

Abstract
Neuronal expression of familial Alzheimer's disease (AD)-mutant human amyloid precursor protein (hAPP) and hAPP-derived amyloid-β (Aβ) peptides causes synaptic dysfunction, inflammation, and abnormal cerebrovascular tone in transgenic mice. Fatty acids may be involved in these processes, but their contribution to AD pathogenesis is uncertain. A lipidomics approach to broadly profile fatty acids in brain tissues of hAPP mice revealed an increase in arachidonic acid and its metabolites, suggesting increased activity of the group IV isoform of phospholipase A2 (GIVA-PLA2). Levels of activated GIVA-PLA2 in the hippocampus were increased in AD patients and hAPP mice. Aβ caused a dose-dependent increase in GIVA-PLA2 phosphorylation in neuronal cultures. Inhibition of GIVA-PLA2 diminished Aβ-induced neurotoxicity. Genetic ablation or reduction of GIVA-PLA2 protected hAPP mice against Aβ-dependent deficits in learning and memory, behavioral alterations, and premature mortality. Inhibition of GIVA-PLA2 may be of benefit in the treatment and prevention of AD.
First described a century ago, Alzheimer's disease (AD) has increased markedly in prevalence, and no effective treatments exist1. Transgenic mice with neuronal expression of familial AD-mutant human amyloid precursor protein (hAPP) show age-dependent deficits in learning and memory, behavioral alterations, and premature mortality2, 3. Amyloid-β (Aβ) peptides, released from hAPP by proteolysis, are thought to mediate these deficits, but the exact mechanisms remain to be fully elucidated1, 3.
Essential fatty acids and their metabolites (Supplementary Fig. 1) participate in processes involved in the pathogenesis of AD, including synaptic plasticity4, inflammation, cerebrovascular function5, 6, and oxidative stress7. Fatty acids are rapidly taken up by the brain, incorporated into phospholipids, and released by phospholipase A2 (PLA2)8. Changing the dietary intake of essential fatty acids modulates the phenotype of hAPP mice9 and may affect cognition and AD progression in humans10.
However, fatty acid metabolism in AD patients and hAPP mice has not yet been broadly profiled. Therefore, we used an unbiased lipidomics approach11 to examine PLA2-dependent fatty acid metabolism in brain tissues of hAPP mice. Our lipidomics profile led us to hypothesize that a specific isoform of PLA2 contributes to Aβ-mediated neuronal deficits. We tested this hypothesis by perturbation analyses in vitro and in vivo.
RESULTS
Increased fatty acids in hAPP mice
To profile essential fatty acid metabolism, we measured the levels of 44 pertinent metabolites in the hippocampus and cortex of transgenic mice with neuronal expression of familial AD-mutant hAPP (line J20, refs.2, 12-15) and nontransgenic (NTG) controls by simultaneous liquid chromatography–tandem triple–quadrupole mass spectrometry (LC-MS/MS) (Supplementary Fig. 1). Hippocampal levels of arachidonic acid (AA) were higher in hAPP mice than in NTG controls, whereas cortical AA levels were comparable (Fig. 1a). Total AA-derived metabolites were also increased in the hippocampus, but not cortex, of hAPP mice (Fig. 1b). Hippocampal, but not cortical, levels of prostaglandin (PG) E2 and PGB2 were higher in hAPP mice than in NTG mice (Fig. 1c). The increase in PGE2 is consistent with the increased hippocampal cyclooxygenase (COX)-2 in another hAPP transgenic model16. Both AA and PG regulate long-term potentiation (LTP)17, which is important for learning and memory. Excessive levels of AA and PG can contribute to excitotoxicity5, which may be involved in AD pathogenesis3, 12, 13. In addition, AA and PG contribute to inflammation, which may also contribute to AD1. Inflammation might be further promoted by leukotrienes (LT), AA-derived fatty acids that are potent chemoattractants for monocytes and granulocytes18. LTB4 levels were higher in hAPP mice than NTG controls in both the hippocampus and cortex (Fig. 1d). Increased LTB4 levels may contribute to the microgliosis seen in hAPP mice and AD patients.
Figure 1.

Increased PLA2-dependent fatty acid levels in brain tissues of hAPP mice. Lipids were extracted from hippocampal or cortical homogenates from hAPP and NTG mice (n=12 per genotype) and analyzed by quantitative LC-MS/MS. Fatty acid levels (pmol) were normalized to total protein (mg). a–f, Hippocampal and cortical levels of (a) AA, (b) total AA-derived metabolites, (c) PGE2 and its nonenzymatic PGB2 degradation product, (d) LTB4, (e) 14,15-EET and its 14,15-DHET metabolite, and (f) total fatty acids. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. NTG or as indicated by bracket (t test; means ± s.e.m.).
Levels of AA, PGD2, PGB2, and LTB4 were not altered in wildtype hAPP transgenic mice from line I5 (Supplementary Fig. 2a–c), which have comparable hAPP levels to those in hAPPJ20 mice but much lower Aβ levels14.
Neurovascular coupling also appears to be affected in AD19, possibly because vasoconstrictive effects of Aβ alter the normal vascular dilation in response to increased neuronal activity20. This effect may be counteracted by PG and epoxyeicosatrienoic acids (EETs); the latter are derived from AA through the activity of cytochrome p450 monooxygenase in astrocytes surrounding blood vessels5, 6. PGE2 and EETs cause cerebrovascular dilation in response to neuronal activity5, 6. The four regioisomers of EETs have similar biological activities and can be metabolized into less active dihydroxyeicosatrienoic acids (DHET), which can serve as markers of EET production6. Cortical levels of 14,15-EET, 14,15-DHET, and 8,9-DHET were higher in hAPP mice than NTG controls (Fig. 1e and Supplementary Fig. 2). hAPP mice also showed a trend toward increased cortical and hippocampal levels of other EET metabolites (Supplementary Fig. 3).
In NTG mice, fatty acid levels were also higher in the hippocampus than cortex (Fig. 1f), suggesting relatively higher PLA2 activity in the hippocampus. The lipidomics profile did not demonstrate significant differences between hAPP and NTG mice in the levels of other fatty acids liberated directly by PLA2: docosahexaenoic (DHA), eicosapentaenoic (EPA), linoleic acid (LA), and α-linolenic acid (ALA) (Supplementary Fig. 4a). Except for 12-hydroxyeicosapentaenoic acid (HEPE), whose hippocampal levels were higher in hAPP mice than NTG controls, we also found no significant differences in other prostaglandins, hydroxyeicosatetraenoic acids HETEs, or more distal metabolites (Supplementary Figs. 4b–c, 5, 6).
