

Abstract
We developed several adenoviral vectors designed to target MDA-7 expression to different subcellular compartments (ie, ER, mitochondria, nucleus, and cytosol) and evaluated their ability to enhance apoptosis. Adenoviral ER-targeted mda-7/IL-24 vector (Ad-ER-mda7) selectively and effectively inhibited the growth and proliferation of lung (A549 and H1299) and esophageal (Seg1 and Bic1) cancer cells by enhancing cell killing. Both Ad-mda7 and Ad-ER-mda7 activated a novel pathway of ER stress-induced apoptosis characterized by unregulated expression of phosphorylated JNK (p-JNK), phosphorylated cJun (p-cJun), and phosphorylated RNA-dependent protein kinase (p-PKR). Caspase-4 activation mediated Ad-mda7- and Ad-ER-mda7-induced cell death. In addition, Ad-mda7- and Ad-ER-mda7-mediated growth inhibition correlated with activation of ER molecular markers PKR and JNK both in vitro (in Ad-mda7- or Ad-ER-mda7-treated lung cancer cells) and in vivo. These findings suggest that vectors targeting the endoplasmic reticulum (Ad-ER-mda7) may be more effective in cancer gene therapy possibly through more effective induction or ER stress pathways.
Keywords: Apoptosis, MDA-7, adenovirus, gene therapy
Introduction
The melanoma differentiation-associated gene 7 (mda-7) is a tumor suppressor gene that induces apoptosis in a wide range of cancer cells, both in vivo and in vitro, when overexpressed through a replication-incompetent adenoviral vector (1-4). The cytotoxic activity of the mda-7 gene product is specific to tumor cells and independent of the status of other tumor suppressor gene products (eg, p53, Rb, ras, or p16INK4) (5). The cDNA of mda-7 encodes an evolutionarily conserved protein (MDA-7) that, despite having only 19% identity with the homodimeric cytokine interleukin-10 (IL-10), has been assigned to the IL-10 family and renamed interleukin-24 (IL-24) (6, 7). This assignment was based on MDA-7′s amino acid identity with IL-10 and incorporation of an IL-10 family motif, its chromosomal localization within an IL-10 family cluster of genes, its translational regulation, and its predicted structural features (eg, a 4-helix bundle structure characteristic of the IL-10 family) (6, 7). Ectopic expression of mda-7 in cancer cells by Ad-mda7 or plasmid DNA vectors results in the overproduction of intracellular MDA-7 protein and its secreted form, IL-24 (1, 5).
In laboratory studies utilizing lung cancer cell lines, we have demonstrated that MDA-7 overexpression leads to the upregulated expression and phosphorylation of the RNA-dependent protein kinase (PKR) necessary for Ad-mda7-induced apoptosis (8, 9). In addition, we and others have demonstrated that MDA-7/IL-24 intracellular-mediated apoptosis may involve the endoplasmic reticulum (ER) signaling pathway. For example, we have demonstrated the consistent overexpression of several ER stress proteins (ie, GRP78/BiP, GADD34, and PP2A) in Ad-mda7-treated lung cancer cells (10). Meanwhile, Fisher’s group has demonstrated that the ER chaperone protein GRP78/BiP, by serving as an intracellular target of MDA-7/IL-24 and thereby mediating MDA-7/IL-24’s activation of its downstream targets p38 MAPK and GADD, selectively mediates apoptosis of prostate and breast cancer cells (11, 12). Molecular chaperones, such as GRP78/BiP and HSP70, play important roles in the unfolded protein response (UPR) pathway by preventing the aggregation of misfolded proteins and shuttling them to the 20S proteasome for degradation (13). Because ER is a principal site for MDA-7/IL-24 protein synthesis and folding (10, 13), the ER stress-mediated cell death pathway can be triggered by disparate perturbations in normal ER function, such as the accumulation of unfolded, misfolded, or excessive MDA-7/IL-24 protein.
Assuming that Ad-mda7 treatment induces the accumulation of MDA-7 proteins and consequently ER and/or cytoplasmic stress, we hypothesized that MDA-7/IL-24’s subcellular location (specifically, in the ER) is an important regulator of its antitumor activity. To test this hypothesis, we designed adenoviral vectors that would target MDA-7 expression to different subcellular compartments (ie, ER, mitochondria, nucleus, and cytosol) and compared their ability to induce apoptosis in lung and esophageal cancer cells. Our results indicated that the adenoviral ER-targeted vector induced much higher levels of cell death than did those targeted elsewhere within the cell and that this vector enhanced cell killing. Our results also showed that caspase-4 and JNK activation mediated the apoptosis induced by both untargeted (Ad-mda7) and ER-targeted (Ad-ER-mda7) vectors.
Materials and Methods
Cell lines and reagents
Human lung (A549 and H1299) and esophageal (Seg1 and Bic1) cancer cell lines were obtained from the American Type Culture Collection (Rockville, MD). PKR+/+ and PKR-/- mouse embryo fibroblasts (MEFs) were obtained from Dr. Glen Barber (University of Miami School of Medicine, Miami, FL) (14). MEF cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 10 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Life Technologies, Inc., Grand Island, NY) in a 5% CO2 atmosphere at 37°C. Caspase-4 and JNK inhibitors were obtained from Calbiochem (San Diego, CA). Final working solutions were diluted in medium to contain <0.01% of dimethyl sulfoxide. All experiments using this compound were performed under subdued lighting conditions.
