

Abstract
The role of innate immunity in the pathogenesis of asthma is unclear. Although increased presence of neutrophils is associated with persistent asthma and asthma exacerbations, how neutrophils participate in the pathogenesis of asthma remains controversial. In this study, we show that the absence of dipeptidyl peptidase I (DPPI), a lysosomal cysteine protease found in neutrophils, dampens the acute inflammatory response and the subsequent mucous cell metaplasia that accompanies the asthma phenotype induced by Sendai virus infection. This attenuated phenotype is accompanied by a significant decrease in the accumulation of neutrophils and the local production of CXCL2, TNF, IL-1β, and IL-6 in the lung of infected DPPI−/− mice. Adoptive transfer of DPPI-sufficient neutrophils into DPPI−/− mice restored the levels of CXCL2 and enhanced cytokine production on day 4 postinfection and subsequent mucous cell metaplasia on day 21 postinfection. These results indicate that DPPI and neutrophils play a critical role in Sendai virus-induced asthma phenotype as a result of a DPPI-dependent neutrophil recruitment and cytokine response.
Allergic asthma is a chronic inflammatory airway disease characterized by bronchial hyperreactivity, eosinophilia, and mucous cell metaplasia. Studies of asthmatic patients and animal models have clearly implicated a role for the acquired immune response, specifically Th2 cells that secrete IL-4, IL-5, and IL-13, in the initiation of the inflammatory cascade leading to eosinophilic airway inflammation (1–7). Recently, noneosinophilic asthma subtypes, and more specifically neutrophilic asthma, have been identified (8, 9). Neutrophils are a major source of oxidative stress and protease-mediated inflammation, and a large body of evidence supports the notion that severe asthma, asthma exacerbations, and fatal attacks of asthma are associated with increased sputum neutrophils (10–13). And whereas eosinophils are sensitive to corticosteroid treatment, neutrophils respond poorly to this treatment modality and may be the underlying cause of steroid-resistant asthma (14, 15). Therefore, new treatment modalities aimed at neutrophils may be beneficial in refractory and severe forms of asthma.
Cathepsin G (CG),3 neutrophil elastase (NE), and proteinase 3 (PR3) are granule-associated serine proteases expressed specifically during the promyelocytic stage of myeloid development (16, 17). Traditionally, these serine proteases are thought to contribute mainly to the degradation of matrix components. However, neutrophil serine proteases can have a major impact on the overall level of inflammation by modulating the release of reactive oxygen species, chemokines, and cytokines (18). We have previously established that the cysteine protease dipeptidyl peptidase I (DPPI, also known as cathepsin C) plays an important and nonredundant role in the processing and activation of all three neutrophil-derived serine proteases in both humans and mice (19). In addition, we have shown that mice deficient in DPPI are resistant to Ab-mediated arthritis (19) and elastase-induced abdominal aortic aneurysm (20) due to a specific defect in neutrophil recruitment.
Paramyxoviral infection in infants has been associated with a significantly increased rate of asthma development (21–23). In addition, viruses are detected in a majority of children and adult asthmatics with acute exacerbations (24, 25). In the mouse, Walter et al. (26–28) have previously shown that a single paramyxoviral infection with the mouse parainfluenza virus type I (Sendai virus (SeV)) can cause a limited bronchiolitis, followed by persistent airway hyperreactivity and mucous cell metaplasia, thus providing an experimental link between paramyxoviral infection and subsequent asthma development. In this study, we hypothesized that neutrophils may affect the development of Sendai-induced asthma phenotype through the release of proteases. Therefore, we examined the DPPI−/− mice that are pan deficient in neutrophil serine proteases in the SeV-induced asthma model.
Materials and Methods
Animals
DPPI−/−, CG−/−, and NE−/− mice were generated in a 129/SvJ strain, as previously described (29, 30). These animals were backcrossed to C57BL/6 mice for >10 generations, using the mutant allele as a marker for selection at each generation. CG−/− and NE−/− mice were intercrossed to generate mice that are deficient in both proteases. Wild-type (WT) C57BL/6 mice were obtained from The Jackson Laboratory along with the C57BL/6-transgenic (UBC-GFP) 30 Scha/J transgenic mouse line, which systemically expresses a GFP cDNA under the control of a chicken β-actin promoter and CMV enhancer. All animals were kept in pathogen-free conditions at Washington University Specialized Research Facility, and all experiments were performed according to protocols approved by the Division of Comparative Medicine at Washington University.
Histological sections
Mouse lungs were fixed in 10% buffered formalin, embedded in paraffin, and stained with Alcian blue/periodic acid-Schiff (PAS) as an indicator of goblet cells. Quantification of PAS+ cells/mm basement membrane was conducted using ImageJ program (http://rsb.info.nih.gov/ij).
Viral infection
Male mice of 6–8 wk of age were anesthetized with ketamine and xylazine, and inoculated intranasally with 1500 50% egg infectious dose of SeV (Fushimi strain) (26) or with UV-inactivated SeV in 45 μl of PBS. Individual mice were weighed before and every other day after infection. Percentage of weight loss was calculated by the following formula: 1 − [weight/original weight] × 100. Experimental infections with SeV were performed in a biohazard containment facility.
Lung and bronchoalveolar lavage (BAL) fluid analysis
After sacrifice, the trachea was cannulated with an 18-gauge catheter, and the lungs of each mouse were lavaged three times with 1 ml of sterile PBS. The postlavage lungs were then minced and incubated with 1.4 mg/ml collagenase type I (Worthington Biomedical) at 37°C for 30 min. Single-cell suspensions were passed through a 70-μm cell strainer to remove debris, and the recovered cells and BAL samples were enumerated for total cell count and stained with anti-Gr1 Abs (neutrophils), anti-F4/80 Abs (macrophages), anti-CD19 (B cells), and anti-CD3 (T cells) and analyzed by flow cytometry. The absolute number of cells was calculated by multiplying the total number of cells from each set of lungs by the percentage of Gr1+, F4/80+, CD19+, and CD3+ cells. All Abs were obtained from BD Biosciences. SeV+ T cells were detected using tetrameric MHC-peptide reagents specific for SeV nucleoprotein NP324–332 provided by the National Institute of Allergy and Infectious Disease Tetramer Core Facility (31).
Intracellular staining
To detect intracellular IFN-γ, cells isolated from infected lungs were incubated with 50 ng/ml PMA (Sigma-Aldrich), 500 ng/ml ionomycin (Sigma-Aldrich), and 10 mg/ml brefeldin A (Sigma-Aldrich) for 4 h at 37°C. Cells were stained with anti-CD4 or anti-CD8 Abs (BD Biosciences), fixed, permeabilized, and stained with anti-IFN-γ, according to manufacturer’s recommendations (BD Biosciences).