Thus, the lipidomics profile revealed a rather selective alteration in PLA2-dependent fatty acid metabolism in hAPP mice, consisting primarily of increased levels of AA and its metabolites. Because different isoforms of PLA2 have specificities for particular fatty acids8, this fatty acid profile implicates a specific isoform of PLA2.
Group IVA-PLA2 is present in mouse neurons
Three isoforms of PLA2 have been reported in the brain (Groups II, IVA, and VIA). GIVA-PLA2 has the strongest preference for AA, is regulated by Ca2+, and has phosphorylation sites for kinases implicated in AD8. GIVA-PLA2-deficient mice fail to generate AA metabolites after brain injury8, 21. In contrast, GVIA-PLA2 is not responsive to Ca2+ and lacks phosphorylation sites for kinases implicated in AD8. The GIIA isoform is absent in our hAPP and NTG mice because of an inbred gene deletion in the C57BL/6 strain21. These data suggested that GIVA-PLA2 may cause the increased AA metabolism in hAPP mice. However, in a previous study, no GIVA-PLA2 immunoreactivity was detected in brains of NTG and hAPP mice22, and analysis of GIVA-PLA2 in AD has yielded inconsistent results 23, 24.
GIVA-PLA2 immunoreactivity was readily detectable in brains of our hAPP mice and NTG controls. Immunostaining with a GIVA-PLA2-specific antibody resulted in a comparable widespread neuronal labeling in brain sections from NTG and hAPP mice on the wildtype GIVA-PLA2 background (Fig. 2a,b), with dense labeling of granule cells of the dentate gyrus and pyramidal neurons in the CA1−3 regions (Fig. 2d,e). No staining was seen in GIVA-PLA2-deficient mice (Fig. 2c,f). Our findings are consistent with detection of neuronal GIVA-PLA2 mRNA25 and immunoreactivity26 in rats.
Figure 2.
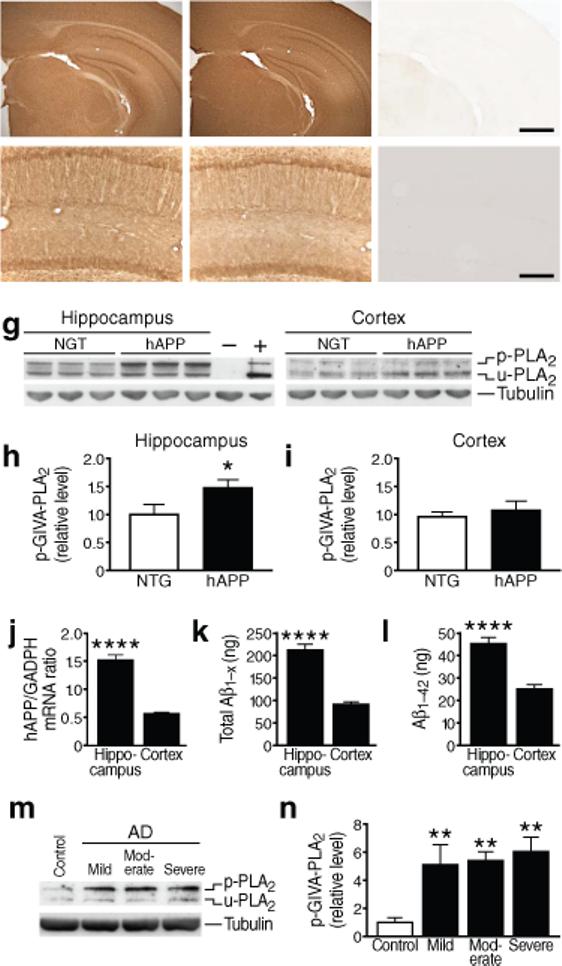
GIVA-PLA2 levels in hAPP mice and humans with AD. (a–f) Coronal sections of cortex and hippocampus (a–c) or hippocampus (d–f) in 6-month-old NTG (a,d), hAPP (b,e), and GIVA-PLA2-deficient (c,f) mice. Scale bar (a–c) 1 mm, (d–f) 250 μm. g–i, Hippocampal and cortical levels of GIVA-PLA2 in mice were determined by western blot analysis with a rabbit polyclonal antibody. g, Representative western blot showing levels of phosphorylated (p) and unphosphorylated (u) GIVA-PLA2. Tubulin served as a loading control, NIH3T3 cells as a positive control (+), and cortex from GIVA-PLA2-deficient mice as a negative control (−). h,i, Hippocampal (h) and cortical (i) GIVA-PLA2 levels determined by densitometric analysis of western blot signals (n=12 mice per genotype and brain region; age: 6 months). j, hAPP mRNA levels in hippocampus and cortex of hAPP mice determined by quantitative RT-PCR (n=7 mice; age: 2−4 months). k, l, Levels of Aβ1-x (k) and Aβ1−42 (l) (ng per g of tissue) in hippocampus and cortex of hAPP mice determined by ELISA (n=8 mice; age, 2−4 months). m–n, Levels of phosphorylated GIVA-PLA2 protein in the CA1 hippocampal region in patients with mild, moderate, or severe AD and in nondemented, age-matched controls (C) were determined by western blot analysis. m, Representative western blot. n, Western blot signals were quantitated densitometrically and normalized to tubulin (n=4−8 cases per group). *P<0.05, ****P<0.0001 (t test; mean ± s.e.m.), **P<0.01 vs. control (Tukey test; mean ± s.e.m.).
GIVA-PLA2 activation in hAPP hippocampus
Next, we determined whether phosphorylated, putatively active GIVA-PLA2 was increased in brains of hAPP mice. Phosphorylated and unphosphorylated forms of GIVA-PLA2 can be detected with the same antibody because phosphorylation leads to an electrophoretic mobility shift. Hippocampal, but not cortical, levels of phosphorylated GIVA-PLA2 were higher in hAPP mice than in NTG controls (Fig. 2g–i), consistent with the differences in fatty acid levels in these regions (Fig. 1f). No GIVA-PLA2 band was seen in brains from GIVA-PLA2-deficient mice (Fig. 2g).
