

Abstract
Maximizing the potential of cancer immunotherapy requires model systems that closely recapitulate human disease to study T cell responses to tumor antigens and to test immunotherapeutic strategies. We have created a new system that is compatible with Cre-LoxP-regulatable mouse cancer models in which the SIY antigen is specifically over-expressed in tumors, mimicking clinically-relevant tumor-associated antigens. To demonstrate the utility of this system, we have characterized SIY-reactive T cells in the context of lung adenocarcinoma, revealing multiple levels of antigen-specific T cell tolerance that serve to limit an effective anti-tumor response. Thymic deletion reduced the number of SIY-reactive T cells present in the animals. When potentially self-reactive T cells in the periphery were activated, they were efficiently eliminated. Inhibition of apoptosis resulted in more persistent self-reactive T cells, but these cells became anergic to antigen stimulation. Finally, in the presence of tumors over-expressing SIY, SIY-specific T cells required a higher level of costimulation to achieve functional activation. This system represents a valuable tool in which to explore sources contributing to T cell tolerance of cancer and to test therapies aimed at overcoming this tolerance.
Keywords: Tumor antigen, T cells, mouse model, lung cancer, immune tolerance
INTRODUCTION
Mouse models have been a mainstay of cancer immunology research. The mouse has been used to probe the function of cells and molecules that influence the ability of tumor-reactive T cells to kill or to otherwise become functionally suppressed, or tolerized(1–3). These studies have largely relied on chemically-induced and spontaneous tumors in immunodeficient mice or on transplanted tumors. Such systems are limited because they fail to reproduce the complex interactions that exist among an emerging tumor, its microenvironment and the multiple elements of an intact immune system. More recently, genetically-engineered cancer models with tissue-specific expression of known antigens have been used to study T cells reactive to tumor antigens(4–9). Interestingly, these studies have led to divergent findings on T cell reactivity and tolerance to tumors, leading to the question of whether to attribute these differences to the immunobiology of specific tissues, the type of cancer under study, and/or the pattern in which antigens are expressed.
Tumor antigens are characterized as either tumor-specific (TSAs) or tumor-associated (TAAs). TSAs are derived from proteins to which the host immune system is naïve, such as mutant, germ cell-restricted, or viral proteins. TAAs, on the other hand, are derived from wild-type somatic proteins over-expressed or inappropriately expressed in tumors (e.g. CEA, ERBB2, hTERT, MUC1, P53). Experimental immune therapies for both TSAs and TAAs are being pursued as neither is clearly a more effective anti-cancer target(10–12). Targeting TSAs might appear preferable because this provides absolute selection for tumor cells; however, TSA expression is often limited to the individual tumors from which they were originally identified(13). Conversely, TAAs are commonly shared among tumor types and represent the majority of known tumor antigens(14). Thus, therapies developed to TAAs may be applicable to patients across cancer types.
Lung cancer is the leading cause of cancer death worldwide. This is due to its high incidence and the failure of existing therapies to effectively treat advanced disease. Thus, there is great need to develop effective novel therapeutic strategies for lung cancer. Relative to other tumor types (e.g. melanoma, ovarian and prostate cancer) little is known about the immune response to lung cancer or the potential for immunotherapy for this cancer type (15–19). To begin to explore the immune response to lung cancer antigens and to build systems for testing immunotherapeutic strategies to treat lung cancer in humans, we have created a mouse model in which the well-characterized T cell antigen SIYRYYGL(20) is over-expressed in autochthonous lung cancer. Based on analysis of T cells reactive to SIYRYYGL in this context, we have uncovered multiple levels of immune tolerance that limits an effective T cell response against a tumor-associated antigen (Fig. S1).
RESULTS
Generation of R26LSL-LSIY
To create a flexible system in which tumor-specific T cell responses could be carefully monitored during tumor progression, we used gene targeting to introduce a fusion of Firefly luciferase and SIYRYYGL (SIY) into the ubiquitously-expressed Rosa26 locus(21) (Fig. S2). Expression of luciferase-SIY fusion protein (termed LSIY) is controlled by a Lox-STOP-Lox, such that efficient expression can only be achieved following Cre-mediated excision of the STOP element(22, 23). The Rosa26-Lox-STOP-Lox-Luciferase-SIY allele is hereafter referred to as R26LSL-LSIY. We chose the synthetic peptide SIYRYYGL because many complementary reagents exist making it a tractable model antigen(20) (Tab. S1). SIY was fused to luciferase to facilitate detection and quantification of antigen expression in vitro and in vivo. Cre-inducible expression from a ubiquitous promoter makes R26LSL-LSIY compatible with various mouse cancer models that bear Cre-regulated tumor-predisposition genes(24).
Adenovirus-Cre (Ad-Cre) infection of R26LSL-LSIY/+ mouse embryonic fibroblasts (MEFs) yielded strong induction of luciferase activity (>103-fold over unrecombined MEFs), verifying Cre-dependent regulation. Interestingly, in the absence of Ad-Cre, we consistently observed slightly elevated luciferase activity (~3-fold) in R26LSL-LSIY/+ MEFs compared to controls (Fig. 1A). This result suggests that R26LSL-LSIY expresses LSIY at low levels.
Figure 1. R26LSL-LSIY exhibits Cre-dependent over-expression.

A. Luciferase activity in R26+/+ and R26LSL-LSIY/+ MEFs uninfected (n = 2 and 12, open bars) or infected with Ad-Cre (n = 3 and 12, filled bars). Each bar shows mean + s.e.m with Student’s t-tests: p < 0.0001 for both R26LSL-LSIY/+ -Cre versus +Cre and -Cre R26+/+ versus R26LSL-LSIY/+
B. Left panel: Number of CD8+1B2+ cells in tail tip bleeds of R26+/+;2C and R26LSL-LSIY/+;2C mice. Each bar shows mean + s.e.m. with p < 0.0001 by Student’s t-test. Right panel: 1B2, CD4, and CD8 staining of thymocytes from representative R26+/+;2C (top) and R26LSL-LSIY/+;2C (bottom) mice.
C. Luciferase activity in tissue lysates from whole thymus, lung, liver, and spleen from R26+/+ (n = 4, open bars), R26LSL-LSIY/+ (n = 5, shaded gray bars), and R261Lox-LSIY/+ (n = 2, filled black bars) mice. Each bar shows mean + s.e.m. with *p < 0.05, **p < 0.005, ***p ≪ 0.001 by Student’s t-test.
D. Representative CFSE dilution in CD8+1B2+cells recovered from mesenteric lymph nodes and spleens of R26+/+ (filled histograms) and R26LSL-LSIY/+ (open histograms) recipients of naïve 2C cells one, two, and three days after i.v. transfer.
2C mice express a dominant TCR (recognized by the clonotypic antibody 1B2) that reacts with SIY bound by H-2Kb haplotype MHC class I(20, 25–27). To test for LSIY expression in vivo, the fate of potentially self-reactive 2C T cells was assessed in R26LSL-LSIY;2C mice. CD8+1B2+ cells were severely reduced in peripheral blood of these mice relative to R26+/+;2C controls (Fig. 1B). Furthermore, the proportion of thymic 1B2+ cells, in particular the CD8+CD4− fraction, was diminished in R26LSL-LSIY/+;2C mice. These data imply negative-selection of SIY-reactive cells during T cell development, or central tolerance, in R26LSL-LSIY mice.