Adenoviral vector construction
Constructions of the Ad-mda7, Ad-LacZ, and Ad-luc vectors have been reported previously (8). Constructions of plasmids targeted to the ER (pCMV/myc/ER/mda-7), mitochondria (pCMV/myc/Mito/mda-7), nucleus (pCMV/myc/Nuc/mda-7), and cytosol (pCMV/myc/Cyto/mda-7) have been reported previously (10). The ER-mda-7 fragment from pCMV/myc/ER/mda-7 was obtained by digestion at the PmI I and Bcl I sites. The Mito-mda-7 fragment from pCMV/myc/Mito/mda-7 was obtained by digestion at the PmI I and Bcl I sites. The Nuc-mda-7 fragment from pCMV/myc/Nuc/mda-7 was obtained by digestion at the HincI I and BcI I sites. The Cyto-mda-7 fragment from pCMV/myc/Cyto/mda-7 was obtained by digestion at the HincI I and BcI I sites.
Individual shuttle vectors were obtained as follows. The PLJ37/pAD-RAP/Shuttle vector was subjected to EcoRV and BcI I digestion and ligated with ER-mda7, Mito-mda7, Nuc-mda-7 or Cyto-mda7 fragments to obtain their respective shuttle vectors (ie, PLJ37/pAD-RAP/ER-mda7/Shuttle vector, PLJ37/pAD-RAP/Mito-mda7/Shuttle vector, PLJ37/pAD-RAP/Nuc-mda7/Shuttle vector, or PLJ37/pAD-RAP/Cyto-mda7/Shutle vector). The resulting shuttle vectors were then subjected to further digestion with BstB I and CIa I to obtain large fragments. Each of the resulting large fragments was then inserted into a pLJ34 vector between the BstB I and CIa I sites to create adenoviral vectors specifically targeted to the ER, mitochondria, nucleus, and cytosol, respectively (ie, Ad-ER-mda7, Ad-Mito-mda7, Ad-Nuc-mda7 and Ad-Cyto-mda7). Adenoviral transduction efficiency in cancer cell lines was determined by infecting cells with Ad-LacZ vectors and quantifying the titers needed to transduce the LacZ gene into at least 70% of the cells.
Immunofluorescent cellular localization studies
A549 lung cancer cells (5 × 104 cells/well) were grown on chamber slides to 70% confluence and then transfected with Ad-luc, Ad-mda7, Ad-ER-mda7, Ad-Mito-mda7, Ad-Nuc-mda7, or Ad-Cyto-mda7 or treated with phosphate buffered saline (PBS) as a negative control. Seventy-two hours later, cells were washed with PBS and fixed with 4% paraformaldehyde/PBS for confocal microscopic analysis as previously described (9). In brief, cells were blocked with 1% normal goat serum for 1 hour and then incubated overnight at a dilution of 1:100 with the primary mouse monoclonal anti-MDA-7 antibody. Next, the slides were washed to remove primary antibody, rinsed with PBS, and placed in a prewarmed staining solution containing ER-Tracker™ red dyes or Mito Tracker Deep Red 633 (M-22426) (Molecular Probes) for approximately 15-20 minutes at 37°C. Then, the slides were washed and incubated with a fluorescein isothiocyanate- or rhodamine-conjugated secondary antibody (Invitrogen, Eugene, OR) for 1 hour. Next, the slides were mounted with ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) and analyzed under an Olympus FluoView FV500 laser confocal microscope (Olympus America, Melville, NY) after adjustment for background staining.
Flow cytometric analysis
Apoptosis was assessed by propidium iodide staining and fluorescence-activated cell sorting (FACS) analysis of cells. In brief, cells were harvested; pelleted by centrifugation; resuspended in PBS containing 50 μg/mL propidium iodide, 0.1% Triton X-100, and 0.1% sodium citrate; vortexed; and then subjected to FACS analysis (Becton-Dickenson FACScan, Mountain View, CA; FL-3 channel).
Immunoblot analyses
72 hours after adenoviral transfection, A549 or H1299 cell extracts were prepared and immunoblot assays performed as previously described (8, 9). Antibodies to JNK, phosphorylated JNK (p-JNK), c-Jun, phosphorylated c-Jun (p-cJun), PKR (K-17), eIF-2α, and β-actin (control) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The caspase-4 was obtained from StressGen. (Victoria, British Columbia, Canada). Antibodies to phosphorylated PKR (pT451) and phosphorylated eIF-2α (Ser51) were obtained from BioSource International (Camarillo, CA). Polyclonal and monoclonal antibodies to MDA-7 were obtained from Introgen Therapeutics, Inc. (Houston, TX).
Analysis of In vivo tumor growth after Ad-mda7 or Ad-ER-mda7 treatment
A549 cells (5 × 106 cells/0.2 mL) were injected subcutaneously into the flanks of female athymic nu/nu mice 4-6 weeks old. Once a tumor grew to approximately 5 mm × 5 mm, PBS, Ad-luc, Ad-mda7, or Ad-ER-mda7 was injected directly into it via a single pass of a 25-gauge hypodermic needle at dose of 3×1012 vp. A second injection was given 3 days later and a third injection 3 days after that. Thus, each mouse received a total of 3 injections (total 9 × 1012 vp) over 6 days. After the third injection, the maximal and minimal diameters of each tumor were measured by slide calipers placed on the skin, every 2 days for 30 days. Tumor volume was calculated by assuming a spherical shape and using the following formula: volume = (a × b2)/2, where a and b are the maximal and minimal diameters, respectively. Results for each treatment group (6-9 mice/group) were averaged and expressed as the mean (SD).
Apoptosis was measured by histological analysis. In brief, tumors were excised 24 hours after the third injection in vivo, fixed in 10% formalin, embedded in paraffin blocks, processed for histological analysis and TUNEL assays. For TUNEL assay, sections were dewaxed, rehydrated (55°C for 15 min), washed in xylene, and then rehydrated through a graded series of ethanol and redistilled water. Tissue sections were then incubated with proteinase K, permeabilized in 0.1% Triton-X100 in 0.1% sodium citrate, and labeled with the TUNEL reaction mixture.
Statistical analysis
Analysis of variance and a two-tailed Student’s t test were used for statistical analysis when appropriate. Significance was set at P<0.05.