To determine if the region-specific increase in GIVA-PLA2 phosphorylation was related to differences in the levels of hAPP transgene products, we measured hAPP mRNA by quantitative reverse transcriptase (RT)-PCR and Aβ by ELISA in 2−4-month-old hAPP mice (before plaque formation). Levels of hAPP mRNA (Fig. 2j), Aβ1-x (Fig. 2k), and Aβ1−42 (Fig. 2l) were indeed higher in hippocampus than cortex in hAPP mice.
Because fresh tissue for reliable lipidomics analysis in human AD patients is not readily available and phosphorylated GIVA-PLA2 levels correlate with AA production8, we measured hippocampal levels of phosphorylated GIVA-PLA2 in human postmortem tissues. Compared with nondemented controls, levels of phosphorylated GIVA-PLA2 were elevated in AD patients, but not in patients with frontotemporal dementia (Fig. 2m–n and Supplementary Fig. 7), who do not have elevated Aβ levels.
GIVA-PLA2 contributes to Aβ-induced neurotoxicity
To determine if exposure to extracellular Aβ was sufficient to activate neuronal GIVA-PLA2, we assessed the effect of synthetic Aβ1−42 oligomers on GIVA-PLA2 phosphorylation in primary neuronal cultures. Treatment with Aβ1−42, but not Aβ42−1 control peptide, caused a dose- and time-dependent increase in phosphorylated GIVA-PLA2 (Fig. 3a). Treatment with cell-secreted hAPP had no such effect (Supplementary Fig. 8a). Similarly, AA release was increased by Aβ1−42, but not by Aβ42−1 or cell-secreted hAPP (Supplementary Fig. 8b). Aβ1−42-induced phosphorylation of GIVA-PLA2 was blocked by the broad–spectrum MAPK inhibitor PD98059 and by the MEK inhibitor SB203580 (Supplementary Fig. 8a), suggesting mediator roles of these kinases. Aβ1−42-dependent AA release was blocked by pretreatment of neuronal cultures with the Ca2+ chelators EGTA and BAPTA, inhibitors of MAPK or MEK, or the GIVA-PLA2 inhibitor arachidonyl trifluoromethyl ketone (AACOCF3) 5, but not by pretreatment with the GVIA-PLA2 inhibitor bromoenolactone (BEL) (Supplementary Fig. 8b).
Figure 3.
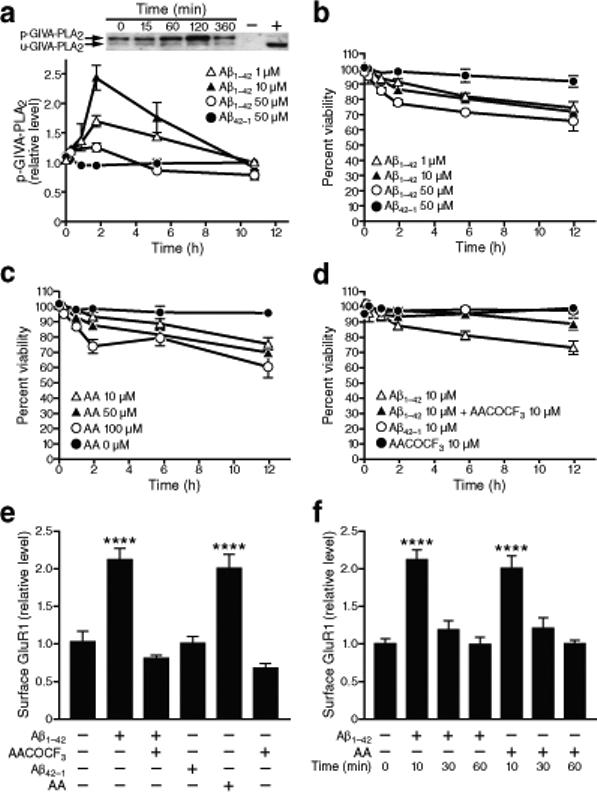
Inhibition of GIVA-PLA2 prevents Aβ1−42 toxicity in primary neuronal cultures. Primary rat neurons were treated with Aβ1−42 oligomers as indicated after 14 days in vitro. Quantitative results were obtained from three wells per condition in five independent experiments and normalized to untreated controls. a, Levels of phosphorylated and unphosphorylated GIVA-PLA2 in cell lysates were determined by western blot analysis. Aβ increased levels of phosphorylated GIVA-PLA2 in a dose- and time-dependent manner (P<0.0001 by two-way ANOVA, mean ± s.e.m.). b–d, Percentage of viable cells determined by trypan blue exclusion and counting of unlabeled cells. b, Aβ caused neuronal cell death in a dose- and time-dependent manner (P<0.001 by repeated-measures ANOVA, mean and s.e.m.). c, AA also led to neuronal death (P<0.01 by repeated-measures ANOVA). d, Pretreatment of cells with AACOCF3, a GIVA-PLA2-specific inhibitor, for 10 min ameliorated Aβ-induced neuronal death (P<0.01 by repeated-measures ANOVA, P<0.001 at 6 and 12 h by paired t test). e, Surface levels of GluR1 were assessed by biotinylation assay 10 min after the indicated treatments. Aβ1−42 (10 μM) increased surface levels of GluR1 compared with Aβ42−1, an effect that could be blocked with AACOCF3 pretreatment and replicated with AA (mean ± s.e.m). f, Surface levels of GluR1 decreased to baseline levels after 30 and 60 min of exposure to Aβ1−42 or AA (mean ± s.e.m.). ****P<0.0001 versus Aβ42−1 (Tukey test).
Aβ1−42 and AA each caused a dose-dependent decrease in neuronal viability (Fig. 3b,c), consistent with previous findings3, 27. Because a nonspecific PLA2 inhibitor protected neurons against Aβ toxicity28, we wondered whether this effect is due to inhibition specifically of GIVA-PLA2. Pretreatment of neuronal cultures with the GIVA-PLA2 inhibitor AACOCF3 reduced Aβ1−42-induced cell death, whereas the GVIA-PLA2 inhibitor BEL had no effect (Fig. 3d and Supplementary Fig. 8c).