Lung SeV titer
Clearance of SeV was assessed in lung homogenates on days 4, 12, and 20 after infection by real-time quantitative RT-PCR, as previously described (26). Briefly, mice were sacrificed on the indicated day, their lungs were harvested, and total RNA was isolated using TRIzol reagent (Invitrogen Life Technologies), as recommended by the manufacturer. Two micrograms of total RNA from each sample was used to synthesize cDNA by reverse transcription on a GeneAmp 9600 thermal cycler system, according to the manufacturer’s recommendations (Applied Biosystems). The reverse-transcription products served as the template for real-time PCR analysis using a fluorogenic probe/primer combination for SeV nucleocapsid protein RNA (nt 519–587 in GenBank accession M30202), according to the manufacturer’s protocol (Applied Biosystems). Results for each sample were normalized to the concentration of GAPDH mRNA measured in the same samples. The copy number of virus-specific RNA in experimental samples was calculated based on a construct standard curve, as previously described (26).
Cytokine ELISA
CXCL1, CXCL2, CXCL15, LPS-induced CXC chemokine (LIX), IL-1β, TNF, and IL-6 levels in BAL fluid were quantified by ELISA (R&D Systems) and conducted as described in the manufacturer’s recommendations.
Adoptive transfer of neutrophils
Neutrophils were isolated from bone marrow by a discontinuous Percoll gradient, as previously described (30). Briefly, the marrow was flushed from bones with HBSS containing 0.1% BSA, pelleted, and resuspended in 3 ml of 45% Percoll (GE Healthcare Bio-Sciences). Solutions of 66, 60, 55, and 50% were prepared by diluting the 100% stock solution of Percoll with HBSS. Commencing with 3 ml of the 66% solution, 2-ml aliquots of each increasing concentration of Percoll solution were carefully layered over one another in a 15-ml conical tube. The bone marrow single-cell suspension in 45% Percoll solution was subsequently layered over the prepared Percoll density gradient, followed by centrifugation at 1800 × g for 30 min at room temperature. Cells were collected from the 66–60% interface, washed with HBSS/0.1% BSA, and layered a second time over another prepared Percoll gradient. Neutrophil purity after two Percoll gradients was consistently 90–95%, as assessed by flow cytometry. The other cells found at this interface include a small percentage of nucleated RBC and B cells (20). Isolated neutrophils were resuspended in PBS (1 × 107 neutrophils in 200 μl of PBS) and injected i.v. into mice on days 1, 2, and 3 following SeV infection.
Statistical analysis
Values for weight losses, viral titers, BAL fluid cell counts, and chemokine/cytokine levels were analyzed using unpaired Student’s t test. Value of p < 0.05 was considered significant.
Results
SeV-induced illness is significantly attenuated in the absence of DPPI
To examine the role of DPPI in SeV infection, WT C57BL/6 and DPPI−/− mice were infected intranasally with 1500 50% egg infectious dose of SeV (26) or UV-inactivated SeV (UV-SeV), and disease manifestation was monitored over time. SeV infection in mice induces an acute and inflammatory response accompanied by weight loss. Although the weight loss resolves by day 21, the mice are left with mucous cell metaplasia and airway hyperreactivity (26). As expected, WT mice exhibited severe weight loss following SeV infection (26). In contrast, DPPI−/− mice experienced significantly less weight loss after intranasal administration of SeV (Fig. 1A). To examine whether DPPI−/− mice were also protected against mucous hyperplasia after the acute inflammatory phase, lungs were harvested on day 21 postinfection (PI) for histology. In WT mice infected with SeV, there was a persistence of peribronchial inflammation accompanied by a significant increase in the number of PAS+ cells compared with UV-SeV-infected WT mice (Fig. 1, B and D). In contrast, these changes were greatly diminished in DPPI−/− mice (Fig. 1C). Quantification of these PAS+ cells confirmed that SeV-infected DPPI−/− mice developed significantly less mucous cell metaplasia on day 21 PI (Fig. 1E). These results suggest that the absence of DPPI reduces the acute illness severity and related airway changes after SeV infection.
FIGURE 1.
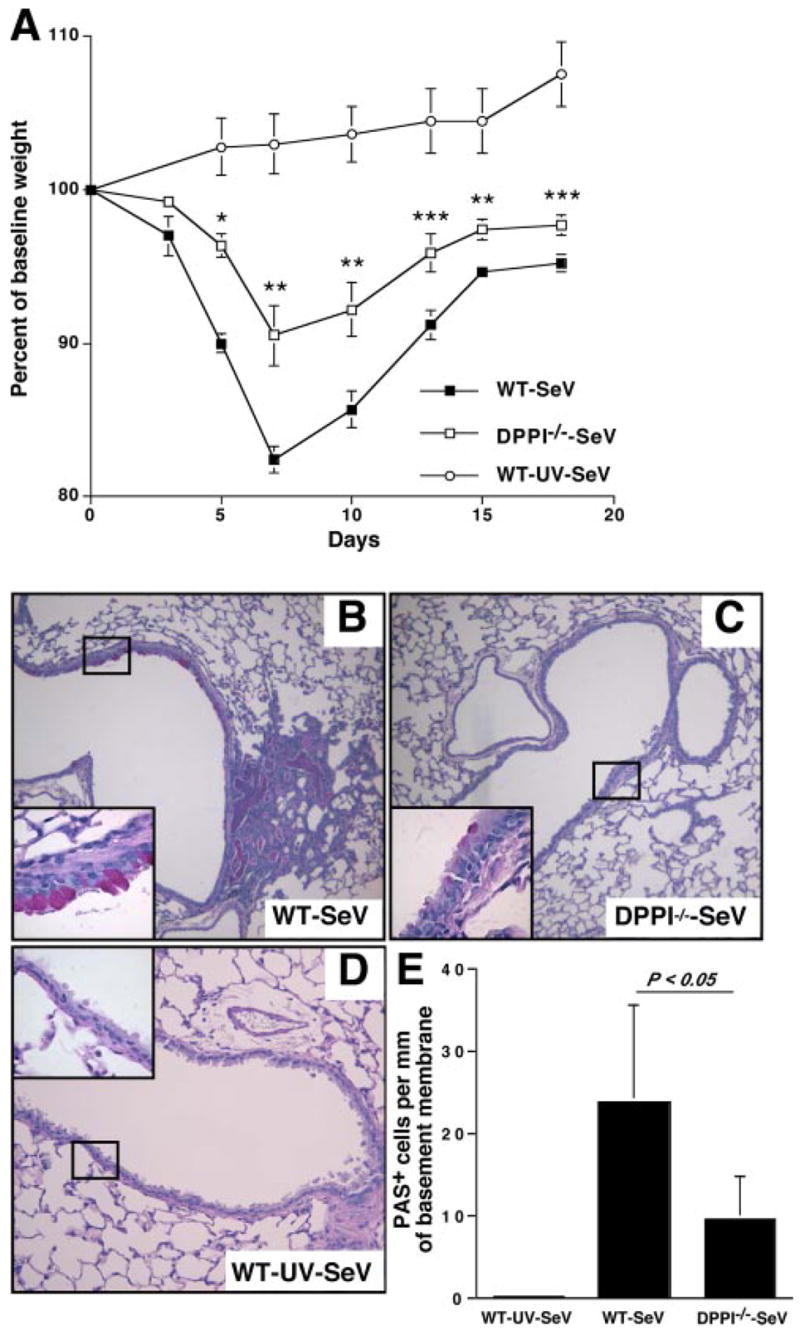
Absence of DPPI protects against SeV-induced severe weight loss and mucous cell hyperplasia. A, WT and DPPI−/− mice were infected with SeV, and their weight was followed over time. Data are presented as weight change from pretreatment baseline weight. Values represent mean ± SEM of five mice per condition from three independent experiments. Mice infected with UV-SeV did not lose weight. **, p < 0.001; **, p < 0.01; ***, p < 0.02. Persistence of peribronchial inflammation and mucous cell metaplasia on day 21 PI. Lung sections were stained with PAS, and representative photomicrographs are shown for SeV-infected WT mice (B), SeV-infected DPPI−/− mice (C), and UV-SeV-infected WT mice (D). Insets, Represent higher magnification of boxed areas. E, Quantification of results shown in B and C using values for PAS+ cells/mm basement membrane. Values represent mean ± SEM (n = 8–9 airways from 3 to 4 mice per condition).