R26LSL-LSIY exhibits Cre-dependent over-expression
Central tolerance could occur due to thymus-restricted or ubiquitous, somatic antigen expression. Various tissues were surveyed to distinguish between these possibilities. Luciferase activity was 3–30-fold higher in both thymic and extra-thymic tissues of R26LSL-LSIY/+ mice relative to background levels in controls (Fig. 1C). By contrast, in R261Lox-LSIY/+ mice, in which the STOP element had been deleted by the Meox2-Cre transgene(28), a 104–105-fold increase in reporter activity was detected in all tissues examined (Fig. S2A; Fig. 1C). These results show that R26LSL-LSIY exhibits low-level, ubiquitous expression of LSIY.
To further characterize the consequences of low-level expression from R26LSL-LSIY, we assessed 2C cell reactivity to R26LSL-LSIY/+ cells. Naïve 2C cells exhibited dose-dependent activation (measured by CD25 surface-expression) and proliferation upon co-culture with R26LSL-LSIY/+, but not with R26+/+, splenocytes (Fig. S3A, B). Additionally, transfer of CFSE-labeled 2C cells into R26LSL-LSIY/+ mice led to progressive dilution of CFSE and upregulation of CD44, a cell-adhesion molecule associated with T cell activation (Fig. 1D; Fig. S3C). These data establish that low-level LSIY expression from R26LSL-LSIY is sufficient to stimulate 2C cells in vitro and in vivo.
Incomplete central tolerance in R26LSL-LSIY mice
Because we detected CD8+ T cells bearing 2C-TCR in blood and peripheral lymphoid organs of R26LSL-LSIY/+;2C mice, central tolerance to SIY must be incomplete (Fig. 1B; data-not-shown). To eliminate the possibility of an artifact of 2C transgenic mice, we infected R26LSL-LSIY/+ and control mice, that have normal TCR repertoires, with Influenza virus engineered to express SIY (WSN-SIY)(29, 30). Seven days after pulmonary WSN-SIY infection of R26+/+ mice, robust induction of SIY-specific CD8+ T cells in lungs and lymphoid tissues was evident by staining with SIY-loaded H-2Kb DimerX reagent (Fig. 2A; data-not-shown). We also detected SIY-reactive T cells in lungs of WSN-SIY-infected R26LSL-LSIY/+ mice, albeit at significantly lower levels compared to controls (Fig. 2A). (SIY-reactive T cells were not detectable in uninfected mice (data-not-shown)).
Figure 2. Central tolerance to SIY is incomplete and functional SIY-reactive T cells can be transiently activated in the periphery.

A. Left panel: Representative plots of CD8 and BD Dimer X SIY/H-2Kb:Ig stained whole lung cell suspensions from R26+/+ and R26LSL-LSIY/+ mice seven days after intranasal WSN-SIY Influenza A infection. Right panel: SIY/H-2Kb:Ig+ fraction of total CD8+ cells from WSN-SIY-infected lungs of R26+/+ (n = 4, open bar) and R26LSL-LSIY/+ (n = 4, filled bar) mice. Each bar shows mean + s.e.m. with p < 0.005 by Student’s t-test.
B. Representative plots of single cell suspensions from lungs (top panels) and mediastinal lymph nodes (bottom panels) of R26+/+ and R26LSL-LSIY/+ mice 7 days post-WSN-SIY infection and uninfected controls. Cells were cultured and stimulated in vitro with 1ug/mL of SIY peptide and an IFN-γ capture assay was performed. CD8+-gated cells are stained for CD107a and secreted IFN-γ.
C. SIY-specific cytotoxicity in mediastinal lymph nodes of R26+/+ and R26LSL-LSIY/+ mice 7 days after pulmonary WSN-SIY vaccination. Mean +/− s.e.m. specific cytolysis are shown for each genotypes.
D. Left panel: BD DimerX SIY/H-2Kb:Ig double-stained splenocytes from mice boosted with DC2.4.LSIY vaccine 5 days prior to analysis. R26+/+ (left) and R26LSL-LSIY/+ (middle) mice were infected intranasally with WSN-SIY >30 days before boost or never infected (right). Right panel: Quantification of percentage of double-positive SIY/H-2Kb:Ig cells among splenocytes in R26+/+ (n = 4, open bar) and R26LSL-LSIY/+ (n = 3, filled bar) mice. Each bar shows mean + s.e.m. with p < 0.05 by Student’s t-test.
Because SIY-reactive T cells in R26LSL-LSIY mice could be self-reactive, we probed the activity of these cells induced by WSN-SIY and found they were functional. When single-cell suspensions from lungs of WSN-SIY-infected mice were stimulated in vitro with SIY, a comparable fraction of cells secreted IFN-γ in both R26+/+ and R26LSL-LSIY/+ cultures (Fig. 2B). Furthermore, by assaying in vivo cytotoxicity (Fig. S4) in WSN-SIY-infected mice, we found SIY-specific cytolysis in proportion to SIY-reactive T cells induced in R26LSL-LSIY/+ mice (Fig. 2C). As we observed long-term health without autoimmunity in WSN-SIY-infected R26LSL-LSIY/+ mice, we investigated SIY-reactive T cell fate. Upon boosting with DC2.4.LSIY cells (a dendritic cell line expressing high levels of costimulatory molecules B7-1 and B7-2(31), modified to additionally express LSIY), we did not detect surviving SIY-reactive T cells in WSN-SIY-infected R26LSL-LSIY/+ mice. In contrast, memory cells were evident in WSN-SIY-infected wild-type mice (Fig. 2D). Together, these results demonstrate that central tolerance to SIY is incomplete in R26LSL-LSIY mice and a transient, but functional, T cell response to SIY can be initiated.
Antigen over-expressing tumors progress normally
R26LSL-LSIY is a novel system in which over-expression of a self-antigen is induced, representing clinically-relevant tumor-associated antigens in human cancer(13). Given the importance of TAAs as targets for cancer immunotherapy, R26LSL-LSIY in Cre-inducible cancer models provides a powerful tool to study T cell-tumor interactions. To explore the utility of this system, we used a model of human lung adenocarcinoma in which oncogenic K-ras is expressed from its endogenous locus after intranasal Ad-Cre administration(22, 23). In this K-rasLSL-G12D model, focal activation of oncogenic K-ras leads to epithelial hyperplasia, which progresses to adenoma and adenocarcinoma over a defined time course resembling human non-small cell lung cancer (NSCLC).
We generated K-rasLSL-G12D/+;R26LSL-LSIY/+ mice and induced lung cancer in these animals and controls using Ad-Cre. K-rasLSL-G12D/+;R26LSL-LSIY/+ mice developed lung tumors histologically indistinguishable from their K-rasLSL-G12D/+;R26+/+ littermates when examined at various times after Ad-Cre (Fig. 3A; data-not-shown). Tumors dissected from K-rasLSL-G12D/+;R26LSL-LSIY/+ mice consistently had 103–104-fold higher luciferase activity than those from K-rasLSL-G12D/+;R26+/+ mice, suggesting that LSIY expression was maintained in established tumors (Fig. 3B). Luciferase activity was also detectable in large tumors several months after tumor initiation via in vivo bioluminescence imaging (Fig. 3C).