How might GIVA-PLA2 activation contribute to Aβ-induced neuronal toxicity? This toxicity is caused, at least partly, by overexcitation3, 29. Oligomeric Aβ generates rapid inward currents in neurons that appear to be necessary for subsequent neuronal toxicity29-31. Oligomeric Aβ-induced neurotoxicity involves AMPA receptors (AMPAR)32, and AMPAR antagonists can block acute Aβ-induced neuronal activation and neurotoxicity31, 33. AMPAR currents are dependent on the number of surface AMPAR32, 34. PLA2 activity leads to phosphorylation of AMPAR in a manner that would predict an increase in their surface levels4, consistent with the ability of GIVA-PLA2 and AA to cause neuronal excitation35 and increase AMPAR activity36. Moreover, controlling for neuronal cell loss, the levels of AMPAR and of AMPAR binding in the hippocampus were higher in AD cases than controls, particularly in the molecular layer of the dentate gyrus37. We therefore hypothesized that GIVA-PLA2 contributes to Aβ-induced neuronal toxicity by increasing surface levels of AMPAR.
Aβ1−42 and AA each increased surface levels of the AMPAR subunits GluR1 (Fig. 3e) and GluR2 (Supplementary Fig. 9a) in cultured neurons within 10 min. Inhibition of GIVA-PLA2 blocked this Aβ1−42-induced increase in GluR1 (Fig. 3e) and GluR2 (Supplementary Fig. 9a). The Aβ-induced increase in surface AMPAR subunits was transient, as surface levels returned to baseline with continued exposure (Fig. 3f and Supplementary Fig. 9b).32, 38, 39. To determine if the early Aβ1−42-induced increase in AMPAR contributed to subsequent neuronal death, we pretreated cells with the AMPAR blocker NBQX. AMPAR blockade significantly decreased neuronal toxicity induced by Aβ1−42 or AA (Supplementary Fig. 8c). Consistent with the Aβ1−42-and GIVA-PLA2-dependent increase in surface AMPAR, treatment of brain slices of NTG wild-type mice with oligomeric Aβ1−42 or AA caused an immediate increase in neuronal activity (Supplementary Fig. 10a–c). Pretreatment of slices with an GIVA-PLA2 inhibitor blocked this Aβ1−42 effect (Supplementary Fig. 10d).
GIVA-PLA2 reduction improves memory in hAPP mice
To determine if GIVA-PLA2 contributes to Aβ-dependent behavioral deficits in vivo, we bred hAPP and NTG mice with GIVA-PLA2-deficient mice to generate six genotypes: hAPP/PLA2+/+, hAPP/PLA2+/−, hAPP/PLA2−/−, PLA2+/+, PLA2+/−, and PLA2−/−. These groups did not differ in background strain, body weight, gross motor function, or swim speed (Supplementary Fig. 11). Removal or reduction of GIVA-PLA2 significantly reduced hippocampal levels of AA and its metabolites in hAPP mice (Supplementary Figure 12). To determine if these changes ameliorated cognitive deficits in hAPP mice, we tested the above groups in the Morris water maze. In the cued component of this test, hAPP mice exhibited deficits in task acquisition that were independent of GIVA-PLA2 levels (Fig. 4a). In the hidden platform (spatial) component of the test, complete or partial removal of GIVA-PLA2 reduced learning deficits in hAPP mice (Fig. 4b). In the probe trial, a putative measure of spatial learning and memory retention, only hAPP/PLA2+/+ mice failed to favor the target platform location (Fig. 4c,d). Removal or reduction of GIVA-PLA2 significantly improved target crossings in hAPP mice in the probe trial (Fig. 4c,d). In addition, hAPP mice with GIVA-PLA2 reduction or removal showed a trend for improved overall target quadrant preference during the final probe trial (data not shown).
Figure 4.

GIVA-PLA2 reduction improves learning and memory in hAPP mice. a,b, Mice (n=8−15 per genotype; age: 4−6 months) were tested in the cued (a) and hidden (b) platform components of the Morris water maze. In both components, hAPP/PLA2+/+ mice differed from all groups without hAPP (P<0.0001, by repeated-measures ANOVA and Tukey test). In the hidden platform component, hAPP/PLA2+/+ mice learned more poorly than hAPP/PLA2+/− mice (P<0.05) and hAPP/PLA2−/− mice (P<0.01, by repeated-measures ANOVA and Tukey test). c, Representative swim paths of individual mice with indicated genotypes during the last probe trial (platform removed). The blue square indicates the target location during hidden platform training. d, Number of times mice crossed this target location during the last probe trial compared with how many times they crossed corresponding locations in non-target quadrants. Only hAPP/PLA2+/+ mice failed to show a clear target location preference. A two-way ANOVA of target/nontarget ratios revealed significant effects of hAPP (P<0.0001) and PLA2 (P<0.001) and a significant interaction between hAPP and PLA2 genotype (P<0.001). *P<0.05, **P<0.01, ***P<0.001 vs. nontarget locations; ###P<0.001 (Tukey test, mean ± s.e.m.).
Deficits in learning and memory in hAPP mice may involve aberrant excitatory and inhibitory network activity3, 13 and correlate well with depletions of Ca2+-regulated, synaptic activity-dependent proteins in the dentate gyrus2. Removal of GIVA-PLA2 prevented depletions of the Ca2+-binding protein calbindin in hAPP mice (Supplementary Fig. 13). Together, these results suggest a causal role of GIVA-PLA2 in Aβ-dependent impairments of learning and memory.
Other behavioral abnormalities and premature mortality
hAPP mice demonstrate hyperactivity that may be related to memory deficits and entorhinal cortex dysfunction14, 15. Complete or partial removal of GIVA-PLA2 significantly decreased hyperactivity in hAPP mice, as reflected in the open field test (Fig. 5a,b) and the Y-maze (Fig. 5c,d). hAPP mice also show a disinhibition-like phenotype in the elevated plus maze, which is widely used to assess anxiety and exploratory behavior14, 15. Both PLA2 and hAPP genotype significantly affected elevated plus maze performance (Fig. 5e–g). Reduction of GIVA-PLA2 dose-dependently diminished the time spent (Fig. 5e) and distance traveled (Fig. 5f) in the open arms of the maze and the number of edge pokes (Fig. 5g) in hAPP mice. In mice without hAPP, removal of GIVA-PLA2 had much more subtle or no effects on these behaviors (Fig. 5a–g).
Figure 5.