Absence of DPPI delays viral clearance, but does not compromise cytotoxic T lymphocyte response following SeV infection
To determine whether the absence of DPPI affects viral clearance, mice were infected with SeV and viral titers determined at various time points PI. SeV titers were measured in lung homogenates obtained on days 0, 4, 12, and 20 PI using quantitative real-time RT-PCR (26). DPPI−/− mice harbored significantly higher viral titers on days 4 and 12 PI (Fig. 2A). Although viral titers were higher at the peak of infection, DPPI−/− mice eventually cleared their viral load by day 20 PI (Fig. 2A). In addition, the number of CD8+ T cells and the frequency of virus-specific CD8+ T cells in DPPI−/− mice were comparable to WT animals at day 8 PI (Fig. 2B). Intracellular staining for IFN-γ in cells isolated from lungs of infected mice on day 8 PI revealed that DPPI−/− mice were not inhibited in their ability to produce IFN-γ (Fig. 3B). These results suggest that the absence of DPPI slightly delays viral clearance, but preserves cytotoxic T cell response to SeV infection.
FIGURE 2.
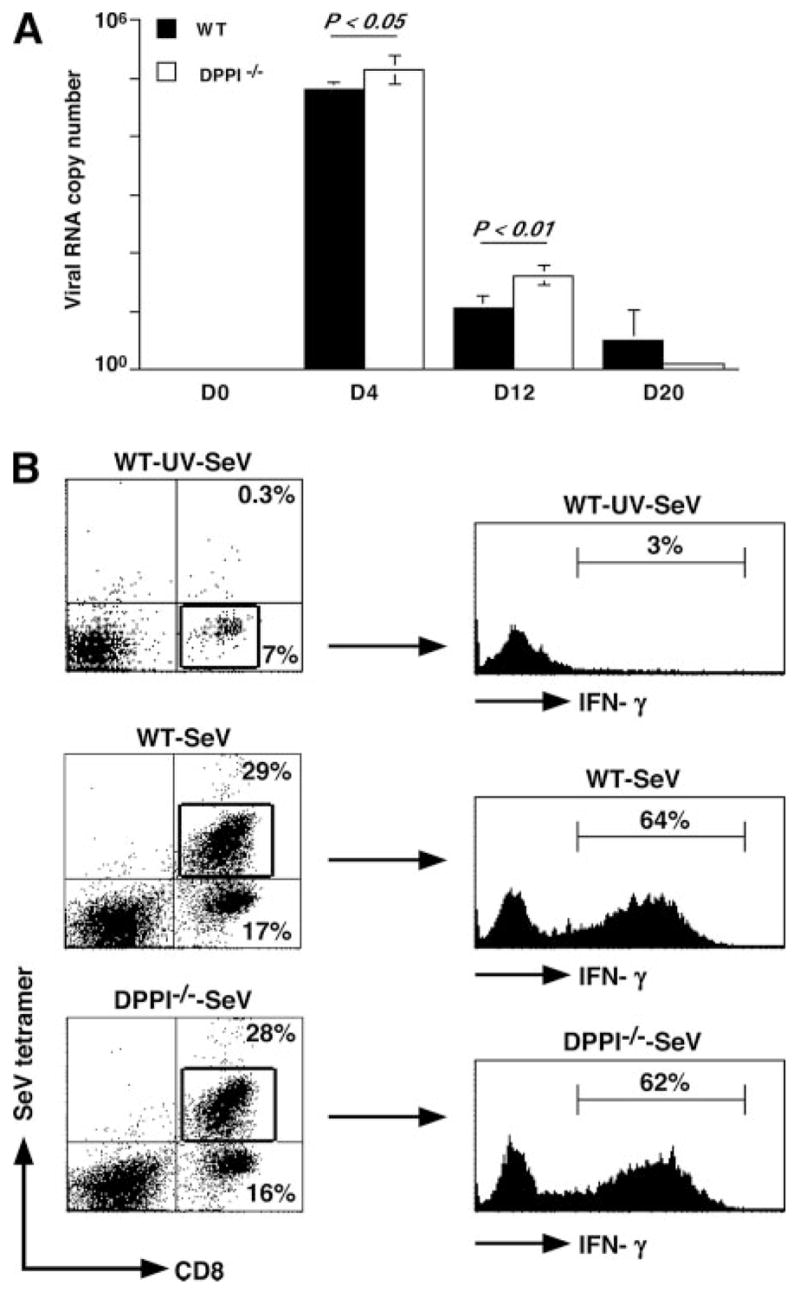
Absence of DPPI delays viral clearance, but does not alter cytotoxic T cell response. A, Lungs were harvested on the indicated days after SeV infection, and viral RNA copy number was determined by real-time RT-PCR for SeV nucleocapsid protein and corrected for GAPDH control. Values represent mean ± SEM of four to five mice per genotype per time point (B). Cells isolated from post-BAL lung homogenates on day 8 PI were surface stained with SeV tetramers and anti-CD8 Abs, and their IFN-γ level was determined by intracellular staining. Note that there was a significant increase in the total number of CD8+ T cells recovered from SeV-infected lungs on day 8 PI compared with control mice (WT-SeV-UV mice = 0.15 ± 0.04 × 106 cells), but no difference was detected between WT (1.58 ± 0.21 × 106 cells) and DPPI−/− mice (1.71 ± 0.51 × 106 cells). Values for CD8+ T cells represent mean ± SEM derived from n = 3–4 mice per condition.
FIGURE 3.