Figure 3. Antigen over-expressing tumors progress normally.

A. Representative H&E-staining lung tumor histology from K-rasLSL-G12D/+;R26+/+ (left) and K-rasLSL-G12D/+;R26LSL-LSIY/+ (right) mice 12 weeks after intranasal Ad-Cre infection. Scale bars indicate 100μm.
B. Luciferase activity of representative individual lung tumors dissected from K-rasLSL-G12D/+;R26+/+ (blue bars) and K-rasLSL-G12D/+;R26LSL-LSIY/+ (red bars) animals 16 weeks after intranasal Ad-Cre infection.
C. In vivo bioluminescence of a K-rasLSL-G12D/+;R26LSL-LSIY/+ mouse > 6 months after intranasal Ad-Cre infection (inset: gross image of lungs after imaging).
D. Quantification of lung tumor burden in K-rasLSL-G12D/+;R26+/+ (n = 3, blue bars) and K-rasLSL-G12D/+;R26LSL-LSIY/+ (n = 3, red bars) animals 12 weeks after intranasal Ad-Cre infection. Each bar shows mean + s.e.m. with p = 0.89 and 0.83 for tumor to lung ratio and average tumor area by Student’s t-test.
Histological examination of tumors from K-rasLSL-G12D/+;R26LSL-LSIY/+ and K-rasLSL-G12D/+;R26+/+ mice at 8, 12, and 16 weeks post-Ad-Cre revealed no evidence of an active anti-tumor T cell response in either genotype (Fig. 3A; data-not-shown). Furthermore, flow cytometry of lungs and lymphoid tissues of K-rasLSL-G12D/+;R26LSL-LSIY/+ mice failed to detect SIY-reactive T cells either early after tumor initiation (within 12 days) or 3–4 months later during tumor progression (data-not-shown). Because this does not eliminate the possibility of an immune response below detection or at an unexamined time, we explored the effects that such a response might have had. Specifically, we compared tumor burdens and sizes between K-rasLSL-G12D/+;R26LSL-LSIY/+ and KrasLSL-G12D/+;R26+/+ littermates, but no significant differences were observed (Fig. 3D). These data suggest that although SIY-reactive T cells are elicited in WSN-SIY-infected R26LSL-LSIY/+ mice (Fig. 2), a response is not generated against SIY-over-expressing lung tumors.
Tumors maintain antigen presentation
Tumor-specific down-regulation of antigen presentation has been proposed as a mechanism to evade recognition by T cells(32–34). To measure MHC class I in tumors, lung tumor cell lines were derived from Ad-Cre-infected K-rasLSL-G12D/+;p53fl/fl;R26LSL-LSIY/+ mice and R26+/+ controls (Fig. S5A, B). By flow cytometry, we detected comparably low levels of H-2Kb, which was induced 40-fold by IFN-γ treatment in all lines (Fig. S5C). This indicates that antigen presentation is functional in tumors. Furthermore, we induced and examined lung tumors in mice on a 2C transgenic background. Tumors in K-rasLSL-G12D/+;R26LSL-LSIY/+;2C mice were highly infiltrated by lymphocytes in contrast to tumors induced in K-rasLSL-G12D/+;R26+/+;2C controls, demonstrating that antigen presentation by SIY-over-expressing tumors is sufficient to recruit 2C cells in vivo (Fig. S5D). These observations imply that loss of SIY presentation is unlikely to account for the unproductive immune response to SIY-over-expressing tumors.
Naïve cells recognize but do not respond effectively to lung tumors
Our data indicate that SIY-reactive T cells are present in K-rasLSL-G12D/+; R26LSL-LSIY/+ animals, but fail to react to SIY-over-expressing lung tumors. There are several non-exclusive explanations for this observation. For example, neither tumor initiation nor progression may be a sufficient stimulus to induce T cell responses to tumor-associated antigens(34). Alternatively, peripheral tolerance to self-antigen may inhibit responses to over-expressed antigens in tumors(35). Finally, tumors over-expressing antigen could be actively suppressing reactive T cells(2, 36). Because the low numbers of endogenous SIY-specific T cells in R26LSL-LSIY mice impedes analyses, we investigated these possible mechanisms by transferring donor 2C cells into tumor-bearing and control animals.
Naïve 2C cells became activated and proliferated in tumor-bearing K-rasLSL-G12D/+;R26LSL-LSIY/+ animals, similar to our observation in tumor-free R26LSL-LSIY/+ mice (Fig. 4A, B; Fig. S3C; Fig. S1F). Only in tumor-bearing K-rasLSL-G12D/+;R26LSL-LSIY/+ mice, however, were 2C cells enriched in lung-draining mediastinal lymph nodes relative to non-draining mesenteric lymph nodes (Fig. 4C). 2C cells are likely retained in mediastinal lymph nodes where SIY antigen presentation is increased due to SIY-over-expressing lung tumors.
Figure 4. Naïve cells recognize, but do not response effectively to lung tumors.
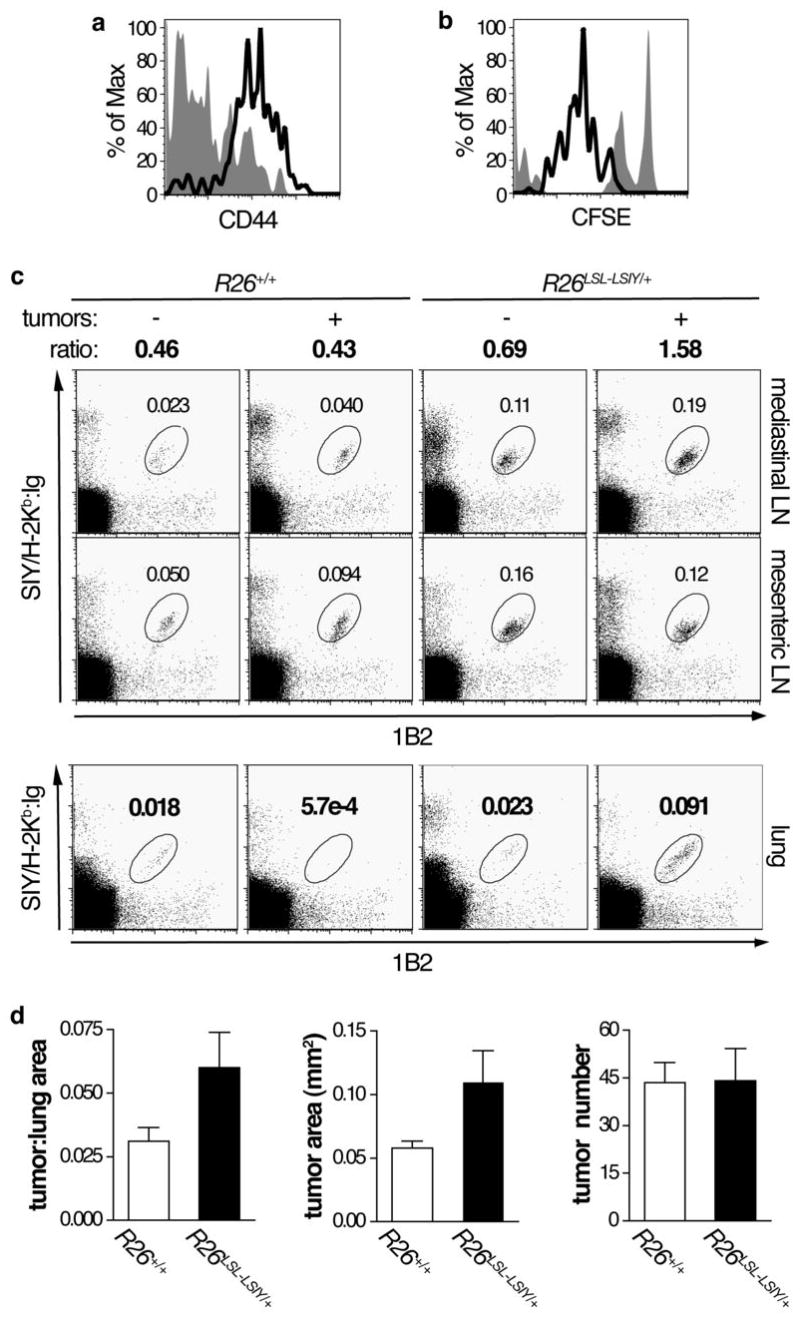
A. Representative plot of CD44 staining in CD8+1B2+ cells 5 days after naïve 2C cell transfer into K-rasLSL-G12D/+;R26+/+ (filled histogram) and K-rasLSL-G12D/+;R26LSL-LSIY/+ (open histogram) animals.