GIVA-PLA2 reduction prevents hyperactivity, abnormal anxiety/exploration-related behavior, and premature mortality in hAPP mice. a–g, Mice (n=8−15 mice per genotype, age: 4−6 months) were tested in the open field (a,b), the Y maze (c,d), and the elevated plus maze (e–g). Genotype effects and interactions were assessed by two-way ANOVA. a, Total movements (P<0.001 for hAPP effect, P<0.0001 for PLA2 effect, P<0.01 for interaction). b, Total rearings (P<0.001 for hAPP effect, P<0.05 for PLA2 effect, and P<0.05 for interaction). c, Total entries (P<0.05 for hAPP effect, P<0.0001 for PLA2 effect, and P<0.001 for interaction). d, Total alternations (P<0.0001 for hAPP effect, P<0.001 for PLA2 effect, and P<0.0001 for interaction). (e) Percent time spent in open arms (P<0.0001 for hAPP effect, P<0.05 for PLA2 effect, and P=0.259 for interaction). (f) Percent distance traveled in open arms (P<0.0001 for hAPP effect, P<0.05 for PLA2 effect, P=0.237 for interaction). g, Edge pokes (P<0.0001 for hAPP effect, P<0.01 for PLA2 effect, P=0.133 for interaction). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. PLA2+/+ mice or as indicated by brackets (Dunnett test, mean ± s.e.m.). h, Kaplan-Meier survival analysis of 189 mice revealed premature mortality in hAPP/PLA2+/+ mice (P<0.001 by log-rank chi-square test), but not in hAPP/PLA2+/− or hAPP/PLA2−/− mice.
hAPP mice also exhibit premature mortality (Fig. 5h), possibly related to their lowered seizure threshold12, 13, 15. Removal or reduction of GIVA-PLA2 significantly improved the survival of hAPP mice (Fig. 5h) without altering the levels of hAPP (Fig. 6a), Aβ1-x (Fig. 6b), Aβ1−42 (Fig. 6c), or the Aβ1−42/Aβ1-x ratio (Fig. 6d). Removal or reduction of GIVA-PLA2 also did not alter Aβ deposition, astroglial activation (GFAP immunostaining), or microglial numbers (Iba-1 immunostaining) (data not shown). These findings suggest that GIVA-PLA2 reduction blocks or counteracts AD-related abnormalities downstream of Aβ production and deposition.
Figure 6.

Reduction or removal of GIVA-PLA2 did not affect hAPP or Aβ levels in hAPP mice. a, Hippocampal hAPP protein levels (relative to GADPH) in hAPP mice with different levels of GIVA-PLA2 expression (n=6 per group; age, 4−6 months) were determined by western blotting and densitometric analysis of signals. b–d, Hippocampal and cortical levels of Aβ1-x (b) and Aβ1−42 (c) and Aβ1−42/Aβ1-x ratios (d) in 4−6-month-old hAPP mice with different levels of GIVA-PLA2 expression (n=6−10 per group; age, 4−6 months) were determined by ELISA and expressed as ng per g of tissue.
DISCUSSION
This study shows that reduction of GIVA-PLA2 can prevent diverse Aβ-dependent functional impairments in a transgenic mouse model of AD. We zeroed in on this enzyme because our unbiased lipidomics analysis revealed a surprisingly selective increase in AA and its metabolites in brain tissues of hAPP mice, suggesting increased activity of this particular PLA2 isoform. We also showed that GIVA-PLA2 is expressed in brains of mice and that hAPP mice and AD patients have increased levels of phosphorylated (putatively activated) GIVA-PLA2 in the hippocampus, which is particularly vulnerable to AD in humans and to Aβ-induced neuronal deficits in transgenic mice2, 3, 12-15.
Because PLA2 releases fatty acids from phospholipids8, the increased GIVA-PLA2 activity we detected in hAPP mice is consistent with the decreased levels of phospholipid-bound AA in AD brains40, further underlining the potential relevance of our findings to the human condition. Increased GIVA-PLA2 activity in hAPP mice may also be related to the increases in isoprostanes (nonenzymatic AA metabolites that form during oxidative stress) in the brain and CSF of AD patients and hAPP mice7 and in 4-hydroxynonenals (oxidative aldehyde products of AA) in brains of AD patients41, 42.
Interestingly, the translocation of GIVA-PLA2 to its phospholipid substrate is primarily regulated by intracellular calcium8, and several lines of evidence suggest that Ca2+-dependent signaling pathways are dysregulated in neurons of hAPP mice, particularly in the hippocampus1-3, 12-15. Because transgene expression and Aβ levels were higher in the hippocampus than the cortex, the regional differences in aberrant GIVA-PLA2 activation may primarily reflect differences in Aβ levels and a threshold effect. Consistent with this interpretation, our in vitro studies demonstrated dose-dependent neuronal GIVA-PLA2 activation by extracellular Aβ1−42.
The concentration of any fatty acid product is dependent on the concentration of its precursor substrate and the enzymatic activity of the protein that metabolizes that precursor. The elevated levels of LTB4 and 14,15 EET in the cortex in the setting of normal AA levels there suggest that the activities of 5-LO, p450, LTA4 hydrolase, and soluble epoxide hydrolase may be increased in the cortex of hAPP-J20 mice compared to NTG mice.
Several lines of evidence suggest that GIVA-PLA2 activation contributes to Aβ1−42-dependent neurotoxicity, which can range from synaptic dysfunction to neuronal cell death. Aβ-induced neurotoxicity requires AMPAR activity, which is regulated by surface AMPAR levels31, 32, 43. Aβ1−42 acutely elevated surface AMPAR levels in cultured neurons, and inhibiting GIVA-PLA2 blocked this process. Treatment with AA also increased surface AMPAR levels. Thus, GIVA-PLA2 and AA may contribute to Aβ neurotoxicity by mediating Aβ-induced increases in surface AMPAR expression (Supplementary Fig. 14). Indeed, increased surface levels of AMPARs would be expected to increase neuronal excitability44. Early neuronal excitability is thought to mediate acute Aβ1−42 toxicity29 and to be essential for delayed Aβ1−42-dependent synaptic deficits30.