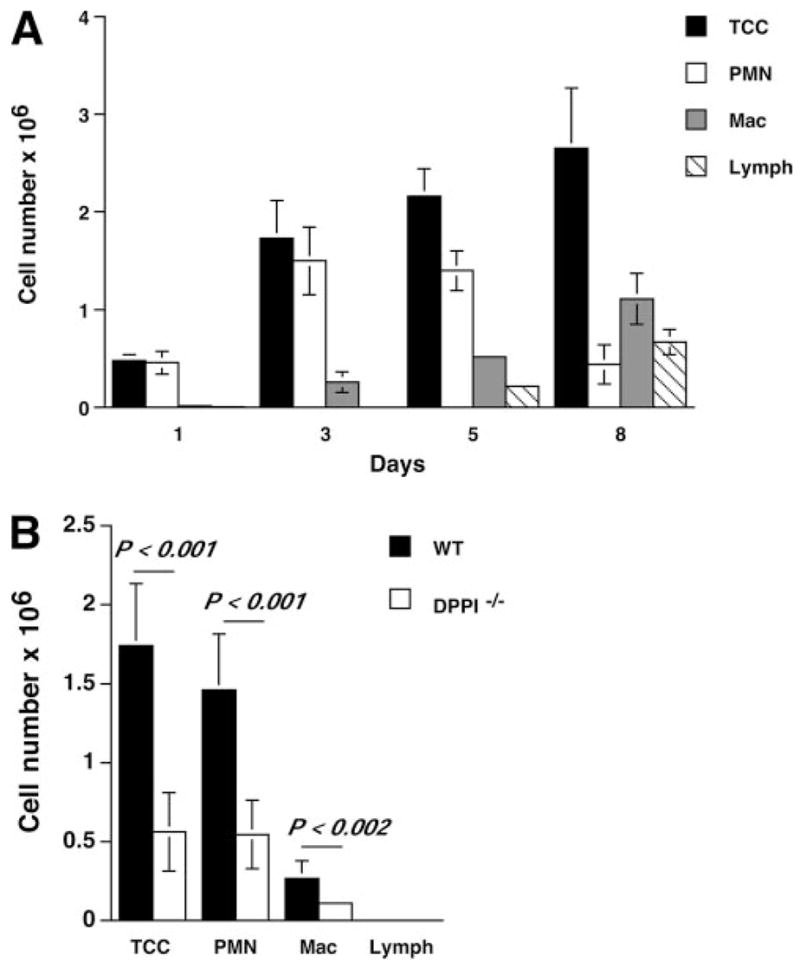
Leukocyte recruitment to the alveolar space is defective in DPPI−/− mice after SeV infection. A, Groups of WT mice were infected with SeV, and their BAL fluid was subjected to total and differential cell counts. B, BAL fluid differential cell counts on SeV PI day 3. The absolute number of each cell type was calculated by multiplying the total number of cells from each lung by the percentage of positive staining cells. Values represent mean ± SEM of at least 10 mice per genotype. TCC, total cell count; PMN, neutrophil; Mac, macrophage; Lymph, lymphocyte.
DPPI regulates neutrophil recruitment following SeV infection
To understand how DPPI modulates the development of immune response to SeV, we examined leukocyte trafficking to the lung and alveolar space during the acute phase of infection. We found that SeV infection led to a predominantly neutrophilic influx into the alveolar space in the early phase of the disease (Fig. 3A). Compared with those obtained from WT animals, BAL samples obtained on day 3 PI from DPPI−/− mice showed significantly reduced total cell numbers, primarily due to a striking decrease in the number of neutrophils (Fig. 3B). In addition, there was also a significant decrease in the number of neutrophils recovered from postlavage lung homogenates of DPPI−/− mice obtained on day 3 PI (data not shown). These results confirm a critical role for DPPI in the early pulmonary recruitment of leukocytes, specifically neutrophils, in response to SeV infection.
The release of chemokines and cytokines into the alveolar space following SeV infection is dampened in the absence of DPPI
A number of studies have shown that respiratory viral infections induce expression of several chemokines that may influence the trafficking of inflammatory leukocytes to the lung and the subsequent cell-mediated immune response (32, 33). We therefore examined whether the alveolar levels of several leukocyte-attracting CXC chemokines, including CXCL1 (also known as KC), CXCL2 (also known as MIP-2), LIX (a murine homologue of human CXCL5/ENA-78), and CXCL15 (also known as lungkine), were altered in the absence of DPPI. All four CXC chemokine levels were elevated in the BAL samples on day 3 PI (Fig. 4). However, CXCL2 levels in the BAL samples of DPPI−/− mice were significantly reduced compared with WT BAL samples (Fig. 4D), whereas the levels of CXCL1, LIX, and CXCL15 were unaltered (Fig. 4, A–C). These results indicate that CXC chemokines may have nonoverlapping roles in regulating leukocyte influx during SeV infection.
FIGURE 4.

Dampened chemokine/cytokine release in DPPI−/− mice following SeV infection. At day 3 PI, BAL fluid was obtained from each group of mice and analyzed for chemokines CXCL1 (A), LIX (B), CXCL15 (C), and CXCL2 (D), and cytokine IL-1β (F), TNF (E), and IL-6 (G) levels. Values represent mean ± SEM of four to five mice per genotype for LIX and CXCL15, and 11–12 mice per genotype for others.
Previous reports have also shown that the temporal production of antiviral cytokine production correlates with viral illnesses (34–36) and that IL-1β and TNF-α are both involved in inducing the severity of the illness and the lung immunopathology (37, 38). Therefore, we measured the levels of IL-1β and TNF released into the alveolar space on day 3 PI. We found that the levels of IL-1β, TNF, along with IL-6 were significantly reduced in DPPI−/− BAL fluid samples (Fig. 4, E–G), suggesting the possibility that dampened cytokine release protected the mice against the severe SeV-induced weight loss and inflammation.
Neutrophils modulate chemokine and cytokine levels in the alveolar space following SeV infection
If a defect in neutrophil recruitment protects DPPI−/− mice against severe SeV-induced lung inflammation, then adoptive transfer of WT neutrophils should restore the inflammatory response. To test this hypothesis, we adoptively transferred WT neutrophils into DPPI−/− mice on days 1, 2, and 3 PI and examined their BAL fluid for cell count, CXCL2, and cytokine levels on day 4 PI. Transfer of WT neutrophils into SeV-infected DPPI−/− mice led to a significant increase in the BAL fluid total cell and neutrophil counts (Fig. 5A), whereas transfer of DPPI−/− neutrophils into DPPI−/− mice did not alter their BAL fluid cell counts. The increase in BAL fluid total cell count also reflected a significantly higher number of macrophages recruited to the alveolar space following WT neutrophil transfer (Fig. 5A), suggesting that WT neutrophils may also provide the signal for the recruitment of other cell types. Because macrophages are a prominent source of chemokines and cytokines, their presence may contribute to the mucous cell metaplasia found on day 21 PI. To further determine whether transferred neutrophils localized to the alveolar space, we used WT GFP-tagged (GFP+) neutrophils for the adoptive transfer. We found that a small, but distinct percentage (4.3%) of the neutrophils recovered from the BAL fluid of reconstituted mice was GFP+ (Fig. 5B). Therefore, it is likely that the presence of these WT neutrophils was sufficient to restore the level of CXCL2 in DPPI−/− lungs toward that of WT animals (Fig. 6A), which then provided the necessary chemotactic gradient to recruit more endogenous DPPI−/− leukocytes to the alveolar space.
FIGURE 5.