B. Representative plot of CFSE dilution in CD8+1B2+ cells 3 days after naïve 2C cell transfer into K-rasLSL-G12D/+;R26+/+ (filled histogram) and K-rasLSL-G12D/+;R26LSL-LSIY/+ (open histogram) animals.
C. Top panel: Representative plots of 1B2 and BD Dimer X SIY/H-2Kb:Ig stained cells recovered from lymph nodes of tumor-free and tumor-bearing R26+/+ and R26LSL-LSIY/+ mice. The ratio of 1B2+SIY/H-2Kb:Ig+ cells in mediastinal to mesenteric lymph nodes is indicated. Double staining with 1B2 and Dimer X to detect 2C cells enhances the signal to noise ratio. Bottom panel: Representative plots of whole lung cell suspensions from the same tumor-free and tumor-bearing R26+/+ and R26LSL-LSIY/+ mice stained with 1B2 and BD Dimer X SIY/H-2Kb:Ig.
D. Tumor to lung area ratio in K-rasLSL-G12D/+;R26+/+ (n = 9, open bars) and K-rasLSL-G12D/+;R26LSL-LSIY/+ (n = 8, filled bars) animals 16 weeks after intranasal Ad-Cre infection, shown in left graft. Animals received naïve 2C cell i.v. transfer at either 4 weeks or 12 weeks after intranasal Ad-Cre. Each bar shows mean + s.e.m. with p = 0.060 by Student’s t-test for tumor burden. The average area of tumors (p = 0.046 by Student’s t-test) in the same animals are shown in the middle graph and the average number of tumors per animal (p = 0.096 by Student’s t-test) is shown in the right graph.
Despite accumulation in high SIY areas, 2C cells did not infiltrate tumors, although 2C cells were detectable in lungs by flow cytometry (Fig. 4C; data-not-shown). Consistent with the absence of tumor-infiltrating lymphocytes, K-rasLSL-G12D/+;R26LSL-LSIY/+ mice displayed no decrease in tumor burden, size, or number relative to K-rasLSL-G12D/+;R26+/+ mice analyzed 4 weeks after 2C transfer (Fig. 4D). These data demonstrate that 2C cells do not hinder tumor growth. Moreover, 2C cells were not detectable in any mice at this time-point whether they bore tumors or not (data-not-shown). These data indicate that 2C cells activated in R26LSL-LSIY mice do not form memory cells, as was observed in WSN-SIY-infected R26LSL-LSIY mice, potentially due to peripheral tolerance where persistent weak TCR stimulation leads to apoptosis(35). Therefore, in the presence of self-antigen, SIY-reactive T cells become activated and divide, but do not significantly kill tumor cells or persist.
SIY-reactive T cells are not cytotoxic in R26LSL-LSIY mice
Failure of T cells to kill SIY-over-expressing tumors could be due to a general defect in SIY-specific cytotoxicity or to a direct inhibition of T cells by tumors. To assess 2C cell functionality, we assayed in vivo cytotoxicity examining the ability of 2C cells to kill non-malignant target cells (Fig. S4; Fig. 5A). As shown in Figure 5C panel 1, DC2.4.LSIY vaccination stimulated 2C cells to become highly cytotoxic in wild-type animals, a positive control for SIY-specific cytotoxicity. As a negative control, naïve 2C cells transferred into R26+/+ without vaccination do not become activated and, thus, only displayed low SIY-specific cytolysis.
Figure 5. A higher threshold for costimulation is required in the presence of tumors over-expressing self-antigen.
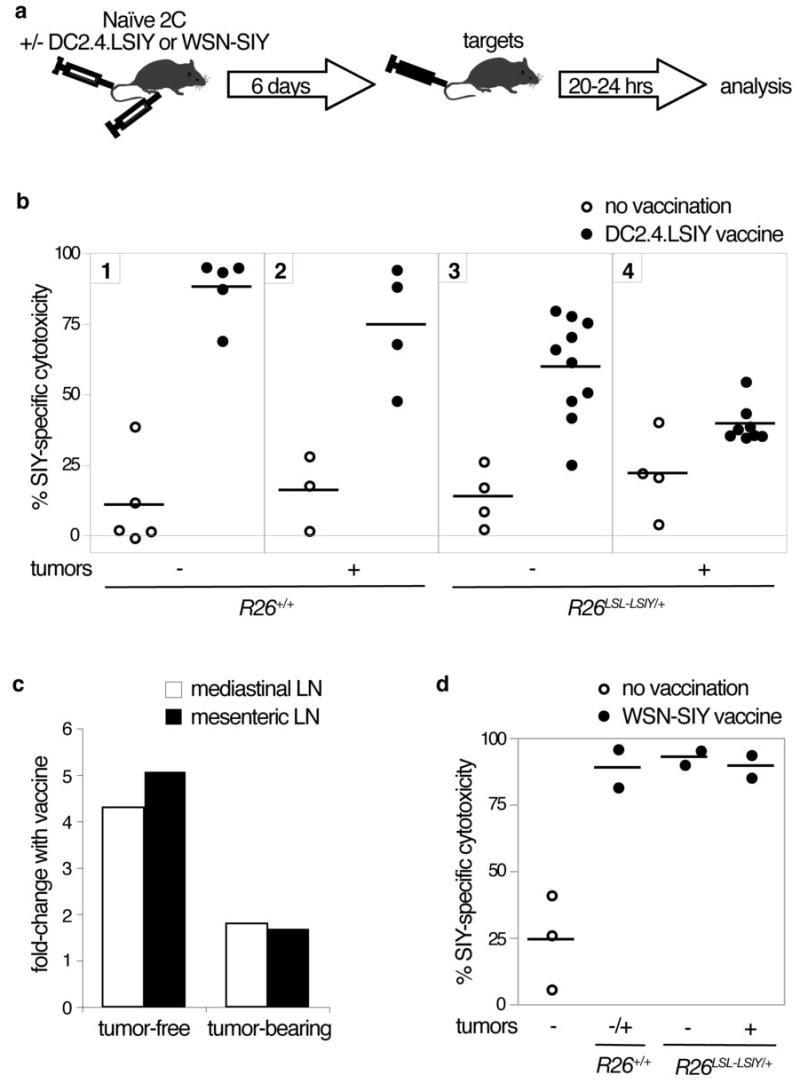
A. Schematic of experimental set-up for the in vivo cytotoxicity assay. Naïve 2C cells are intravenously injected into recipients with or without intraperitoneal DC2.4.LSIY or intranasal WSN-SIY vaccination. Six days later, differentially-labeled R261LoxLSIY/+ target and R26+/+ control cells are intravenously injected and animals are analyzed 20–24 hours later.