In vivo and in vitro evidence suggests that Aβ can elicit not only neuronal overexcitation, but also synaptic depression, and that both of these mechanisms may contribute to AD-related cognitive impairments3, 12, 13, 30, 32, 45. Our results raise the possibility that the decreases in surface AMPAR observed after longer exposure to Aβ32, 39 may result from feedback inhibition (e.g., synaptic scaling46) triggered by the acute increase in surface AMPAR expression and associated increases in neuronal excitability (Supplementary Fig. 14). Additional potential causes include long-term depression–like mechanisms32, 39, 43, and impairments of neuronal functions.
It is difficult to extrapolate from in vitro to in vivo conditions, and activation of GIVA-PLA2 leads to the production of many fatty acids with diverse biological activities8, 18. Thus, it was not clear a priori whether GIVA-PLA2 activation in hAPP mice or AD is beneficial or detrimental. For instance, AA and its metabolites participate in synaptic plasticity4, excitotoxicity47, cerebrovascular regulation5, 6, oxidative stress7, 41, 42, and inflammation5, all of which have been implicated in the pathogenesis of AD1, 3, 5.
To determine if reducing GIVA-PLA2 activity is beneficial or detrimental in the presence of abnormally elevated Aβ levels, we crossed hAPP mice onto a GIVA-PLA2-deficient background. Elimination or partial reduction of GIVA-PLA2 was well tolerated and effectively reduced learning and memory deficits, behavioral alterations, and premature mortality in hAPP mice. These findings suggest that GIVA-PLA2 contributes to the pathogenesis of these abnormalities and might be a useful target for therapeutic interventions in AD.
Methods
Mice and tissue
We analyzed sex-balanced groups of 2−6-month-old heterozygous transgenic and NTG mice from line J202, 12-15. PLA2−/− mice were generated by Joseph Bonventre (Harvard Medical School, Boston)21. All mice were on a C57/BL6 background. All experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco. Anesthetized mice were perfused transcardially with saline. Mouse brains were processed for immunohistochemistry12, protein12 and lipid analysis11, 48 as described.
Lipid analysis
Frozen brain regions were pulverized in liquid nitrogen and spiked with deuteriated and odd chain metabolite surrogates48 prior to homogenization in ice cold methanol. Samples were diluted to 10% methanol with phosphate-buffered saline (pH7.4) and lipids were isolated by solid-phase extraction on Oasis HLB cartridges (Waters). Residues were dried under vacuum and reconstituted in the presence of internal standards allowing surrogate recoveries to be determined48. Analytes were separated by reverse-phase liquid chromatography on a Waters UPLC equipped with a 2.1 × 150 mm, 1.7-μm C18-BEH Acquity column. A gradient of mobile phases A (water/0.1% acetic acid, wt/wt) and B (80:15 (v/v) acetonitrile/methanol/0.1% acetic acid, wt/wt) was used. Analytes were detected by negative-mode electrospray ionization with a tandem quadrupole mass spectrometer (QuattroMicro, Waters) operated in multireaction monitoring mode, and quantified against five-point calibration curves bracketing the observed concentrations. Quantitative analyte measurements were adjusted according to the recovery of structurally similar surrogate metabolites that were spiked into the samples before lipid extraction. Less than 2% of the spiked epoxide and hydroxyprostane rings were lost during sample preparation. The dehydration products of PGE2 and PGD2 (i.e., PGB2 and PGJ2) were quantified and combined with their parent PGs before statistical analysis.
Primary neuronal culture
Rat cortices (embryonic day 18) were digested with papain. Cells were plated in polylysine-coated wells and maintained in serum-free neurobasal medium supplemented with B27 and antibiotics. Half the medium was changed after 7 days in culture and 1 day before use. Cells were used after 14 days in culture. More than 95% of cells were neurons, as determined by staining with antibody against the neuron-specific marker NeuN (data not shown). Cell viability was assessed with trypan blue49. Neuronal cell treatments included Aβ1−42, AA, AACOCF3, and BEL (Cayman Chemical), cell-secreted hAPP695 obtained from medium of Lipofectamine–tranfected HEK cells, NBQX, PD98059, and SB203580 (Sigma).
Aβ preparation
To prepare Aβ peptides, 1 mg of Aβ1−42 (Biopeptide) was solubilized in a glass tube with ice-cooled 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma), incubated for 1 h at room temperature, allowed to evaporate over 24 h, and spun for 10 min on a speed vac. The remaining precipitate was solubilized with 100 μM DMSO in neurobasal medium, incubated at 4°C for 48 h, and centrifuged at 14,000 × g for 10 min at 4°C. Soluble oligomers were collected and resuspended in neurobasal medium. Concentrations of monomeric Aβ were determined by absorbance at 595 nm after addition of Coomassie reagent (Sigma).
Quantitative RT-PCR
RT reactions contained 300 ng of total RNA (DNase-treated) and random hexamer plus oligo d(T) primers. Diluted reactions were analyzed with SYBR green PCR reagents and an ABI Prism 7700 (Applied Biosystems). cDNA levels of hAPP and mGAPDH were determined relative to standard curves from pooled samples. The slope of standard curves, control reactions without RT, and dissociation curves of products indicated adequate PCR quality. The following hAPP primers were used: hAPP 5RF primer sequence (5'−3'): GAGGAGGATGACTCGGATGTCT; hAPP 6RR primer sequence (5'−3'): AGCCACTTCTTCCTCCTCTGCTA.
Aβ ELISAs
Snap-frozen brain tissues were homogenized with guanidine buffer followed by ELISA measurements of human Aβ peptides15. The ELISA for Aβ1−42 detects this specific peptide. The ELISA for Aβ1-x recognizes N-terminal fragments of Aβ containing the first 28 amino acids.
Immunoblotting
Frozen samples were homogenized in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, protease inhibitor cocktail (Roche), 10 μM pepstatin, 5 mM EDTA, 400 μM PMSF, and 0.2% Triton X-100), sonicated on ice, and centrifuged (5000 × g, 15 min). Protein concentration was determined by Bradford protein assay. Protein (40 μg) was loaded into each well of a 4−12% gradient SDS-PAGE gel. Gels were transferred to nitrocellulose membranes. Membranes were incubated with rabbit anti-GIVA-PLA2 (1:200, Santa Cruz Biotechnology), rabbit anti-phospho-GIVA-PLA2 (1:200, Abd Serotec), rabbit anti-calbindin (1:1000, Swant) in blocking buffer for 1 h. Secondary goat anti-rabbit antibodies (Chemicon) were used at 1:10,000 dilution. Protein bands were visualized with an ECL system (Pierce) and quantified densitometrically with Image J (NIH) software.
