google Scholar]
- 15.Pizzagalli F, Varga Z, Huber RD, et al. Identification of steroid sulfate transport processes in the human mammary gland. J Clin Endocrinol Metab. 2003;88:3902–12. doi: 10.1210/jc.2003-030174. [DOI] [PubMed] [Google Scholar]
- 16.Alcorn J, Lu X, Moscow JA, McNamara PJ. Transporter gene expression in lactating and nonlactating human mammary epithelial cells using real-time reverse transcription-polymerase chain reaction. J Pharmacol Exp Ther. 2002;303:487–96. doi: 10.1124/jpet.102.038315. [DOI] [PubMed] [Google Scholar]
- 17.Konig J, Seithel A, Gradhand U, Fromm MF. Pharmacogenomics of human OATP transporters. Nauyn Schmiedebergs Arch Pharmacol. 2006;372:432–43. doi: 10.1007/s00210-006-0040-y. [DOI] [PubMed] [Google Scholar]
- 18.Nozawa T, Suzuki M, Takahashi K, et al. Involvement of estrone-3-sulfate transporters in proliferation of hormone-dependent breast cancer cells. J Pharmacol Exp Ther. 2004;311:1032–37. doi: 10.1124/jpet.104.071522. [DOI] [PubMed] [Google Scholar]
- 19.Nozawa T, Suzuki M, Yabuuchi H, et al. Suppression of cell proliferation by inhibition of estrone-3-sulfate transporter in estrogen-dependent breast cancer cells. Pharm Res. 2005;22:1634–41. doi: 10.1007/s11095-005-7096-0. [DOI] [PubMed] [Google Scholar]
- 20.Chetrite GS, Cortes-Prieto J, Philippe JC, Wright F, Pasqualini JR. Comparison of estrogen concentrations, estrone sulfatase and aromatase activities in normal, and in cancerous, human breast tissues. J Steroid Biochem Mol Biol. 2000;72:23–27. doi: 10.1016/s0960-0760(00)00040-6. [DOI] [PubMed] [Google Scholar]
- 21.Tirona RG, Lee W, Leake BF, et al. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med. 2003;9:220–4. doi: 10.1038/nm815. [DOI] [PubMed] [Google Scholar]
- 22.Barwick JL, Quattrochi LC, Mills AS, et al. Trans-species gene transfer for analysis of glucocorticoid-inducible transcriptional activation of transiently expressed human CYP3A4 and rabbit CYP3A6 in primary cultures of adult rat and rabbit hepatocytes. Mol Pharmacol. 1996;50:10–16. [PubMed] [Google Scholar]
- 23.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol. 1999;56:1329–39. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 24.Healan-Greenberg C, Waring JF, Kempf DJ, et al. A human immunodeficiency virus protease inhibitor is a novel functional inhibitor of human pregnane X receptor. Drug Metab Dispos. 2008;36:500–7. doi: 10.1124/dmd.107.019547. [DOI] [PubMed] [Google Scholar]
- 25.Moore LB, Parks DJ, Jones SA, et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;19;275:15122–27. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 26.Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation: Estrogen receptor alpha and estrogen receptor beta: regulation by selective estrogen receptor modulators and importance in breast cancer. Cancer Res. 2000;2:335–44. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Russo J, Hu YF, Silva ID, Russo IH. Cancer risk related to mammary gland structure and development. Microsc Res Tech. 2001;52:204–23. doi: 10.1002/1097-0029(20010115)52:2<204::AID-JEMT1006>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 28.Cordera F, Jordan VC. Steroid receptors and their role in the biology and control of breast cancer growth. Semin Oncol. 2006;33:631–41. doi: 10.1053/j.seminoncol.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 29.Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J Natl Cancer Inst. 2004;96:1856–65. doi: 10.1093/jnci/djh336. [DOI] [PubMed] [Google Scholar]
- 30.Beattie MS, Costantino JP, Cummings SR, et al. Endogenous sex hormones, breast cancer risk, and tamoxifen response: an ancillary study in the NSABP Breast Cancer Prevention Trial (P-1) J Natl Cancer Inst. 2006;98:110–15. doi: 10.1093/jnci/djj011. [DOI] [PubMed] [Google Scholar]
- 31.Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998;19:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]