Results
Mda-7 adenoviral vectors induce targeted intracellular expression of MDA-7 protein
As confirmed by immunofluorescent confocal microscopic analysis of A549 lung cancer cells 72 hours after transfection, the adenovirus-mediated intracellular targeting vectors Ad-ER-mda7, Ad-Mito-mda7, Ad-Nuc-mda7, and Ad-Cyto-mda7 successfully targeted MDA-7 protein expression to specific subcellular regions of the transduced cancer cells (Fig. 1A). The precise subcellular localization of the targeted proteins was confirmed by comparison their expression patterns with those of molecular markers known to reside in these compartments. For example, nuclear targeted MDA-7 colocalized with DAPI-stained nuclei, and ER-targeted MDA-7 colocalized with ER-Tracker™ red dye. To determine whether these intracellularly targeted mda-7 vectors caused MDA-7 secretion, posttransfection MDA-7 expression in lung cancer A549 cell lysates and supernatants was assessed by Western blot analysis. These analyses confirmed that each intracellularly targeted adenoviral vector successfully promoted the expression of correctly sized MDA-7 protein (Fig. 1B) (10), thereby demonstrating that Ad-mda7 transduction did cause the secretion of MDA-7 protein. Unexpectedly, however, the ER-targeted MDA-7 protein was shown to be secreted even though it contained an ER retention signal.
Figure 1.
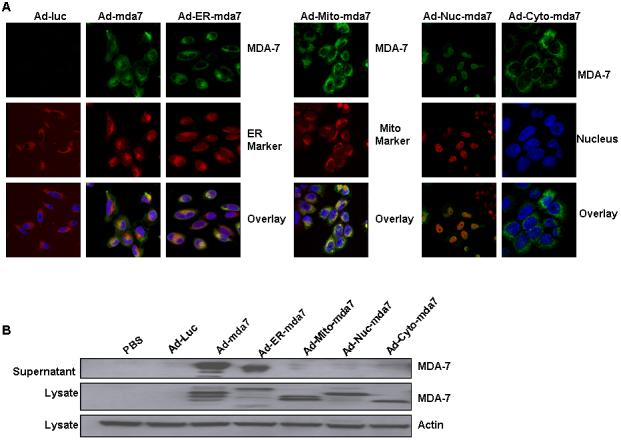
Immunofluorescent confocal microscopic analysis of intracellularly targeted mda-7 adenoviral vectors. A, Analysis of antibody against MDA-7 stained green, endoplasmic reticulum (ER) stained red with ER-Tracker™ dyes, and nuclei stained blue with DAPI demonstrated colocalization of both MDA-7 protein and the ER marker in Ad-ER-mda7-treated A549 cancer cells 72 hours after transfection. Analysis of antibody against MDA-7 stained green, mitochondria (Mito) stained red with Mito Tracker Deep Red 633 (M-22426), and nuclei stained blue with DAPI demonstrated colocalization of both MDA-7 protein and the Mito marker in Ad-Mito-mda7-treated A549 cancer cells 72 hours after transfection. Analysis of antibodies against MDA-7 stained green and nuclei (Nuc) stained red or blue with DAPI demonstrated nuclear localization of MDA-7 protein in Ad-Nuc-mda7-treated A549 cancer cells 72 hours after transfection. Analysis of antibodies against MDA-7 stained green and nuclei (Nuc) stained blue with DAPI demonstrated cytosolic localization of MDA-7 protein in Ad-cyto-mda7-treated A549 cancer cells 72 hours after transfection. B, Western blot analysis of MDA-7 expression in A549 cell lysates and supernatant 72 hours after treatment with PBS, Ad-Luc (2500 vp/ or 24.5 moi), Ad-mda7 (2500 vp/ or 25.5 moi), Ad-ER-mda7 (2500 vp/ or 24.27 moi), Ad-Mito-mda7 (2500 vp/ or 22.1 moi), Ad-Nuc-mda7 (2500 vp/ or 26.59 moi), or Ad-Cyto-mda7 (2500 vp/ or 24 moi). β-Actin expression was analyzed as a control.
Adenoviral ER-targeted vector induces activation of the ER stress-mediated cell death pathway
As shown by flow cytometric analysis of human lung (A549 and H1299) and esophageal (Seg1 and Bic-1) cancer cells after transfection, all of the intracellularly targeted mda-7 adenoviral vectors except Ad-Nuc-mda7 vector induced cell death within 72 hours of transfection (Fig. 2A). Of those vectors that did induce apoptosis, the ER-targeted MDA-7 adenoviral vector (Ad-ER-mda7) was much more lethal than either the mitochondria-targeted (Ad-Mito-mda7) or cytosol-targeted mda-7 (Ad-Cyto-mda7) vectors and also more lethal than the untargeted mda-7 vector (Ad-mda7). Moreover, as shown by flow cytometric analysis of normal human bronchial epithelial [NHBE] cells and WI-38 normal human fibroblasts 72 hours after their adenoviral exposure, only one of the intracellularly targeted adenoviral mda7 vectors (Ad-Mito-mda7) was toxic to normal cells (Fig. 2A).
Figure 2.