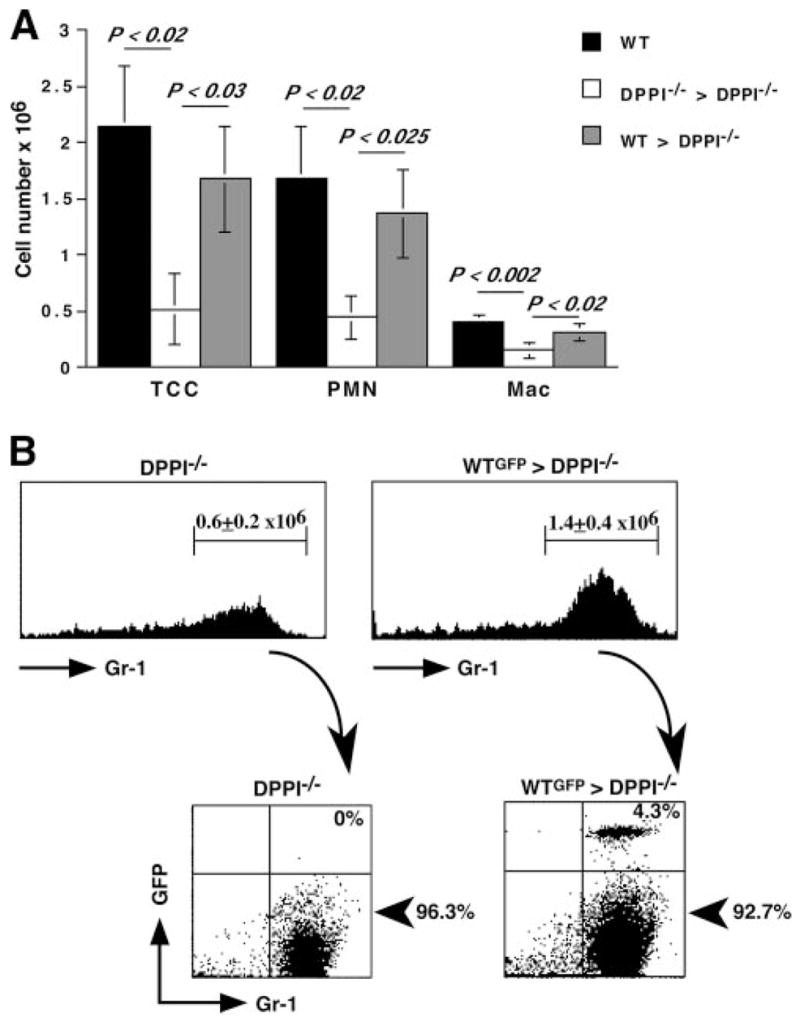
Leukocyte recruitment into the alveolar space is dependent on WT neutrophils. A, Mice were infected with SeV on day 0, followed by i.v. transfer of 107 purified WT or DPPI−/− neutrophils on days 1, 2, and 3 PI. On day 4 PI, mice were sacrificed and their lungs were lavaged. Adoptive transfer of WT neutrophils into DPPI−/− mice after SeV infection led to an absolute increase in the number of cells recruited to the alveolar space. In contrast, transfer of DPPI−/− neutrophils failed to increase the number of cells localizing to the alveolar space. Values represent mean ± SEM (n = 4–5 mice per condition). B, To confirm that adoptively transferred neutrophils localized to the lungs, GFP+ WT neutrophils were purified and adoptively transferred into DPPI−/− mice on days 1, 2, and 3 PI. Twenty-four hours after the last transfer, mice were sacrificed and the cells recovered from their BAL fluid were analyzed by flow cytometry. Transfer of GFP+ WT neutrophils into DPPI−/− mice led to an increase in the total number of neutrophils that localized to the alveolar space, as revealed by flow cytometric analysis of Gr1-positive cells. Of these, only a small percentage of cells was from the donor pool, as revealed by the presence of GFP+ GR1+ cells. Representative flow cytometry plots are shown. The absolute number of neutrophils was calculated by multiplying the total number of cells obtained from the BAL fluid by the percentage of GR1+ cells. Values represent mean ± SEM of four mice per condition. TCC, total cell count; PMN, neutrophil.
FIGURE 6.
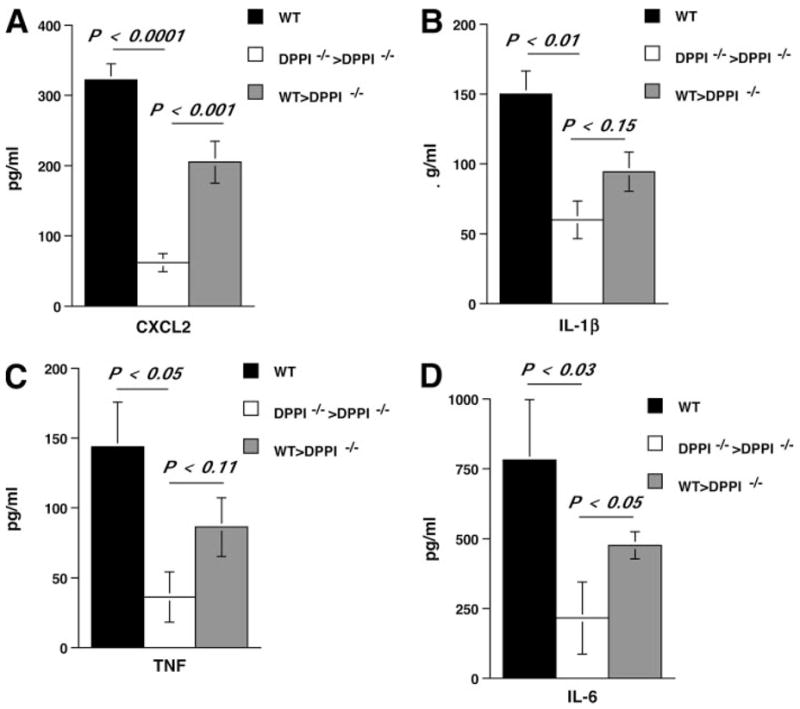
Adoptive transfer of WT neutrophils restores CXCL2 levels. Mice were infected on day 0 and received 107 purified WT or DPPI−/− neutrophils i.v. on days 1, 2, and 3 PI. On day 4 PI, the mice were sacrificed and their lungs were lavaged. Adoptive transfer of WT neutrophils, but not DPPI−/− neutrophils, led to a significant increase in the levels of CXCL2 (A) and enhanced the levels of IL-1β (B), TNF (C), and IL-6 (D) in the alveolar space. Values represent mean ± SEM of three to four mice per condition.
Transfer of WT neutrophils also increased the levels of IL-1β, TNF, and IL-6 (Fig. 6, B–D) in DPPI−/− BAL samples; however, the differences did not reach statistical significance for IL-1β and TNF. In contrast, transfer of DPPI−/− neutrophils into SeV-infected DPPI−/− mice did not alter the levels of chemokines or cytokines. Taken together, these results are consistent with a role for neutrophils and DPPI in modulating CXCL2 and cytokine release in the inflammatory response associated with an acute paramyxoviral infection.