B. SIY-specific cytotoxicity in mediastinal lymph nodes draining the lung seven days after naïve 2C cell transfer into recipients with (closed circles) or without (open circles) DC2.4.LSIY vaccination.
C. Fold change in SIY-specific cytotoxicity in mediastinal (open bars) and mesenteric lymph nodes (filled bars) of tumor-free and tumor-bearing R26LSL-LSIY/+ mice when vaccinated with DC2.4.LSIY over unvaccinated mice.
D. SIY-specific cytotoxicity in mediastinal lymph nodes draining the lung seven days after naïve 2C cell transfer into recipients with (closed circles) or without (open circles) WSN-SIY vaccination
The assay was applied to tumor-free R26LSL-LSIY/+ mice. Despite inducing activation markers and proliferating, 2C cells exhibited no more cytotoxicity in these animals than 2C cells in R26+/+ mice, indicating that self-antigen-induced activation fails to yield full effector function (Fig. 4A, B; Fig. 5B). In tumor-bearing K-rasLSL-G12D/+;R26LSL-LSIY/+ animals, SIY-specific cytotoxicity was similarly defective (Fig. 5B). These data demonstrate generalized impairment in T cell cytotoxicity to antigenic targets, rather than specific inhibition of tumoricidal activity in R26LSL-LSIY mice.
TAA-over-expressing tumors suppress T cell cytotoxicity
Peptide presentation without costimulation often yields unproductive activation and T cell deletion analogous our observations(37). To determine whether DC2.4.LSIY could provide the necessary costimulation to functionally activate 2C cells in animals in which SIY is a self-antigen, naïve 2C cells were transferred into R26LSL-LSIY/+ animals concurrently with DC2.4.LSIY vaccination (Fig. 5A). With this treatment, 2C cells exhibited high levels of SIY-specific cytotoxicity in tumor-free animals, despite low-level ubiquitous SIY (Fig. 5B, C). This result demonstrates that self-antigen presented with ample costimulation can yield functional activation of potentially self-reactive T cells.
When 2C cells were transferred into tumor-bearing K-rasLSL-G12D/+;R26LSL-LSIY/+ mice with DC2.4.LSIY vaccination, however, only marginal induction of SIY-specific cytotoxicity was observed (Fig. 5B, C). Thus, although costimulation enabled 2C cells to become highly cytotoxic to self-antigen in tumor-free animals, it failed to similarly induce SIY-specific cytotoxicity in the presence of tumors over-expressing SIY. Importantly, 2C transfer and DC2.4.LSIY vaccination of tumor-bearing K-rasLSL-G12D/+;R26+/+ mice resulted in robust SIY-specific cytotoxicity (Fig. 5B). This result strongly supports a mechanism by which tumors induce suppression of immune responses towards antigens over-expressed in those tumors. Furthermore, we observed comparably low cytotoxicity in mediastinal lymph nodes draining tumor-bearing lungs and mesenteric lymph nodes not draining tumors suggesting that tumor-induced inhibition of TAA-specific T cells occurs systemically (Fig. 5C; data-not-shown).
Functional activation of TAA-reactive T cells requires stronger costimulation
The balance of immune stimulatory and inhibitory factors determines whether immune reactivity or tolerance is generated. Therefore, we reasoned that stronger immunological stimuli might induce SIY-specific cytotoxicity in tumor-bearing R26LSL-LSIY/+ mice. DC2.4.LSIY vaccination failed to expand endogenous SIY-reactive T cells to detectable levels in R26LSL-LSIY/+ mice (data-not-shown) whereas WSN-SIY was more potent (Fig. 2). Thus, we used WSN-SIY instead of DC2.4.LSIY to vaccinate naïve 2C cell recipients (Fig. 5A). In contrast to the case with DC2.4.LSIY vaccination, WSN-SIY vaccination caused 2C cells to exhibit high SIY-specific cytotoxicity in both tumor-free and tumor-bearing R26LSL-LSIY/+ mice (Fig. 5D). This result indicates that the threshold costimulation required to induce SIY-specific cytotoxicity is increased in the presence of SIY-over-expressing tumors. Despite being highly cytotoxic, however, 2C cells did not seem to exert a significant antitumor effect in WSN-SIY-infected K-rasLSL-G12D/+;R26LSL-SIY/+ mice, although the number of animals tested was small (data not shown). This phenomenon is likely a result of the relatively brief lifespan of 2C cells in R26LSL-LSIY mice.
SIY-reactive T cells blocked from death succumb to anergy
We have shown that SIY-reactive T cells can be functionally activated in tumor-bearing R26LSL-LSIY/+ mice, but as these cells are short-lived, T cell death remains an obstacle to effective anti-tumor immunity. To overcome this barrier, we over-expressed anti-apoptotic Bcl2 by infecting SIY-stimulated 2C cells with a retrovirus carrying Bcl2 and EGFP (MIG-Bcl2). By observing enrichment of EGFP+ cells in culture, we confirmed that Bcl2-over-expressing 2C cells had enhanced survival over uninfected cells when deprived of IL-2(38) (Fig. 6A). When MIG-Bcl2-infected 2C cells were transferred into R26LSL-LSIY/+ and R26+/+ mice, Bcl2-over-expressing cells again became enriched over uninfected cells in both genotypes (Fig. 6B). Despite this enrichment, 2C cells in R26LSL-LSIY/+ mice was still significantly reduced and decreased over time compared to 2C cells in R26+/+ recipients, suggesting that induction of both intrinsic and extrinsic apoptotic pathways account for loss of 2C cells.
Figure 6. Inhibition of apoptosis results in T cell anergy in the presence of self-antigen.