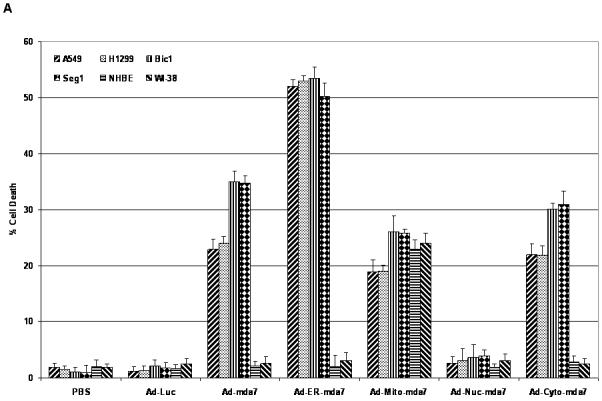
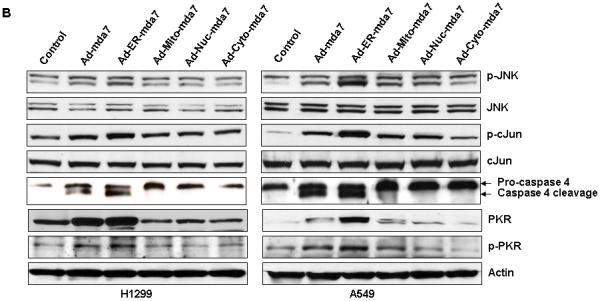
Ability of adenoviral endoplasmic reticulum (ER) targeting mda-7 vector (Ad-ER-mda7) to induce signaling along the ER stress-mediated cell death pathway. A, Flow cytometric analysis of apoptosis in A549, H1299, Seg1, Bic1, NHBE and WI-38 cells 72 hours after treatment with PBS, Ad-Luc (2500 vp/ or 24.5 moi), Ad-mda7 (2500 vp/ or 25.5 moi), Ad-ER-mda7 (2500 vp/ or 24.27 moi), Ad-Mito-mda7 (2500 vp/ or 22.1 moi), Ad-Nuc-mda7 (2500 vp/ or 26.59 moi), or Ad-Cyto-mda7 (2500 vp/ or 24 moi). Experiments involving each cell line were performed in triplicate. B. Western blot analysis of phosphorylated JNK (pJNK), JNK, phosphorylated c-Jun (p-cJun), cJun, caspase-4, phosphorylated PKR (p-PKR) and PKR protein expression in A549 and H1299 cell lysates 72 hours after treatment with Ad-Luc (2500 vp/ or 24.5 moi), Ad-mda7 (2500 vp/ or 25.5 moi), Ad-ER-mda7 (2500 vp/ or 24.27 moi), Ad-Mito-mda7 (2500 vp/ or 22.1 moi), Ad-Nuc-mda7 (2500 vp/ or 26.59 moi), or Ad-Cyto-mda7 (2500 vp/ or 24 moi). β-Actin expression was analyzed as a control.
We hypothesize that the ER stress (accumulation of MDA-7 proteins in ER) is essential to Ad-ER-mda7 mediated cell death activity in cancer cells. To test our hypothesis, we determined the most important ER stress markers (JNK, phospho-JNK, cJun, and phospho-cJun) in A549 and H1299 lung cancer cells following PBS, Ad-mda7, Ad-ER-mda7, Ad-Mito-mda7, Ad-Nuc-mda7 and Ad-Cyto-mda7 treatment for 72 hrs by immunoblot analysis (15). As shown in Figure 2B, Expression of p-JNK and p-cJun was upregulated after Ad-mda7 and Ad-ER-mda7 transduction but not after Ad-Mito-mda7, Ad-Nuc-mda7, or Ad-Cyto-mda7 transduction (Fig. 2B), suggesting that activation of JNK or c-JUN was the mechanism by which Ad-mda7 and Ad-ER-mda7 mediated cell killing. . Caspase-4 is an ER-resident caspase that is processed in human cells in response to ER stress and is required for ER stress-induced cell death (15). We next investigated whether caspase-4 was involved in Ad-mda7 and Ad-ER-mda7-induced cell death. Caspase-4 was cleaved to its active form in A549 and H1299 human lung cancer cells treated with Ad-mda7 or Ad-ER-mda7 but not in those treated with Ad-Mito-mda7, Ad-Nuc-mda7, or Ad-Cyto-mda7 (Fig. 2B). To more directly determine the importance of JNK and caspase-4 activation to Ad-mda7 and Ad-ER-mda7-mediated cell death, A549 cells were exposed to Ad-mda7 and Ad-ER-mda7 in the absence or presence of a chemical JNK inhibitor (SP600125, 10 μmol/L) or a peptide inhibitor of caspase-4 (Ac-LEVD-CHO, 10 μmol/L) and subjected to FACS analysis to determine the extent of the resulting cell death. In both case, inhibitory treatment reduced the amount of cell death induced by both Ad-mda7 (from 22.3% to 17% after JNK inhibition and from 22.3% to 13% after caspase-4 inhibition) and Ad-ER-mda7 (from 52% to 41% after JNK inhibition and from 52% to 27% after caspase-4 inhibition (Supplemental data 1). Conversely, neither inhibitor reduced the amount of cell death induced by Ad-Mito-mda7 or Ad-Cyto-mda7 (Supplemental data 1). Together, this suggested that both Ad-mda7- and Ad-ER-mda7-mediated cell killing involved the activation of JNK and caspase-4.
We have demonstrated that MDA-7 overexpression in human lung cancer cells leads to the upregulation and phosphorylation of PKR necessary for Ad-mda7-induced apoptosis (8, 9). We next determined if PKR activation might also play a role in Ad-ER-mda7 -infected cancer cells. Human lung cancer cells revealed that both Ad-mda7 and Ad-ER-mda7 upregulated and promoted the phosphorylation of PKR (Fig. 2B). In contrast, Ad-Mito-mda7, Ad-Nuc-mda7 and Ad-Cyto-mda7 did not. This contribution of PKR activation to Ad-ER-mda7’s apoptotic activity was confirmed in experiments using MEFs from PKR knockout mice. That Ad-mda7-induced cell killing was dependent on PKR was confirmed by the fact that only MEFs from PKR+/+ (wild-type) mice underwent apoptosis induction after Ad-mda7 treatment (Supplemental data 2) (8). Ad-ER-mda7 mediated cell killing was also dependent on PKR with background level of apoptosis occurring in PKR null and 20% apoptosis occurring in wildtype MEFs (Supplemental data 2). Conversely, neither Ad-Mito-mda7- nor Ad-Cyto-mda7-induced apoptosis appeared to be dependent on PKR genomic status, apoptosis occurring at similar rates in both PKR-/- and PKR+/+ MEFs (Supplemental data 2).