- 32.Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos. 2001;29:1467–72. [PubMed] [Google Scholar]
- 33.Guo GL, Staudinger J, Ogura K, Klaassen CD. Induction of rat organic anion transporting polypeptide 2 by pregnenolone-16alpha-carbonitrile is via interaction with pregnane X receptor. Mol Pharmacol. 2002;61:832–9. doi: 10.1124/mol.61.4.832. [DOI] [PubMed] [Google Scholar]
- 34.Pasqualini JR, Chetrite GS. Recent insight on the control of enzymes involved in estrogen formation and transformation in human breast cancer. J Steroid Biochem Mol Biol. 2005;93:221–36. doi: 10.1016/j.jsbmb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharyya S, Tobacman JK. Steroid sulfatase, arylsulfatases A and B, galactose-6-sulfatase, and iduronate sulfatase in mammary cells and effects of sulfated and non-sulfated estrogens on sulfatase activity. J Steroid Biochem Mol Biol. 2007;103:20–34. doi: 10.1016/j.jsbmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki T, Miki Y, Nakata T, et al. Steroid sulfatase and estrogen sulfotransferase in normal human tissue and breast carcinoma. J Steroid Biochem Mol Biol. 2003;86:449–54. doi: 10.1016/s0960-0760(03)00356-x. [DOI] [PubMed] [Google Scholar]
- 37.Foster PA, Chander SK, Parsons MF, et al. Efficacy of three potent steroid sulfatase inhibitors: pre-clinical investigations for their use in the treatment of hormone-dependent breast cancer. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-007-9769-3. Epub. [DOI] [PubMed] [Google Scholar]
- 38.Salhab M, Reed MJ, Al Sarakbi W, Jiang WG, Mokbel K. The role of aromatase and 17-beta-hydroxysteroid dehydrogenase type 1 mRNA expression in predicting the clinical outcome of human breast cancer. Breast Cancer Res Treat. 2006;99:155–62. doi: 10.1007/s10549-006-9198-8. [DOI] [PubMed] [Google Scholar]
- 39.Gunnarsson C, Jerevall PL, Hammar K, et al. Amplification of HSD17B1 has prognostic significance in postmenopausal breast cancer. Breast Cancer Res Treat. 2008;108:35–41. doi: 10.1007/s10549-007-9579-7. [DOI] [PubMed] [Google Scholar]
- 40.Mnif W, Pascussi JM, Pillon A, et al. Estrogens and antiestrogens activate hPXR. Toxicol Lett. 2007;170:19–29. doi: 10.1016/j.toxlet.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Jacobs MN, Nolan GT, Hood SR. Lignans, bacteriocides and organochlorine compounds activate the human pregnane X receptor (PXR) Toxicol Appl Pharmacol. 2005;209:123–33. doi: 10.1016/j.taap.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 42.Blais L, Desgagne A, LeLorier J. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors and the risk of cancer: a nested case-control study. Arch Intern Med. 2000;160:2363–68. doi: 10.1001/archinte.160.15.2363. [DOI] [PubMed] [Google Scholar]
- 43.Cauley JA, Zmuda JM, Lui LY, et al. Lipid-lowering drug use and breast cancer in older women: a prospective study. J Womens Health. 2003;12:749–56. doi: 10.1089/154099903322447710. [DOI] [PubMed] [Google Scholar]
- 44.Boudreau DM, Gardner JS, Malone KE, et al. The association between 3-hydroxy-3-methylglutaryl conenzyme A inhibitor use and breast carcinoma risk among postmenopausal women: a case-control study. Cancer. 2004;100:2308–16. doi: 10.1002/cncr.20271. [DOI] [PubMed] [Google Scholar]
- 45.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell MJ, Esserman LJ, Zhou Y, et al. Breast cancer growth prevention by statins. Cancer Res. 2006;66:8707–14. doi: 10.1158/0008-5472.CAN-05-4061. [DOI] [PubMed] [Google Scholar]
- 47.Ho RH, Tirona RG, Leake BF, et al. Drug and bile Acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 48.Zhou J, Liu M, Zhai Y, Xie W. The anti-apoptotic role of Pregnane X Receptor in human colon cancer cells. Mol Endocrinol. 2008;22:868–80. doi: 10.1210/me.2007-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]