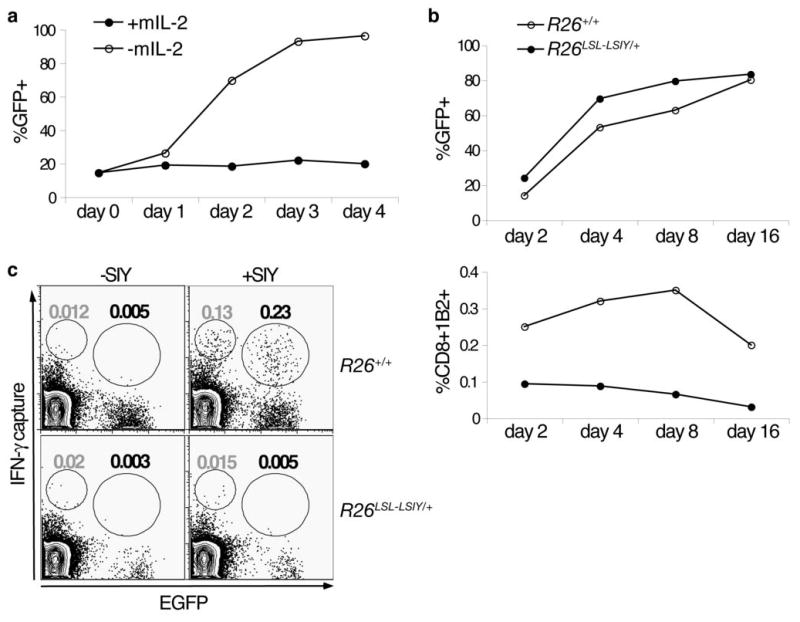
A. In vitro activated 2C cells infected with MIG-Bcl2 propagated in vitro in the presence (filled circles) or absence (open circles) of survival cytokine IL-2. CD8+1B2+ cells were analyzed by flow cytometry for the percentage of EGFP+ cells each day for 4 days after withdrawal of IL-2.
B. In vitro activated 2C cells infected with MIG-Bcl2 intravenously transferred into either R26+/+ (open circles) or R26LSL-LSIY/+ (filled circles) recipient mice. Spleens were harvested and analyzed by flow cytometry at 2, 4, 8, and 16 days after transfer. EGFP+ cells as a percentage of total CD8+1B2+ (top panel) and CD8+1B2+ cells as a percentage of total splenocytes (bottom panel) are shown.
C. Representative plots of CD8+-gated splenocytes from R26+/+ and R26LSL-LSIY/+ mice 8 days after receiving MIG-Bcl2 infected activated 2C cells. Cells were stimulated in vitro with 1ug/mL of SIY peptide (right panels) or unstimulated (left panels) and an IFN-γ capture assay was performed. Population gates represent percentage of EGFP− IFN-γsecreting (gray gate value) and EGFP+ IFN-γ-secreting (black gate value) CD8+ splenocytes.
Next, we examined the function of persisting Bcl2-over-expressing 2C cells. Upon in vitro SIY stimulation of splenocytes recovered from 2C recipients, we observed that 2C cells from R26LSL-LSIY/+ mice had impaired IFN-γ secretion compared to controls (Fig. 6C). Therefore, although Bcl2 over-expression can partially protect 2C cells from apoptosis in the presence of self-antigen, additional mechanisms act to induce anergy in these cells.
DISCUSSION
We have described a novel system that allows inducible expression of a defined antigen in mice to mimic tumor-associated antigens (TAAs) in human cancer. We have characterized T cells reactive to the SIY TAA in lung adenocarcinoma and uncovered multiple levels of immune tolerance that limit an effective response against TAAs (model depicted in Fig. S1). Due to low-level antigen expression, SIY-reactive T cells were subjected to central tolerance, eliminating the majority of potentially tumor- and self-reactive T cells during development. In the periphery, SIY-reactive cells that escaped thymic selection died soon after activation, likely a mechanism to limit auto-reactivity. Self-reactive T cells that were transiently blocked from apoptosis became anergic. Finally, in mice bearing autochthonous SIY-over-expressing lung tumors, strong immunological stimuli were required to activate transferred 2C cells to become highly cytotoxic toward targets. This is noteworthy because a weaker vaccine was able to impart cytotoxic activity on self-reactive T cells only in the absence of SIY-over-expressing tumors. Thus, in the context of a tumor-associated antigen, reactive T cells must overcome developmental negative selection, peripheral self-tolerance, and tumor-induced inhibition as barriers to effective anti-tumor immunity.
Our data suggest that neither prophylactic nor therapeutic vaccines to TAAs will be effective in preventing or treating lung cancer. Protection provided by prophylactic vaccination against an antigen relies on formation and maintenance of memory cells that can respond efficiently to tumors bearing that antigen(39). In R26LSL-LSIY animals, where SIY is a self-antigen, we have demonstrated that SIY-reactive T cells do not persist and cannot result in immunological memory or protection to SIY (Fig. 2D; data-not-shown). In contrast, when targeting antigens to which the host is not tolerant, prophylactic vaccines protect against even large numbers of potentially tumorigenic cells(9).
Therapeutic vaccines boost immune responses to tumor antigens in patients with established cancers(39). To be effective, this requires both the presence of antigen-specific T cells and the ability to properly activate these cells. Based on our model, both elements are hampered in the context of TAAs. We have shown that central tolerance results in significant diminution of SIY TAA-specific T cells (Fig. 1C, D). Additionally, we have demonstrated that DC2.4.LSIY vaccination yields only marginal induction of SIY-specific cytotoxicity in mice bearing tumors over-expressing SIY (Fig. 5). Furthermore, although WSN-SIY can endow 2C cells with high cytotoxicity, activated 2C cells are anergized and die in the presence of self-antigen (Fig. 2D; Fig. 6). Our data is consistent with observations from human trials in which therapeutic vaccines have resulted in only rare cases of objective clinical response(40). Importantly, therapeutic vaccines may still be effective in combination with other therapeutic strategies(41).
Immune ignorance of tumors, immune avoidance by tumors, and active suppression of anti-tumor immunity are the prevailing explanations for defective T cell responses in cancer(1, 2, 34). Loss of antigen expression or presentation is often described as a means of immune avoidance(32, 33). We have demonstrated sustained LSIY expression in lung cancer, even at advanced stages (Fig. 3B, C). Moreover, we have shown that antigen presentation was not different between R26LSL-LSIY/+ lung tumor cell lines and controls (Fig. S5). Furthermore, we have demonstrated the inability of 2C cells to efficiently kill non-malignant cells expressing high SIY in SIY-over-expressing tumor-bearing hosts (Fig. 5). These data exclude immune avoidance by tumors as a major contributor to deficient anti-tumor T cell responses.
We cannot exclude the possibility of immune ignorance playing a role in early stages of tumorigenesis, but our data argue for active tumor-induced immune suppression. Inefficient 2C cell killing in mice bearing SIY-over-expressing tumors was observed even with coincident antigen presentation and costimulation provided by DC2.4.LSIY (Fig. 5). This treatment is normally immune-activating and, moreover, is capable of overcoming self-tolerance in tumor-free R26LSL-LSIY/+ mice. This implies that even if tumors are naturally ignored in our model, tumor-induced T cell suppression can dominate over immune-stimulatory signals.
There are many mechanisms by which SIY-reactive T cells may be suppressed in K-rasLSL-G12D/+;R26LSL-LSIY/+ mice(2, 36). Suppression can occur by contact with tumors or by intermediaries, but since we observe systemic antigen-specific T cell suppression, direct contact between T cells and tumors is unlikely to be the dominant mechanism. Tumor suppression by intermediaries may involve cellular and/or soluble inhibitors of T cell cytotoxicity. These factors may be present normally to limit self-reactivity, but are increased in animals with self-antigen over-expressing tumors and thus induce greater suppression of antigen-reactive T cells. Consistent with this idea, we have shown that high levels of costimulation provided by WSN-SIY was required to effectively induce cytotoxicity in 2C cells (Fig. 5).