Immunohistochemistry
Sections were stained for GIVA-PLA2, Aβ, and GFAP with an avidinbiotin/peroxidase system (Vector Laboratories). For antigen retrieval, sections were boiled in 50 mM citric acid for 15 min. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 15 min. Sections were blocked at room temperature for 1 h in 10% serum and 1% nonfat milk, and incubated overnight with a rabbit anti-GIVA-PLA2 antibody clone N-216 (1:100; Santa Cruz Biotechnology), mouse anti-Aβ1−5 (1:500, 3D6 antibody, Elan Pharmaceuticals), or rabbit anti-GFAP (1:500, Dako) at 4°C in blocking solution. Secondary biotinylated antibody was used at a dilution of 1:200 (Vector Laboratories). The chromagen was diaminobenzidine.
Biotinylation assay for AMPAR
Primary neuronal cultures were treated with Aβ or AA in artificial CSF (aCSF) or with vehicle for 10, 30, or 60 min and rinsed with ice-cold aCSF, and surface receptors were bound with EZ-link sulfo-NHS-SS-Biotin (Pierce). After rinsing with ice-cold glycine and then with ice-cold aCSF, cells were treated with immunoblotting lysis buffer (above) and scraped from the 24-well plate. Surface receptors were precipitated with avidinsepharose beads (Pierce). Samples from two wells were combined for western blot analysis with antibodies against GluR1 (1:1000, Chemicon) or GluR2 (1:1000, Chemicon)50.
Behavioral testing
To minimize effects of social and environmental stressors on behavior, mice were housed singly under conditions of controlled temperature with a standard 12 h light/12h dark cycle. Before each behavioral assessment of mice in the elevated plus maze, open field, and Y-maze tests, residues were removed and the apparatus was cleaned with ethanol-wipes to standardize odors.
Water-maze test
As described previously2, 12, mice were trained to locate first a visible platform (days 1−3) and then a hidden platform (days 4−8) in a pool (122-cm diameter) filled with opaque water (18°C). Training consisted of two daily sessions 2−3 h apart, each consisting of two (visible) or three (hidden) 60-sec trials. During hidden platform training, the platform location was kept constant (in the center of the target quadrant) and the starting point was changed between trials. For all trials, the time (latency) and path length to reach the platform and the swim speed were recorded with a Noldus Instruments EthoVision video tracking system (San Diego Instruments). On days 6, 9, and 11 (before the first trial of the day), the platform was removed for a 60-sec probe trial, during which the proportion of time spent in the different quadrants and the number of target crossings were recorded. Mice that demonstrated a thigmotactic swim pattern during training were excluded from analysis (0−2 mice per genotype).
Open field activity
To assess activity levels, mice were placed individually into well-lit automated activity cages with rows of infrared photocells on each side interfaced with a computer (San Diego Instruments). Open field activity was recorded for two consecutive 5-min periods. Beam breaks were recorded automatically and used to calculate total fine and ambulatory movements, path lengths, and number of rearings at the center and periphery of the field.
Y maze
Exploratory behavior was tested in a Y-shaped maze consisting of three identical arms, made of dark opaque polyvinyl plastic, with equal angles between each arm. For each trial, mice were placed individually into one arm and allowed to explore the maze for 6 min, during which the sequence and number of arm entries were recorded manually.
Elevated plus maze
Exploratory behavior and anxiety were assessed with an elevated, plus-shaped maze consisting of two open arms and two closed arms equipped with rows of infrared photocells interfaced with a computer (Hamilton-Kinder). In brief, mice were placed individually in the center of the maze and allowed to explore freely for 10 min. Beam breaks were recorded automatically and used to calculate the number of entries, distance traveled, and total time spent in each arm and the number of extensions over the edges of the open arms (edge pokes).
Slice preparation and electrophsyiology
Four-month-old C57/BL6J mice (n=4) were used for in vitro recordings. Mice were deeply anesthetized and decapitated. The brain was rapidly removed and placed in cold (∼4°C) oxygenated slicing solution containing (in mM): 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, 11 glucose, and 234 sucrose. A block of brain was fastened to the stage of a Vibratome-3000 (Vibratome, St. Louis, MO) with cyanoacrylate and 350 μm coronal slices were cut in slicing solution. Slices were incubated for ≥30 min at 30°C in standard aCSF containing (in mM): 2.5 KCl, 126 NaCl, 10 glucose, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, and 26 NaHCO3 (pH ∼ 7.4 when gassed with a mixture of 95% O2-5% CO2). Single slices were transferred to a submerged recording chamber where they were maintained at 30°C and perfused at a rate of ∼2 mL/min. Recordings were made from neurons identified under infrared differential interference contrast video microscopy with a Zeiss Axioskop 2 FS plus microscope. Patch pipettes (4−6 MΩ) were pulled from borosilicate glass World Precision Instrument and filled with (in mM) 5 KCl, 135 K-gluconate, 2 NaCl, 10 HEPES, 4 EGTA, 4 MgATP, and 0.3 Na3GTP (pH ∼7.3, 287 mosmol). Access resistance (RA) was monitored during recordings and neurons were discarded if RA > 15 MΩ. Drugs were applied locally via a local perfusion system (Automate Scientific).
Data analysis
For western blots, protein levels were normalized to mean levels in NTG controls. Unpaired, two-tailed t tests were used to assess differences between means for LC-MS/MS and western blot analyses and the effect of AACOCF3 on cell viability. Two-way ANOVA was used for western blot analysis of GIVA-PLA2 after Aβ treatments. One-way ANOVA with repeated measures was used for experiments assessing the dose- and time-dependent effects of Aβ and AA on cell viability. Behavioral data were assessed by two-way ANOVA with repeated measures when indicated. Tukey or Dunnett posthoc tests were used for multiple comparisons. Null hypotheses were rejected at the 0.05 level.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the ADRC at UCSF for postmortem brain tissues, T. Wu, A. Thwin and H. Solanoy for technical support, C. McCulloch for help with statistical analysis, G. Howard and S. Ordway for editorial review, J. Carroll and C. Goodfellow for preparation of graphics, and D. McPherson for administrative assistance. The study was supported by NIH Grants AG011385, AG022074, and NS041787 to L.M., AG028233 to R.S.-M, and NIH/NCRR CO6RR018928 to the J. David Gladstone Institutes.