In vivo regression of tumors follows intratumoral injection of Ad-mda7 or Ad-ER-mda7 in nu/nu mice
As shown by in vivo experiments in a subcutaneous nu/nu tumor mouse model and in confirmation of previous studies showing that intratumoral administration of Ad-mda7 inhibits the growth of lung tumor xenografts (16), intratumorally injected Ad-mda7 and Ad-ER-mda7 were both significantly more growth inhibitory than either PBS or Ad-luc (controls) (P=0.01 [Ad-mda7] and P=0.005 [Ad-ER-mda7] versus controls) (Fig. 3A). In addition, intratumorally injected Ad-ER-mda7 was significantly more growth inhibitory than Ad-mda7 (P=0.02) (Fig. 3A). No significant tumor inhibition was observed in either Ad-luc-treated or PBS-treated mice. Analysis of A549 lung tumor tissue showed that (a) the growth inhibition observed in Ad-mda7- or Ad-ER-mda7-treated mice was caused by MDA-7 protein expression and (b) this MDA-7 protein expression in both cases was associated with higher levels of p-JNK and PKR expression than those seen in control tumor tissues (Fig. 3B). TUNEL assays demonstrated increased TUNEL positive staining cells in Ad-mda7- or Ad-ER-mda7-treated mice tissue (Fig. 3B). Thus, Ad-ER-mda7 effectively inhibited lung tumor growth in vivo, and intratumoral injection of Ad-mda7 or Ad-ER-mda7 caused no significant systemic toxicity. Together, these results demonstrated the potential antitumor effect of Ad-mda7 or Ad-ER-mda7 when injected directly into tumors.
Figure 3.
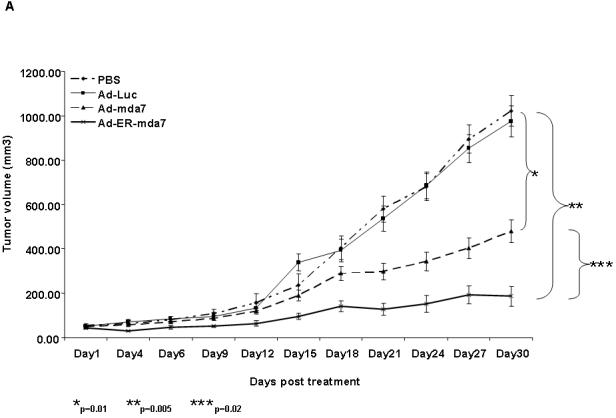
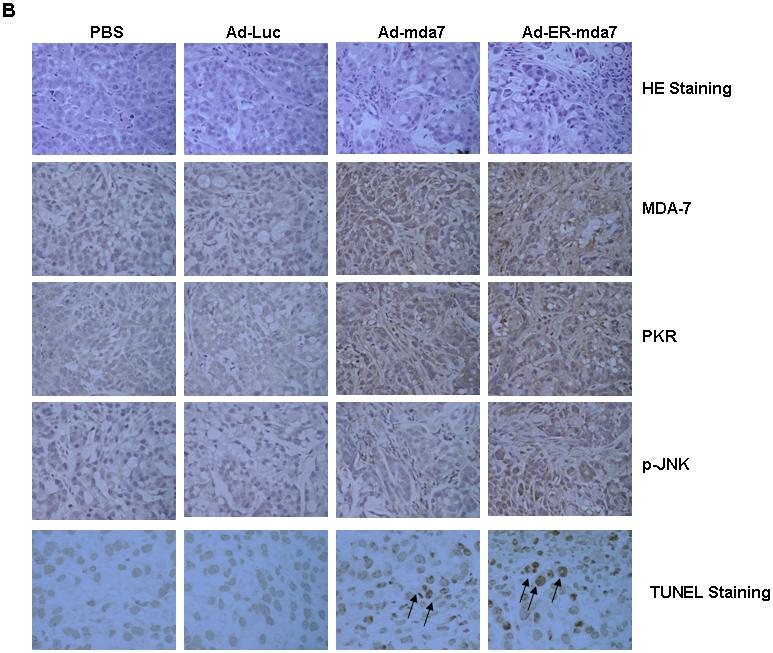
In vivo regression of tumors after intratumoral injection of Ad-mda7 or Ad-ER-mda7 in nu/nu mice. A, A549 tumor growth was significantly inhibited after injection with Ad-mda7 or Ad-ER-mda7 as opposed to PBS or Ad-luc. In each case, mice received a total of 3 injections over 6 days. Tumor volumes (vertical axis) were measured in 6-9 mice per group over 30 days; data represent the mean (SD). B. Histology of A549 tumors 24 hours after last treatment with PBS, Ad-luc, Ad-mda7, or Ad-ER-mda7 (hematoxylin and eosin staining, ×40 magnification). Analysis of A549 lung tumor tissues showed that the growth inhibition observed in Ad-mda7- or Ad-ER-mda7-treated mice was caused by MDA-7 protein expression and apoptosis. In addition, MDA-7 protein expression was associated with increased p-JNK and PKR expression in tumor tissues treated with Ad-mda7 or Ad-ER-mda7 as opposed to controls (PBS or Ad-luc).
Discussion
In the present study, we have demonstrated that Ad-mda7 or Ad-ER-mda7 treatment results in ER stress-induced apoptosis. We have also demonstrated that Ad-mda7- or Ad-ER-mda7-mediated growth inhibition in vivo correlates with the activation of ER molecular markers such as PKR and JNK observed in vitro in Ad-mda7- or Ad-ER-mda7-treated cancer cells.