We immunophenotyped animals to screen for potential cellular mediators of T cell suppression. Two putative immunosuppressive populations, Gr1+CD11b+ myeloid suppressor cells and CD4+CD25+FoxP3+ T regulatory cells, were reproducibly expanded in lungs and lymphoid tissues of tumor-bearing mice relative to tumor-free mice (Fig. S6). Interestingly, both immune cell types are overrepresented in NSCLC patients and appear to mediate systemic immune suppression(42–44). Further studies are needed to determine if either of these cell types are playing a causal role in tumor-induced T cell suppression in our model.
Melanoma, ovarian and prostate cancers are more often treated with immunotherapy in the clinic than other cancers. This may reflect an inherent susceptibility of these cancers to immune attack or simply our limited experience in using the immune system to treat other cancers. Our observations of tolerance in the presence of tumors are similar to those made in TRAMP mice in which prostate-specific expression of SV40 TAg leads to spontaneous prostate cancer(4, 5). In contrast to our model, however, priming with a DC vaccine yielded more effective T cells that significantly reduced tumor burden in TRAMP mice(4). Interestingly, T cell tolerance was not observed in a mouse model of insulinoma also driven by SV40 TAg(8). To learn more about tumor-immune interactions, it will be important to compare immune responses among different cancer types and even among tumors bearing different oncogenic alterations as particular pro-oncogenic events have been described to specifically modulate immune responses(36). Numerous Cre-regulated oncogenes and tumor suppressor genes have been generated in the context of human cancer models(24). Because R26LSL-LSIY is expressed ubiquitously and independent of oncogenic events, our model tumor antigen can be studied in several cancer models to gain greater understanding of context-dependent effects on anti-tumor immunity. Data derived from these studies will help define the best strategies to successfully stimulate the immune system to recognize and eliminate cancers of different origins.
METHODS
Mice
2C mice were provided by J. Chen, Trp53flox mice by A. Berns, and Meox-Cre and Rag2− mice were purchased from Jackson Laboratories. K-rasLSL-G12D mice were generated in our laboratory(22, 23). R26LSL-LSIY mice and controls used were either pure 129S4/SvJae or enriched on C57BL/6 3–13 generations. Donor 2C cells for transfer experiments were from pure C57BL/6 2C;Rag2−/− mice and recipients were always C57BL/6 enriched. To induce tumors, mice were infected with Adenovirus-Cre intra-nasally as previously described(22). WSN-SIY was provided by J. Chen and infections were done by intra-nasal instillation similar to Adenovirus-Cre. For tumor studies, lung histology was prepared and analyzed by Bioquant Image Analysis software as previously described(45). Animal studies were approved by MIT’s Committee for Animal Care and conducted in compliance with Animal Welfare Act Regulations and other federal statutes relating to animals and experiments involving animals and adheres to the principles set forth in the 1996 National Research Council Guide for Care and Use of Laboratory Animals (institutional animal welfare assurance number, A-3125-01).
Cells
MEFs were derived from e13.5 embryos and grown as previously described(23). KP/KPLSIY cell lines were derived from K-rasLSL-G12D/+;p53flox/flox mice(45) 16 weeks post-Ad-Cre. Individual tumors were plucked from lungs, minced, trypsinized and cultured in MEF growing media until lines were established. DC2.4.LSIY cells were generated by infection of DC2.4 cells(31) with a lentivirus bearing Luciferase-SIY and selection of a single positive clone. In vitro activated 2C cells were made by stimulating 2C splenocytes/lymphocytes suspensions with 1μg/mL SIY in DMEM-10 full medium overnight, then culturing with 10ng/mL mIL-2 (R&D Systems cat#402-ML) for 6 days. Bcl2 over-expression was achieved by spin-infecting in vitro activated 2C cells twice (at days 3 and 4 of activation) with a pMSCV-IRES-GFP retrovirus carrying Bcl2, prepared as described(46).
Luciferase detection
Cells were lysed with Passive Lysis Buffer (Promega cat#E1941) and assayed with Luciferase Assay Reagent (Promega cat#E1501) according to manufacturer’s instructions. Tissues and tumors were mechanically disrupted before lysis and tissue debris was pelleted before assay. In vivo bioluminescence imaging was performed on a NightOWLII LB983 (Berthold Technologies). Mice were shaved and intra-peritoneally injected with Beetle Luciferin (Promega cat#E1602) within 20 minutes prior to imaging.
Cell transfer and DC vaccination
Single-cell suspensions from lymph nodes and spleens of 2C;Rag2−/−, R261Lox-LSIY/+ and R26+/+ mice were used for 2C transfer, in vivo cytotoxicity assay’s target and control cells, respectively. Cells were counted by hemocytometer, resuspended in RPMI and transferred into recipients via tail vein. For CFSE-labeling, cells were resuspended at 4–6×106 cells/mL RPMI with 5% fetal calf serum containing 5μM (or 1μM for control cells in in vivo cytotoxicity assay) CFSE (Molecular Probes cat# C1157) for 10 minutes at 37°C. Cells were then washed twice in RPMI with 5% fetal calf serum, followed by washes in serum-free RPMI before intravenous transfer into recipients. 3×106 cells were injected for naïve 2C transfer, 12×106 cells for in vitro activated 2C, and 6×106 cells each of CFSE-labeled target and control in vivo cytotoxicity cells. DC2.4.LSIY cells were washed and resuspended in RPMI for intra-peritoneal vaccination with 5×105 cells/mouse.
Cell isolation
Single-cell suspensions from lymph nodes, spleen and thymus were generated by mechanical disruption. For lung preparations, tissue samples were minced and digested at 37°C for 30 minutes in 125U/mL of collagenase-typeI (Gibco) in phosphate-buffered saline before mechanical disruption and passage through a 70μm-pore filter (BD Falcon). Peripheral blood was collected by tail tip bleeds into 40μL of 50mM EDTA to prevent coagulation. Red blood cells were lysed with an aqueous solution containing 0.83% NH4Cl, 0.1% KHCO3, and 0.000037% Na2EDTA at pH 7.2–7.4. Single-cell suspensions were passed through a 35μm-pore cell-strainer cap (BD Falcon) before culture, intravenous transfer, or staining for flow cytometry.
Reagents and flow cytometry
SIYRYYGL peptide was synthesized by MIT’s Biopolymers Laboratory. Recombinant murine Interferon-γ (cat#315-05) was purchased from PeproTech Inc. The following antibodies were purchased from BD Pharmingen: αCD4(H129.19), αCD8(53-6.7), αCD11b(M1/70), αCD16/CD32(2.4G2), αCD25(7D4 and PC61), αCD44(IM7), αCD69(H1.2F3), αCD107a(1D4B), αGr-1(RB6-8C5), αH-2Kb(AF6-88.5), and αIgG1(X56). Phycoerythrin-conjugated and unconjugated DimerX I reagents were purchased from BD Pharmingen and prepared according to manufacturer’s instructions. Intracellular Foxp3 staining was performed with FITC-α-mouse/rat Foxp3 staining set from eBioscience and secreted IFN-γ capture/detection was performed with Mouse IFN-γ Secretion Assay Detection kit (order#130-090-516) from Miltenyi Biotec, according to manufacturers’ instructions. Biotinylated 1B2 monoclonal antibody was provided by J. Chen. Streptavidin-allophycocyanin and streptavidin-phycoerythrin conjugates were purchased from BD Pharmingen. Cells were read on a FACSCalibur (BD Biosciences) and analyzed using Flowjo 8.1 software (Tree Star Inc). Dead cells were excluded by 1μg/mL propidium iodide staining (Sigma).