DPPI-sufficient neutrophils contribute to mucous cell metaplasia post-SeV infection
To understand whether adoptive transfer of DPPI-sufficient neutrophils also restores the SeV-induced airway changes, DPPI−/− mice were infected with SeV, followed by adoptive transfer of WT or DPPI−/− neutrophils on days 1, 2, and 3 PI. The mice were allowed to recover from their illness, and on day 21 PI the animals were sacrificed and their lungs were harvested and analyzed. We noted a trend toward higher number of PAS+ cells in DPPI−/− mice that received WT neutrophils (Fig. 7A) compared with mice that received DPPI−/− neutrophils (Fig. 7B). However, this difference did not reach statistical significance (Fig. 7C). Nonetheless, the higher number of PAS+ cells in mice that receive WT neutrophils suggests that neutrophils recruited to the lung following SeV infection most likely contribute to mucous cell metaplasia.
FIGURE 7.

WT neutrophils contribute to mucous cell metaplasia post-SeV infection. DPPI−/− mice were infected with SeV on day 0, followed by i.v. transfer of 107 WT or DPPI−/− neutrophils on days 1, 2, and 3 PI. On day 21 PI, mice were sacrificed and their lungs were harvested for analysis. Lung sections from DPPI−/− mice that received WT neutrophils showed an increase in the number of PAS+ cells (A) compared with mice that received DPPI−/− neutrophils (B), although this increase did not reach statistical significance (C). Values represent mean ± SEM of three mice per condition.
The absence of CG and NE does not protect mice from the acute lung immunopathology following SeV infection
Because DPPI is critical for the activation of neutrophil serine proteases CG and NE, we asked whether these proteases are also required for leukocyte recruitment and chemokine/cytokine release following SeV infection. Previous studies have shown that these proteases are important in collagen-induced arthritis and neutrophil recruitment in s.c. air pouch model (19, 30). Surprisingly, we found that mice deficient in both CG and NE were just as susceptible to SeV-induced illness and weight loss as WT mice (Fig. 8A). Moreover, analysis of BAL fluid revealed comparable cell counts, CXCL2, IL-1β, TNF, and IL-6 (Fig. 8, B–F) levels between WT and serine protease-deficient mice. These results indicate that although neutrophils and DPPI play an important role in the early immune response to SeV infection, CG and NE are dispensable.
FIGURE 8.

Absence of CG and NE does not protect mice against SeV-induced weight loss and proinflammatory chemokine/cytokine release. Mice deficient in both CG and NE (CGNE) had the same weight loss curve as WT mice infected with SeV (A) and released the same levels of chemokines and cytokines into the alveolar space on day 3 PI (B–F). Values represent mean ± SEM of four mice per genotype. TCC, total cell count; PMN, neutrophil; Mac, macrophage; Lymph, lymphocyte.
Discussion
Although increased presence of neutrophils is associated with persistent asthma and asthma exacerbations, how neutrophils participate in the pathogenesis of asthma remains controversial. Neutrophils represent a rich source of proteases, and the release of these enzymes from activated neutrophils has been implicated in the tissue damage that accompanies airway inflammation and remodeling (39). In addition, neutrophils are implicated in the production of proinflammatory chemokines and cytokines that recruit additional leukocytes and perpetuate the inflammatory cycle (18, 40). Hence, neutrophils and neutrophil-derived mediators may be partially responsible for the persistence of inflammation and tissue injury in chronic airway diseases. In these studies, we showed that neutrophil-associated DPPI exacerbates asthmatic phenotype by amplifying the production of chemokines and cytokines, whereas the absence of DPPI protects mice against severe airway inflammation without interfering with their ability to clear virus.
SeV infection leads to the induction of an inflammatory infiltrate that comprises mainly of neutrophils in the early phase, followed by recruitment of monocytes and lymphocytes in the later phase of the disease (26). Although neutrophils are traditionally viewed as the first line of defense against bacterial infections, they can be involved in virally induced pathology (41, 42). In this study, we demonstrated that neutrophils were the first cell type recruited to the alveolar space in response to SeV infection. In the absence of DPPI, there was a significant decrease in neutrophil influx into the lungs, and this was accompanied by a decrease in CXCL2, TNF, IL-1β, and IL-6 levels. Yet, despite a reduction in the early inflammatory response to SeV infection, mortality and viral clearance were not significantly altered in the absence of DPPI. We further demonstrated that adoptive transfer of DPPI-sufficient neutrophils into DPPI−/− mice restored the level of CXCL2 in the lungs and enhanced the levels of other proinflammatory cytokines. In addition, adoptive transfer of DPPI-sufficient neutrophils led to an increase in the number of PAS+ cells, indicating that neutrophils are most likely responsible for the postviral mucous cell metaplasia. However, because a small number of B cells were also found in the neutrophil preparations, we cannot completely rule out that DPPI-sufficient B cells may contribute to the increase in CXCL2 level or the mucous cell hyperplasia following adoptive transfer. In summary, blocking DPPI-dependent cell functions in the early phase of infection may ameliorate the subsequent asthma phenotype without compromising the host’s defense ability.
Recruitment of neutrophils to sites of inflammation requires a series of coordinated events, including rolling, adhesion, diapedesis, and transmigration. Because neutrophils circulating in the blood make contact with activated endothelium, multiple stimuli may cause them to become activated, slow down, and sense the local environment (43). The presence of chemokines in this environment causes neutrophils to adhere and migrate across the endothelium following a chemotactic gradient (44). In this study, we showed that in vivo DPPI-sufficient neutrophils recruited to the lungs in the early phase of SeV infection amplified the inflammatory response by enhancing the release of CXCL2 into the alveolar space and perpetuating leukocyte recruitment. CXCL2 is the mouse homologue of human IL-8, a potent neutrophil chemoattractant that is consistently associated with neutrophilic inflammation during asthma exacerbation (45, 46). IL-8 antagonists have been developed; however, in early clinical trials, blockade of IL-8 by itself has no significant effect on inflammation (47). Thus, blockade of neutrophil-derived proteases, such as DPPI, may present an alternative way to reduce chemoattractant levels without compromising the host defense.
Neutrophil granules contain a family of serine proteases that are activated by DPPI and are physiologically active upon secretion (18). We have previously shown that extracellular, membrane-bound neutrophil serine proteases can modulate neutrophil effector functions, including CXCL2 release (30), and mice deficient in CG and NE are protected against Ab-mediated inflammatory arthritis (19). Thus, we were surprised to find that mice deficient in both CG and NE had normal chemokine and cytokine release into the alveolar space following SeV infection. However, it has been shown that the closely related serine protease PR3 directly activates protease-activated receptor 2 on human fibroblasts and epithelial cells, resulting in the release of proinflammatory chemokines and cytokines (48, 49). Protease-activated receptor 2 activation stimulates the production of multiple proinflammatory mediators, including IL-6 and IL-8 (50). In addition, PR3 has been implicated in the alternative processing of TNF and IL-1β, independent of TNF-converting enzyme and caspase 1 (51). Therefore, these alternative pathways may explain why DPPI-deficient mice are relatively protected against SeV infection, whereas mice deficient in CG and NE are not. Our laboratory is currently generating the PR3-deficient mice to test this hypothesis.