The replication-incompetent adenoviral vector Ad-mda7 is minimally toxic to normal cells but a potent inducer of apoptosis in a variety of cancer cell lines (1-5). As we and our collaborators have demonstrated, mda-7 gene transfer via Ad-mda7 induces tumor-specific apoptotic, growth-inhibitory, and antiangiogenic effects independently of the status of other tumor suppressor genes (17-20). In addition to its direct cytotoxic effects, Ad-mda7 transduction elicits the secretion of MDA-7, a protein that is also known as IL-24 and has unique apoptotic properties (21-23). In phase I trials in patients resistant to conventional therapy, intratumoral Ad-mda7 injection has produced minimal adverse effects and exhibited clinical activity in a subset of patients heavily pretreated with chemotherapeutics and radiation (24). Previously, we have shown that Ad-mda7 transduction of human lung cancer cells can increase expression of stress-related proteins, including GRP78/BiP, GADD34, PP2A, caspase-7, and XBP-1, consistent with activation of the UPR pathway, a key sensor of ER-mediated stress (25). The UPR pathway is an ER-to-nucleus signal transduction pathway that regulates the expression of a wide variety of target genes and is responsible for maintaining cellular homeostasis (13). Thus activation of UPR signaling appears to be important in mediating apoptosis induced by Ad-mda7.
In the present study, we utilized a set of intracellularly targeted mda-7 adenoviral vectors that we designed to drive the expression of protein in specific subcellular compartments (ie, ER, mitochondria, nucleus, and cytoplasm). Of all these vectors, the ER-targeted Ad-ER-mda7 vector was by far the most lethal to cells.
Interestingly, our demonstration that Ad-ER-mda7 transduction can still induce the secretion of MDA-7 protein warrants further studies to determine whether a secreted extracellular form from Ad-ER-mda7 could induce STAT3 activation and apoptosis in cancer cells. Indeed, after Ad-mda7 transduction of lung cancer cells, MDA-7 protein exists in both an intracellular form and a secreted extracellular form (1). The secreted extracellular form can bind to 2 different receptors: the type 1 IL-20 heterodimeric receptor complex comprising IL-20R1/IL-20R2 and the type 2 complex comprising IL-22R1/IL-20R2, which leads to activation of the STAT signaling pathways (12).
We next examined the expression of several downstream proteins including JNK, c-jun, and caspase-4. The ER stress-mediated cell death pathway involves recruitment of the cytosolic adaptor TRAF2 to the ER membrane, where TRAF2 activates the apoptosis-signaling kinase 1 (ASK1). Activation of ASK1 leads in turn to activation of JNK and mitochondria-dependent caspase (26). Meanwhile, human caspase-4, which is 48% homologous to murine caspase-12, has been shown to be localized to the ER membrane and to be specifically activated by and required for ER stress-induced apoptosis (13). In this light, we demonstrated that JNK and c-Jun activation is essential for Ad-mda7- and Ad-ER-mda7-mediated cell death, that Ad-mda7 and Ad-ER-mda7 treatment induces caspase-4 cleavage, and that p-JNK and caspase-4 are major drivers of apoptosis in Ad-mda7- and Ad-ER-mda7-transduced A549 lung cancer cells. Like Ad-mda7, which has been reported to utilize caspase-dependent and independent pathways of activation (8), our studies suggest that Ad-ER-mda7-induced apoptosis is partially dependent on caspase. Taken together, our data indicate that induction of ER stress-induced apoptosis by JNK and caspase-4 mediates Ad-mda7 and Ad-ER-mda7-induced apoptosis.
PKR is well characterized in the literature as an antiviral immune mediator that responds to viral double-stranded RNA (dsRNA) by activating eIF-2α, inhibiting protein synthesis inhibition, and blocking viral protein production (27). Apparently, its activation in HeLa, Cos1, U937 and NIH3T3 tumor cells leads to apoptosis induction (28, 29). Building upon our previous demonstrations that Ad-mda7 transfection of lung cancer cells leads to the upregulation and phosphorylation of PKR necessary for Ad-mda7-induced apoptosis (8, 9), we have now demonstrated that Ad-ER-mda7 transduction does the same. We also found that upregulation and phosphorylation of PKR is specific for Ad-mda7 and Ad-ER-mda7 since we saw no change in PKR expression after transduction with Ad-Mito-mda7, Ad-Nuc-mda7, or Ad-Cyto-mda7. In turn, we also conclude that the PKR upregulation and phosphorylation induced by Ad-mda7 and Ad-ER-mda7 is not solely a response to adenoviral transfection since all of the other vectors we tested failed to induce PKR induction. Indeed, PKR induction appears to be critical for Ad-mda7 or Ad-ER-mda7 apoptosis since normal MEFs lacking PKR were unable to undergo apoptosis as opposed to MEFs expressing wild-type PKR. This inhibition of apoptosis appeared to be specific to both Ad-mda7 and Ad-ER-mda7 since transduction of MEFs lacking PKR with the Ad-Mito-mda7 vector did not impair apoptosis. In light of our previous identification of MDA-7 as a novel binding partner for PKR (9), further study is warranted to determine whether Ad-ER-mda7-induced MDA-7 might also interact with PKR.
Ad-ER-mda7 appears to be extremely growth inhibitory. This was shown by our experiments in an in vivo xenograft tumor model in which intratumoral administration of Ad-mda7 or Ad-ER-mda7 in mice bearing subcutaneous tumors significantly inhibited tumor growth and by tumor tissue studies showing an association between MDA-7 protein expression and the increased induction of PKR and p-JNK expression in Ad-mda7- or Ad-ER-mda7-treated tumor tissue as opposed to untreated (control) tissues.