References
- 1.Roberson ED, Mucke L. 100 years and counting: Prospects for defeating Alzheimer's disease. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palop JJ, et al. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc. Natl. Acad. Sci. USA. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 4.Menard C, Patenaude C, Massicotte G. Phosphorylation of AMPA receptor subunits is differentially regulated by phospholipase A2 inhibitors. Neurosci. Lett. 2005;389:51. doi: 10.1016/j.neulet.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res. Brain Res. Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 7.Praticò D, Uryu K, Leight S, Trojanowswki JQ, Lee VMY. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68−69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 9.Lim GP, et al. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005;25:3032–3040. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freund-Levi Y, et al. ω-3 Fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD Study: A randomized double-blind trial. Arch. Neurol. 2006;63:1402–1408. doi: 10.1001/archneur.63.10.1402. [DOI] [PubMed] [Google Scholar]
- 11.Yoshikawa K, Kita Y, Kishimoto K, Shimizu T. Profiling of eicosanoid production in the rat hippocampus during kainic acid-induced seizure: Dual phase regulation and differential involvement of COX-1 and COX-2. J. Biol. Chem. 2006;281:14663–14669. doi: 10.1074/jbc.M511089200. [DOI] [PubMed] [Google Scholar]
- 12.Roberson ED, et al. Reducing endogenous tau ameliorates amyloid ®-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 13.Palop J, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meilandt WJ, et al. Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer's disease. J. Neurosci. 2008;28:5007–5017. doi: 10.1523/JNEUROSCI.0590-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng I, et al. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 16.Hwang DY, et al. Alterations in behavior, amyloid β-42, caspase-3, and COX-2 in mutant PS2 transgenic mouse model of Alzheimer's disease. FASEB J. 2002;16:805–813. doi: 10.1096/fj.01-0732com. [DOI] [PubMed] [Google Scholar]
- 17.Williams JH, Errington ML, Lynch MA, Bliss TVP. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature. 1989;341:739–742. doi: 10.1038/341739a0. [DOI] [PubMed] [Google Scholar]
- 18.Funk CD. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 19.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 20.Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 21.Bonventre JV, et al. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 22.Stephenson D, et al. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia. 1999;27:110–128. doi: 10.1002/(sici)1098-1136(199908)27:2<110::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 23.Colangelo V, et al. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: Transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 2002;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 24.Gattaz WF, Maras A, Cairns NJ, Levy R, Forstl H. Decreased phospholipase A2 activity in Alzheimer brains. Biol. Psychol. 1995;37:13–17. doi: 10.1016/0006-3223(94)00123-K. [DOI] [PubMed] [Google Scholar]
- 25.Kishimoto K, Matsumura K, Kataoka Y, Morii H, Watanabe Y. Localization of cytosolic phospholipase A2 messenger RNA mainly in neurons in the rat brain. Neuroscience. 1999;92:1061–1077. doi: 10.1016/s0306-4522(99)00051-2. [DOI] [PubMed] [Google Scholar]
- 26.Sandhya TL, Ong WY, Horrocks LA, Farooqui AA. A light and electron microscopic study of cytoplasmic phospholipase A2 and cyclooxygenase-2 in the hippocampus after kainate lesions. Brain Res. 1998;788:223–231. doi: 10.1016/s0006-8993(97)01552-7. [DOI] [PubMed] [Google Scholar]
- 27.Malaplate-Armand C, et al. Soluble oligomers of amyloid-β peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 2006;23:178–189. doi: 10.1016/j.nbd.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Kriem B, et al. Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-β peptide. FASEB J. 2004;19:85–87. doi: 10.1096/fj.04-1807fje. [DOI] [PubMed] [Google Scholar]
- 29.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Cell. doi: 10.1016/j.tins.2008.06.005. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snyder EM, et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 31.Brorson JR, et al. The Ca ■ 2+ ■ influx induced by β-amyloid peptide 25−35 in cultured hippocampal neurons results from network excitation. J. Neurobiol. 1995;26:325–338. doi: 10.1002/neu.480260305. [DOI] [PubMed] [Google Scholar]
- 32.Hsieh H, et al. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanchard BJ, Thomas VL, Ingram VM. Mechanism of membrane depolarization caused by the Alzheimer Aβ1−42 peptide. Biochem. Biophys. Res. Commun. 2002;293:1197–1203. doi: 10.1016/S0006-291X(02)00346-7. [DOI] [PubMed] [Google Scholar]
- 34.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 35.Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- 36.Gaudreault SB, Chabot C, Gratton JP, Poirier J. The caveolin scaffolding domain modifies 2-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor binding properties by inhibiting phospholipase A2 activity. J. Biol. Chem. 2004;279:356–362. doi: 10.1074/jbc.M304777200. [DOI] [PubMed] [Google Scholar]
- 37.Carter TL, et al. Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer's disease patients according to Braak stage. Exp. Neurol. 2004;187:299–309. doi: 10.1016/j.expneurol.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 38.Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc. Natl. Acad. Sci. USA. 2007;104:353–358. doi: 10.1073/pnas.0608807104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamenetz F, et al. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 40.Prasad MR, Mark AL, Mustafa Y, Harbhajan D, William RM. Regional membrane phospholipid alterations in Alzheimer's disease. Neurochem. Res. 1998;V23:81–88. doi: 10.1023/a:1022457605436. [DOI] [PubMed] [Google Scholar]
- 41.Cutler RG, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid β-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J. Neurosci. 1997;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shankar GM, et al. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 45.Hsia A, et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc. Natl. Acad. Sci. USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 47.Brady KM, Texel SJ, Kishimoto K, Koehler RC, Sapirstein A. Cytosolic phospholipase A2 α modulates NMDA neurotoxicity in mouse hippocampal cultures. Eur. J. Neurosci. 2006;24:3381–3386. doi: 10.1111/j.1460-9568.2006.05237.x. [DOI] [PubMed] [Google Scholar]
- 48.Luria A, et al. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J. Biol. Chem. 2007;282:2891–2898. doi: 10.1074/jbc.M608057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by β-amyloid peptides in hippocampal neurons. J. Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin JW, et al. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat. Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.