In summary, neutrophils appear to play a critical role in SeV-induced asthma phenotype as a result of a DPPI-dependent neutrophil recruitment and cytokine response. The process of neutrophil recruitment and activation is largely redundant. Therefore, targeting a specific mediator is not likely to succeed. Additionally, a neutrophil-depleting strategy may put patients at an unacceptably high risk of infection. In this study, we have identified DPPI as a single important molecule involved in the recruitment of neutrophils following a respiratory viral infection, without being critical for the clearance of virus. And the absence of DPPI diminishes chronic asthma phenotype. Therefore, the possibility of delivering a DPPI inhibitor directly to the lung may be effective in limiting airway inflammation precipitated by respiratory viral infections while minimizing systemic side effects.
Acknowledgments
We thank Ying Hu and Michelle Rohlfing for their technical expertise.
Footnotes
This work was supported by grants from the Sandler Program for Asthma Research and the National Institutes of Health (National Institute of Allergy and Infectious Diseases and National Heart, Lung, and Blood Institute).
Abbreviations used in this paper: CG, cathepsin G; BAL, bronchoalveolar lavage; DPPI, dipeptidyl peptidase I; LIX, LPS-induced CXC chemokine; NE, neutrophil elastase; PAS, periodic acid-Schiff; PI, postinfection; PR3, proteinase 3; SeV, Sendai virus; WT, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Durham SR, Ying S, Kimmitt P, Barkans J, Assoufi B, Pfister R, Menz G, Robinson DS, Kay AB, Corrigan CJ. IL-4 and IL-5 mRNA and protein in bronchial biopsies from patients with atopic and nonatopic asthma: evidence against “intrinsic” asthma being a distinct immunopathologic entity. Am J Respir Crit Care Med. 1996;154:1497–1504. doi: 10.1164/ajrccm.154.5.8912771. [DOI] [PubMed] [Google Scholar]
- 3.Temann UA, Prasad B, Gallup MW, Basbaum C, Ho SB, Flavell RA, Rankin JA. A novel role for murine IL-4 in vivo: induction of MUC5AC gene expression and mucin hypersecretion. Am J Respir Cell Mol Biol. 1997;16:471–478. doi: 10.1165/ajrcmb.16.4.9115759. [DOI] [PubMed] [Google Scholar]
- 4.Webb DC, McKenzie AN, Koskinen AM, Yang M, Mattes J, Foster PS. Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J Immunol. 2000;165:108–113. doi: 10.4049/jimmunol.165.1.108. [DOI] [PubMed] [Google Scholar]
- 5.Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. 1998;188:1307–1320. doi: 10.1084/jem.188.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mattes J, Yang M, Mahalingam S, Kuehr J, Webb DC, Simson L, Hogan SP, Koskinen A, McKenzie AN, Dent LA, et al. Intrinsic defect in T cell production of interleukin (IL)-13 in the absence of both IL-5 and eotaxin precludes the development of eosinophilia and airways hyperreactivity in experimental asthma. J Exp Med. 2002;195:1433–1444. doi: 10.1084/jem.20020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whittaker L, Niu N, Temann UA, Stoddard A, Flavell RA, Ray A, Homer RJ, Cohn L. Interleukin-13 mediates a fundamental pathway for airway epithelial mucus induced by CD4 T cells and interleukin-9. Am J Respir Cell Mol Biol. 2002;27:593–602. doi: 10.1165/rcmb.4838. [DOI] [PubMed] [Google Scholar]
- 8.Pavord ID, Brightling CE, Woltmann G, Wardlaw AJ. Non-eosinophilic corticosteroid unresponsive asthma. Lancet. 1999;353:2213–2214. doi: 10.1016/S0140-6736(99)01813-9. [DOI] [PubMed] [Google Scholar]
- 9.Douwes J, Gibson P, Pekkanen J, Pearce N. Non-eosinophilic asthma: importance and possible mechanisms. Thorax. 2002;57:643–648. doi: 10.1136/thorax.57.7.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll N, Carello S, Cooke C, James A. Airway structure and inflammatory cells in fatal attacks of asthma. Eur Respir J. 1996;9:709–715. doi: 10.1183/09031936.96.09040709. [DOI] [PubMed] [Google Scholar]
- 11.Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med. 1999;160:1532–1539. doi: 10.1164/ajrccm.160.5.9806170. [DOI] [PubMed] [Google Scholar]
- 12.Ordonez CL, Shaughnessy TE, Matthay MA, Fahy JV. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: clinical and biologic significance. Am J Respir Crit Care Med. 2000;161:1185–1190. doi: 10.1164/ajrccm.161.4.9812061. [DOI] [PubMed] [Google Scholar]
- 13.Kamath AV, I, Pavord D, Ruparelia PR, Chilvers ER. Is the neutrophil the key effector cell in severe asthma? Thorax. 2005;60:529–530. doi: 10.1136/thx.2005.043182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keatings VM, Jatakanon A, Worsdell YM, Barnes PJ. Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPD. Am J Respir Crit Care Med. 1997;155:542–548. doi: 10.1164/ajrccm.155.2.9032192. [DOI] [PubMed] [Google Scholar]
- 15.Tillie-Leblond I, Gosset P, Tonnel AB. Inflammatory events in severe acute asthma. Allergy. 2005;60:23–29. doi: 10.1111/j.1398-9995.2005.00632.x. [DOI] [PubMed] [Google Scholar]
- 16.Hanson RD, Connolly NL, Burnett D, Campbell EJ, Senior RM, Ley TJ. Developmental regulation of the human cathepsin G gene in myelomonocytic cells. J Biol Chem. 1990;265:1524–1530. [PubMed] [Google Scholar]
- 17.Zimmer M, Medcalf RL, Fink TM, Mattmann C, Lichter P, Jenne DE. Three human elastase-like genes coordinately expressed in the myelomonocyte lineage are organized as a single genetic locus on 19pter. Proc Natl Acad Sci USA. 1992;89:8215–8219. doi: 10.1073/pnas.89.17.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 19.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagano MB, Bartoli MA, Ennis TL, Mao D, Simmons PM, Thompson RW, Pham CT. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci USA. 2007;104:2855–2860. doi: 10.1073/pnas.0606091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. 2000;161:1501–1507. doi: 10.1164/ajrccm.161.5.9906076. [DOI] [PubMed] [Google Scholar]
- 22.Sigurs N. Epidemiologic and clinical evidence of a respiratory syncytial virus-reactive airway disease link. Am J Respir Crit Care Med. 2001;163:S2–S6. doi: 10.1164/ajrccm.163.supplement_1.2011109. [DOI] [PubMed] [Google Scholar]
- 23.Sigurs N. Clinical perspectives on the association between respiratory syncytial virus and reactive airway disease. Respir Res. 2002;3(Suppl 1):S8–S14. doi: 10.1186/rr186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnston SL. Mechanisms of asthma exacerbation. Clin Exp Allergy. 1998;28(Suppl 5):181–186. doi: 10.1046/j.1365-2222.1998.028s5181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan WC, Xiang X, Qiu D, Ng TP, Lam SF, Hegele RG. Epidemiology of respiratory viruses in patients hospitalized with near-fatal asthma, acute exacerbations of asthma, or chronic obstructive pulmonary disease. Am J Med. 2003;115:272–277. doi: 10.1016/s0002-9343(03)00353-x. [DOI] [PubMed] [Google Scholar]