Acknowledgments
We thank D. Crowley for histological preparations, R. Bronson for histopathological analyses, and G. Paradis for flow cytometry help. We thank H. Ploegh and M. Winslow for critical reading of the manuscript and members of the Jacks and Chen labs for experimental advice and assistance. This work was supported by grant 1 U54 CA126515-01 from the National Institutes of Health and partially by Cancer Center Support (core) grant P30-CA14051 from the National Cancer Institute and the Margaret A. Cunningham Immune Mechanisms in Cancer Research Fellowship Award (A.F.C.) from the John D. Proctor Foundation. T.J. is a Howard Hughes Investigator and a Daniel K. Ludwig Scholar.
References
- 1.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117(5):1137–46. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson MJ, Shafer-Weaver K, Greenberg NM, Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. J Immunol. 2007;178(3):1268–76. doi: 10.4049/jimmunol.178.3.1268. [DOI] [PubMed] [Google Scholar]
- 5.Drake CG, Doody AD, Mihalyo MA, et al. Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell. 2005;7(3):239–49. doi: 10.1016/j.ccr.2005.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huijbers IJ, Krimpenfort P, Chomez P, et al. An inducible mouse model of melanoma expressing a defined tumor antigen. Cancer Res. 2006;66(6):3278–86. doi: 10.1158/0008-5472.CAN-05-3216. [DOI] [PubMed] [Google Scholar]
- 7.Muller-Hermelink N, Braumuller H, Pichler B, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. 2008;13(6):507–18. doi: 10.1016/j.ccr.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen LT, Elford AR, Murakami K, et al. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J Exp Med. 2002;195(4):423–35. doi: 10.1084/jem.20010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437(7055):141–6. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 10.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117(6):1466–76. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. 2005;54(3):187–207. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevanovic S. Identification of tumour-associated T-cell epitopes for vaccine development. Nat Rev Cancer. 2002;2(7):514–20. doi: 10.1038/nrc841. [DOI] [PubMed] [Google Scholar]
- 15.Raez LE, Fein S, Podack ER. Lung cancer immunotherapy. Clin Med Res. 2005;3(4):221–8. doi: 10.3121/cmr.3.4.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuyama T, Hanagiri T, Takenoyama M, et al. Identification of a new cancer/germline gene, KK-LC-1, encoding an antigen recognized by autologous CTL induced on human lung adenocarcinoma. Cancer Res. 2006;66(9):4922–8. doi: 10.1158/0008-5472.CAN-05-3840. [DOI] [PubMed] [Google Scholar]
- 17.Fukuyama T, Ichiki Y, Yamada S, et al. Cytokine production of lung cancer cell lines: Correlation between their production and the inflammatory/immunological responses both in vivo and in vitro. Cancer Sci. 2007;98(7):1048–54. doi: 10.1111/j.1349-7006.2007.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizukami M, Hanagiri T, Shigematsu Y, et al. Effect of IgG produced by tumor-infiltrating B lymphocytes on lung tumor growth. Anticancer Res. 2006;26(3A):1827–31. [PubMed] [Google Scholar]
- 19.Woo EY, Yeh H, Chu CS, et al. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168(9):4272–6. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- 20.Udaka K, Wiesmuller KH, Kienle S, Jung G, Walden P. Self-MHC-restricted peptides recognized by an alloreactive T lymphocyte clone. J Immunol. 1996;157(2):670–8. [PubMed] [Google Scholar]
- 21.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 22.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5(4):375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 24.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7(9):645–58. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 25.Kranz DM, Sherman DH, Sitkovsky MV, Pasternack MS, Eisen HN. Immunoprecipitation of cell surface structures of cloned cytotoxic T lymphocytes by clone-specific antisera. Proc Natl Acad Sci U S A. 1984;81(2):573–7. doi: 10.1073/pnas.81.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saito H, Kranz DM, Takagaki Y, Hayday AC, Eisen HN, Tonegawa S. A third rearranged and expressed gene in a clone of cytotoxic T lymphocytes. Nature. 1984;312(5989):36–40. doi: 10.1038/312036a0. [DOI] [PubMed] [Google Scholar]
- 27.Sha WC, Nelson CA, Newberry RD, Kranz DM, Russell JH, Loh DY. Selective expression of an antigen receptor on CD8-bearing T lymphocytes in transgenic mice. Nature. 1988;335(6187):271–4. doi: 10.1038/335271a0. [DOI] [PubMed] [Google Scholar]
- 28.Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26(2):113–5. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Schulman J, Itamura S, Palese P. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J Virol. 1993;67(11):6667–73. doi: 10.1128/jvi.67.11.6667-6673.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen CH, Ge Q, Talay O, Eisen HN, Garcia-Sastre A, Chen J. Loss of IL-7R and IL-15R expression is associated with disappearance of memory T cells in respiratory tract following influenza infection. J Immunol. 2008;180(1):171–8. doi: 10.4049/jimmunol.180.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158(6):2723–30. [PubMed] [Google Scholar]
- 32.Algarra I, Garcia-Lora A, Cabrera T, Ruiz-Cabello F, Garrido F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: implications for tumor immune escape. Cancer Immunol Immunother. 2004;53(10):904–10. doi: 10.1007/s00262-004-0517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–39. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- 35.Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity. 2005;22(3):275–84. doi: 10.1016/j.immuni.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6(10):715–27. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 37.Hernandez J, Aung S, Redmond WL, Sherman LA. Phenotypic and functional analysis of CD8(+) T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J Exp Med. 2001;194(6):707–17. doi: 10.1084/jem.194.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charo J, Finkelstein SE, Grewal N, Restifo NP, Robbins PF, Rosenberg SA. Bcl-2 overexpression enhances tumor-specific T-cell survival. Cancer Res. 2005;65(5):2001–8. doi: 10.1158/0008-5472.CAN-04-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lollini PL, Cavallo F, Nanni P, Forni G. Vaccines for tumour prevention. Nat Rev Cancer. 2006;6(3):204–16. doi: 10.1038/nrc1815. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlom J, Arlen PM, Gulley JL. Cancer vaccines: moving beyond current paradigms. Clin Cancer Res. 2007;13(13):3776–82. doi: 10.1158/1078-0432.CCR-07-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755–66. [PubMed] [Google Scholar]
- 43.Perrot I, Blanchard D, Freymond N, et al. Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol. 2007;178(5):2763–9. doi: 10.4049/jimmunol.178.5.2763. [DOI] [PubMed] [Google Scholar]
- 44.Woo EY, Chu CS, Goletz TJ, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61(12):4766–72. [PubMed] [Google Scholar]
- 45.Jackson EL, Olive KP, Tuveson DA, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65(22):10280–8. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 46.Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med. 2000;6(9):1029–35. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]