In summary, by targeting the ER, Ad-ER-mda7 can induce cell death to a much greater extent through the ER-stress mediated cell death pathway involving caspase-4, JNK and PKR activation. Ad-mda7 or Ad-ER-mda7 treatment results in ER stress-induced apoptosis and to tumor growth inhibition in vivo. Going forward, our preliminary identification of the molecular mechanisms responsible for Ad-mda7-induced apoptosis of human cancer cells bodes well for the rational development of strategies for enhancing the antitumor effects of MDA-7, and our development of intracellularly targeted mda-7 adenoviral vectors will allow for the clinical optimization of this gene transfer therapy and the identification of patients unresponsive to Ad-mda7 therapies who might benefit from targeted therapy.
Supplementary Material
Supplemental data 1 Flow cytometric analysis of cell death in A549 cancer cells 72 hours after treatment with PBS, Ad-Luc (2500 vp/ or 24.5 moi), Ad-mda7 (2500 vp/ or 25.5 moi), Ad-ER-mda7 (2500 vp/ or 24.27 moi), Ad-Mito-mda7 (2500 vp/ or 22.1 moi), Ad-Nuc-mda7 (2500 vp/ or 26.59 moi), or Ad-Cyto-mda7 (2500 vp/ or 24 moi) with or without caspase-4 (Ac-LEVD-CHO, 10 μmol/L) or JNK chemical inhibitor (10 μmol/L). Experiments were performed in triplicate; data represent the mean (SD).
Supplemental data 2 Flow cytometric analysis of cell death in PKR+/+ and PKR-/- mouse embryo fibroblasts (MEFs) 72 hours after treatment with PBS, Ad-Luc (2500 vp/ or 24.5 moi), Ad-mda7 (2500 vp/ or 25.5 moi), Ad-ER-mda7 (2500 vp/ or 24.27 moi), Ad-Mito-mda7 (2500 vp/ or 22.1 moi), Ad-Nuc-mda7 (2500 vp/ or 26.59 moi) or Ad-Cyto-mda7 (2500 vp/ or 24 moi). Experiments were performed in triplicate; data represent the mean (SD).
Acknowledgements
Grant support: National Cancer Institute and National Institutes of Health grants P01 CA78778-01A1 (J.A. Roth and S.G. Swisher), SPORE 2P50-CA70970-04, SBIR S/C 1R43 CA86587-1 (S.G. Swisher and S. Chada), and supporting Core Grant CA16672 (M. D. Anderson Cancer Center); gifts from Tenneco and Exxon to the Division of Surgery for its Core Laboratory Facility; a grant from the Tobacco Settlement Funds as appropriated by the Texas State Legislature (Project 8); a grant from the W. M. Keck Foundation; and a sponsored research agreement with Introgen Therapeutics, Inc. (SR93-004-1); and by support from the Homer Flower Gene Therapy Fund, the Charles Rogers Gene Therapy Fund and the George P. Sweeney Esophageal Research Fund;
We thank Bingbing Wang for her technical assistance and Debbie Smith for her assistance in preparing the manuscript.
Abbreviations
- mda-7
melanoma differentiation-associated gene 7
- MDA-7
melanoma differentiation-associated gene 7 protein
- Ad-mda7
adenoviral mda-7
- VP
viral particle
- PKR
RNA-dependent protein kinase
- UPR
unfolded protein response
References
- 1.Chada S, Ramesh R, Mhashilkar A. Cytokine/chemokine-based gene therapy for cancer. Curr Opin Mol Ther. 2003;5:463–74. [PubMed] [Google Scholar]
- 2.Gopalan B, Litwak A, Sharma S, et al. Activation of the Fas/Fasl signaling pathway by mda-7/IL-24 kills human ovarian cancer cells. Cancer Res. 2005;65:3017–24. doi: 10.1158/0008-5472.CAN-04-3758. [DOI] [PubMed] [Google Scholar]
- 3.Saeki T, Mhashilkar A, Chada S, Branch C, Roth JA, Ramesh R. Tumor-suppressive effects by adenovirus-mediated mda-7 gene transfer in non-small cell lung cancer in vitro. Gene Ther. 2000;7:2051–7. doi: 10.1038/sj.gt.3301330. [DOI] [PubMed] [Google Scholar]
- 4.Gopalkrishnan RV, Sauane M, Fisher PB. Cytokine and tumor cell apoptosis inducing activity of mda-7/IL-24. Int Immunopharmacol. 2004;4:635–47. doi: 10.1016/j.intimp.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Mhashilkar AM, Schrock RD, Hindi M, et al. Melanoma differentiation associated gene (mda7): a novel anti-tumor gene for cancer gene therapy. Mol Med. 2001;7:271–82. [PMC free article] [PubMed] [Google Scholar]
- 6.Dumoutier L, Leemans C, Lejeune D, Kotenko SV, Renauld JC. Cutting edge: STAT activation by IL-19, IL-20 and mda-7 through IL-20 receptor complexes of two types. J Immunol. 2001;167:3545–9. doi: 10.4049/jimmunol.167.7.3545. [DOI] [PubMed] [Google Scholar]
- 7.Fickenscher H, Hor S, Kupers H, Knappe A, Wittmann S, Sticht H. The interleukin-10 family of cytokines. Trends Immunol. 2002;23:89–96. doi: 10.1016/s1471-4906(01)02149-4. [DOI] [PubMed] [Google Scholar]
- 8.Pataer A, Vorburger SA, Barber GN, et al. Adenoviral transfer of the melanoma differentiation-associated gene 7 (mda7) induces apoptosis of lung cancer cells via up-regulation of the double-stranded RNA dependent protein kinase (PKR) Cancer Res. 2002;62:2239–43. [PubMed] [Google Scholar]