- 26.Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest. 2002;110:165–175. doi: 10.1172/JCI14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyner JW, Uchida O, Kajiwara N, Kim EY, Patel AC, O’Sullivan MP, Walter MJ, Schwendener RA, Cook DN, Danoff TM, Holtzman MJ. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med. 2005;11:1180–1187. doi: 10.1038/nm1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grayson MH, Ramos MS, Rohlfing MM, Kitchens R, Wang HD, Gould A, Agapov E, Holtzman MJ. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol. 2007;179:1438–1448. doi: 10.4049/jimmunol.179.3.1438. [DOI] [PubMed] [Google Scholar]
- 29.Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci USA. 1999;96:8627–8632. doi: 10.1073/pnas.96.15.8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raptis SZ, Shapiro SD, Simmons PM, Cheng AM, Pham CT. Serine protease cathepsin G regulates adhesion-dependent neutrophil effector functions by modulating integrin clustering. Immunity. 2005;22:679–691. doi: 10.1016/j.immuni.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683–691. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 32.Wareing MD, Lyon AB, Lu B, Gerard C, Sarawar SR. Chemokine expression during the development and resolution of a pulmonary leukocyte response to influenza A virus infection in mice. J Leukocyte Biol. 2004;76:886–895. doi: 10.1189/jlb.1203644. [DOI] [PubMed] [Google Scholar]
- 33.Schaller M, Hogaboam CM, Lukacs N, Kunkel SL. Respiratory viral infections drive chemokine expression and exacerbate the asthmatic response. J Allergy Clin Immunol. 2006;118:295–302. doi: 10.1016/j.jaci.2006.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Reeth K. Cytokines in the pathogenesis of influenza. Vet Microbiol. 2000;74:109–116. doi: 10.1016/s0378-1135(00)00171-1. [DOI] [PubMed] [Google Scholar]
- 35.Jafri HS, Chavez-Bueno S, Mejias A, Gomez AM, Rios AM, Nassi SS, Yusuf M, Kapur P, Hardy RD, Hatfield J, et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis. 2004;189:1856–1865. doi: 10.1086/386372. [DOI] [PubMed] [Google Scholar]
- 36.Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, Sambhara S, Tumpey TM, Katz JM. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. 2007;81:2736–2744. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol. 2005;79:6441–6448. doi: 10.1128/JVI.79.10.6441-6448.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sedgwick JB, Menon I, Gern JE, Busse WW. Effects of inflammatory cytokines on the permeability of human lung microvascular endothelial cell monolayers and differential eosinophil transmigration. J Allergy Clin Immunol. 2002;110:752–756. doi: 10.1067/mai.2002.128581. [DOI] [PubMed] [Google Scholar]
- 39.Gualano RC, Vlahos R, Anderson GP. What is the contribution of respiratory viruses and lung proteases to airway remodelling in asthma and chronic obstructive pulmonary disease? Pulm Pharmacol Ther. 2006;19:18–23. doi: 10.1016/j.pupt.2005.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eyles JL, Roberts AW, Metcalf D, Wicks IP. Granulocyte colony-stimulating factor and neutrophils: forgotten mediators of inflammatory disease. Nat Clin Pract Rheumatol. 2006;2:500–510. doi: 10.1038/ncprheum0291. [DOI] [PubMed] [Google Scholar]
- 41.Smith PK, Wang SZ, Dowling KD, Forsyth KD. Leukocyte populations in respiratory syncytial virus-induced bronchiolitis. J Paediatr Child Health. 2001;37:146–151. doi: 10.1046/j.1440-1754.2001.00618.x. [DOI] [PubMed] [Google Scholar]
- 42.Bataki EL, Evans GS, Everard ML. Respiratory syncytial virus and neutrophil activation. Clin Exp Immunol. 2005;140:470–477. doi: 10.1111/j.1365-2249.2005.02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 44.Burg ND, Pillinger MH. The neutrophil: function and regulation in innate and humoral immunity. Clin Immunol. 2001;99:7–17. doi: 10.1006/clim.2001.5007. [DOI] [PubMed] [Google Scholar]
- 45.Norzila MZ, Fakes K, Henry RL, Simpson J, Gibson PG. Interleukin-8 secretion and neutrophil recruitment accompanies induced sputum eosinophil activation in children with acute asthma. Am J Respir Crit Care Med. 2000;161:769–774. doi: 10.1164/ajrccm.161.3.9809071. [DOI] [PubMed] [Google Scholar]
- 46.Teran LM, Johnston SL, Schroder JM, Church MK, Holgate ST. Role of nasal interleukin-8 in neutrophil recruitment and activation in children with virus-induced asthma. Am J Respir Crit Care Med. 1997;155:1362–1366. doi: 10.1164/ajrccm.155.4.9105080. [DOI] [PubMed] [Google Scholar]
- 47.Leitner JM, Mayr FB, Firbas C, Spiel AO, Steinlechner B, Novellini R, Jilma B. Reparixin, a specific interleukin-8 inhibitor, has no effects on inflammation during endotoxemia. Int J Immunopathol Pharmacol. 2007;20:25–36. doi: 10.1177/039463200702000104. [DOI] [PubMed] [Google Scholar]
- 48.Uehara A, Sugawara S, Muramoto K, Takada H. Activation of human oral epithelial cells by neutrophil proteinase 3 through protease-activated receptor-2. J Immunol. 2002;169:4594–4603. doi: 10.4049/jimmunol.169.8.4594. [DOI] [PubMed] [Google Scholar]
- 49.Uehara A, Sugawara Y, Sasano T, Takada H, Sugawara S. Proinflammatory cytokines induce proteinase 3 as membrane-bound and secretory forms in human oral epithelial cells and antibodies to proteinase 3 activate the cells through protease-activated receptor-2. J Immunol. 2004;173:4179–4189. doi: 10.4049/jimmunol.173.6.4179. [DOI] [PubMed] [Google Scholar]
- 50.Shpacovitch VM, Brzoska T, Buddenkotte J, Stroh C, Sommerhoff CP, Ansel JC, Schulze-Osthoff K, Bunnett NW, Luger TA, Steinhoff M. Agonists of proteinase-activated receptor 2 induce cytokine release and activation of nuclear transcription factor κB in human dermal microvascular endothelial cells. J Invest Dermatol. 2002;118:380–385. doi: 10.1046/j.0022-202x.2001.01658.x. [DOI] [PubMed] [Google Scholar]
- 51.Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. Converting enzyme-independent release of tumor necrosis factor α and IL-1β from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA. 1999;96:6261–6266. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]