- 9.Pataer A, Vorburger SA, Chada S, et al. Melanoma differentiation-associated gene-7 protein physically associates with the double-stranded RNA-activated protein kinase PKR. Mol Ther. 2005;11:717–23. doi: 10.1016/j.ymthe.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 10.Sieger KA, Mhashilkar AM, Stewart A, et al. The tumor suppressor activity of MDA-7/IL-24 is mediated by intracellular protein expression in NSCLC cells. Mol Ther. 2004;9:355–67. doi: 10.1016/j.ymthe.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Gupta P, Walter MR, Su ZZ, et al. BiP/GRP78 Is an Intracellular Target for MDA-7/IL-24 Induction of Cancer-Specific Apoptosis. Cancer Res. 2006;66:8182–91. doi: 10.1158/0008-5472.CAN-06-0577. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar D, Su ZZ, Lebedeva IV, et al. mda-7 (IL-24): signaling and functional roles. Biotechniques. 2002;(Suppl):30–9. [PubMed] [Google Scholar]
- 13.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–84. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 14.Balachandran S, Kim CN, Yeh WC, Mak TW, Bhalla K, Barber GN. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998;17:6888–902. doi: 10.1093/emboj/17.23.6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 16.Saeki T, Mhashilkar A, Swanson X, et al. Inhibition of human lung cancer growth following adenovirus-mediated mda-7 gene expression in vivo. Oncogene. 2002;21:4558–66. doi: 10.1038/sj.onc.1205553. [DOI] [PubMed] [Google Scholar]
- 17.Jiang H, Lin JJ, Su ZA, Goldstein NI, Fisher PB. Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene. 1995;11:2477–86. [PubMed] [Google Scholar]
- 18.Sarkar D, Su ZZ, Lebedeva IV, et al. mda-7 (IL-24): signaling and functional roles. Biotechniques. 2002;(Suppl):30–9. [PubMed] [Google Scholar]
- 19.Chada S, Mhashilkar AM, Ramesh R. Bystander activity of ad-mda7: mda-7 protein kills melanoma cells via an IL-20 receptor-dependent but STAT3-independent mechanism. Mol Ther. 2004;10:1085–95. doi: 10.1016/j.ymthe.2004.08.020. al. [DOI] [PubMed] [Google Scholar]
- 20.Chada S, Bocangel D, Ramesh R, et al. mda-7/IL24 kills pancreatic cancer cells by inhibition of the Wnt/PI3K signaling pathways: identification of IL-20 receptor-mediated bystander activity against pancreatic cancer. Mol Ther. 2005;11:724–33. doi: 10.1016/j.ymthe.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 21.Caudell EG, Mumm JB, Poindexter N, et al. The protein product of the tumor suppressor gene, melanoma differentiation-associated gene 7, exhibits immunostimulatory activity and is designated IL-24. J Immunol. 2002;168:6041–46. doi: 10.4049/jimmunol.168.12.6041. [DOI] [PubMed] [Google Scholar]
- 22.Ramesh R, Mhashilkar AM, Tanaka F, et al. Melanoma Differentiation-associated Gene 7/Interleukin (IL)-24 Is a Novel Ligand That Regulates Angiogenesis via the IL-22 Receptor. Cancer Res. 2003;63:5105–13. [PubMed] [Google Scholar]
- 23.Caudell EG, Mumm JB, Poindexter N, et al. The protein product of the tumor suppressor gene, melanoma differentiation-associated gene 7, exhibits immunostimulatory activity and is designated IL-24. J Immunol. 2002;168:6041–46. doi: 10.4049/jimmunol.168.12.6041. [DOI] [PubMed] [Google Scholar]
- 24.Gopalkrishnan R. INGN-241. Introgen. Curr Opin Investig Drugs. 2002;3:1773–7. [PubMed] [Google Scholar]
- 25.Chada S, Sutton RB, Eckmekcioglu S, Ellerhorst J, Mumm JB, Leitner W, et al. MDA-7/IL-24 is a unique cytokine-tumor suppressor in the IL-10 family. Int Immunopharmacol. 2004;4:649–67. doi: 10.1016/j.intimp.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Urano F, Bertolotti A, Ron D. IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci. 2000;21:3697–702. doi: 10.1242/jcs.113.21.3697. [DOI] [PubMed] [Google Scholar]
- 27.Barber GN. Host defense, viruses and apoptosis. Cell Death Differ. 2001;8:113–26. doi: 10.1038/sj.cdd.4400823. [DOI] [PubMed] [Google Scholar]
- 28.Jagus R, Joshi B, Barber GN. PKR, apoptosis and cancer. Int J Biochem Cell Biol. 1999;31:123–38. doi: 10.1016/s1357-2725(98)00136-8. [DOI] [PubMed] [Google Scholar]
- 29.Williams BR. Signal integration via PKR. Sci STKE. 2001;89:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data 1 Flow cytometric analysis of cell death in A549 cancer cells 72 hours after treatment with PBS, Ad-Luc (2500 vp/ or 24.5 moi), Ad-mda7 (2500 vp/ or 25.5 moi), Ad-ER-mda7 (2500 vp/ or 24.27 moi), Ad-Mito-mda7 (2500 vp/ or 22.1 moi), Ad-Nuc-mda7 (2500 vp/ or 26.59 moi), or Ad-Cyto-mda7 (2500 vp/ or 24 moi) with or without caspase-4 (Ac-LEVD-CHO, 10 μmol/L) or JNK chemical inhibitor (10 μmol/L). Experiments were performed in triplicate; data represent the mean (SD).
Supplemental data 2 Flow cytometric analysis of cell death in PKR+/+ and PKR-/- mouse embryo fibroblasts (MEFs) 72 hours after treatment with PBS, Ad-Luc (2500 vp/ or 24.5 moi), Ad-mda7 (2500 vp/ or 25.5 moi), Ad-ER-mda7 (2500 vp/ or 24.27 moi), Ad-Mito-mda7 (2500 vp/ or 22.1 moi), Ad-Nuc-mda7 (2500 vp/ or 26.59 moi) or Ad-Cyto-mda7 (2500 vp/ or 24 moi). Experiments were performed in triplicate; data represent the mean (SD).