

Abstract
Our experiments investigated associations of specific isoforms of protein kinase C (PKC) with individual proteins in the cardiac troponin complex. Troponin I (cTnI) associated with PKC ε and ζ and troponin T (cTnT) associated with PKC α, δ, and ε. Based on its association with cTnI, we hypothesized that PKCζ is a major regulator of myofilament protein phosphorylation. To test this, we infected adult cardiac myocytes with adenoviral constructs containing DsRed monomer-tagged wild type (WT) and the following constitutively active forms of PKCζ: the pseudo-substrate region (A119E), 3′-phospho-inositide-dependent kinase-1 (T410E), and auto-phosphorylation (T560E). The A119E and T410E mutants displayed increased localization to the Z-discs compared with WT and T560E. Immunoprecipitations were performed in myocytes expressing PKCζ using PKC phospho-motif antibodies to determine the phosphophorylation of cTnI, cTnT, tropomyosin, myosin-binding protein C, and desmin. We did not find serine (Ser) phosphorylation of cTnI or cTnT. However, we observed a significant decrease in threonine (Thr) phosphorylation of cTnI and cTnT notably by PKCζ T560E. Ser phosphorylation of tropomyosin was increased by all three active mutants of PKCζ. Ser/Thr phosphorylation of myosin-binding protein C increased primarily by PKCζ A119E. Both PKCζ A119E and T410E mutants increased desmin Ser/Thr phosphorylation. To explain the apparent Thr dephosphorylation of cTnI and cTnT, we hypothesized that PKCζ exists as a complex with p21-activated kinase-1 (Pak1) and protein phosphatase 2A (PP2A), and this was confirmed by immunoprecipitation Western blot. Our data demonstrate that PKCζ is a novel regulator of myofilament protein phosphorylation.
The protein kinase C (PKC)2 pathway participates in cardiac myofilament protein phosphorylation in association with hypertrophic signaling (1, 2). The eleven or so PKC isoforms are classified as conventional (α, β, βII, γ), novel (δ, ε, η, θ), and atypical (ζ, ι/λ). Conventional isoforms are regulated by Ca2+ and diacylglycerol (DAG); novel PKCs are activated by DAG alone; and atypical PKC isoforms are neither Ca2+ nor DAG-dependent. However, all three types are regulated by phosphatidylserine, 3′-phosphos-inositide-dependent kinase-1 (PDK1), and auto-phosphorylation (3). Using transgenic approaches, up-regulation of conventional (β and βII) or novel (δ and ε) PKC isoforms has been shown to increase cardiac troponin I (cTnI) and/or cardiac troponin T (cTnT) phosphorylation in addition to promoting hypertrophy (4–7). The atypical PKCζ has also been found in the heart (8), but its role in cardiac function remains unknown.
PKC also regulates myofilament activity. Although PKC phosphorylation sites have been identified in cTnI and cTnT (9), their functional significance has only been elucidated recently. Phosphorylation of cTnI, the major inhibitor of the actin-myosin cross-bridge reaction, regulates the activity and Ca2+ sensitivity of tension and actomyosin MgATPase rate in vitro (10) and alters maximum tension level and thin filament sliding speed, which then ultimately affect force development, myofilament activation, cross-bridge cycling rate, and cardiac dynamics (10, 11). In cTnT, which transduces the Ca2+ signal between the troponin complex and tropomyosin, PKC phosphorylation inhibits tension development, Ca2+ sensitivity, and cooperativity (12). However, the exact PKC isoforms that functionally modulate thin filament proteins remain unclear.
In the current study, we demonstrate isoform-specific interactions with PKC to cTnI and cTnT. We identified the atypical PKCζ isoform to associate specifically with cTnI in untreated adult rat ventricular cardiac myocytes. To determine whether PKCζ modulates myofilament protein phosphorylation, we used adenoviral expression of PKCζ in adult rat ventricular myocytes. Because the upstream regulators of PKCζ are unclear and the atypical PKC isoforms are neither DAG nor Ca2+-dependent (3), we generated three constitutively active forms with mutations in the pseudo-substrate (A119E) domain (13), the 3′-phospho-inositide-dependent kinase-1 (PDK1) phosphorylation site (T410E), and the auto-phosphorylation site (T560E) (14). We determined the localization of active PKCζ in adult rat ventricular myocytes and the states of Ser and Thr phosphorylation by PKCζ of the thin filament proteins cTnI, cTnT, tropomyosin (Tm), the thick filament protein myosin-binding protein-C (MyBP-C), and the intermediate filament/Z-disc protein desmin. Our data indicate that the activation of PKCζ is a significant control mechanism regulating both phosphorylation and dephosphorylation of myofilament proteins.
EXPERIMENTAL PROCEDURES
Isolation and Culturing of Adult Rat Cardiac Ventricular Myocytes
All experiments were performed in compliance with animal care policies of the Animal Care Committee at the University of Illinois at Chicago. Isolated adult rat cardiac ventricular myocytes from 200 –250 g male Sprague-Dawley rats (Harlan) were isolated as described previously (15). Rod-shaped Ca2+ tolerant myocytes were counted and assayed for viability by trypan blue exclusion assay. Myocytes were plated in M199 (Mediatech) and supplemented with 5 mmol/liter creatine, 2 mmol/liter L-carnitine, 5 mmol/liter taurine (Sigma), 50 units of penicillin, and 50 units of streptomycin (Mediatech) at a density of 1.4 × 105 cells per 35-mm dish (Falcon) or a one-chambered slide (Nalge) coated with 15 μg of mouse laminin (Invitrogen). After two hours of plating at 37 °C, 5% CO2 unattached cells were removed by two washes with media, leaving only attached rod-shaped viable myocytes.
Gluathione S-Transferase (GST)-Troponin Expression and Purification
cDNAs encoding cTnI, cTnT, and cTnC were amplified by polymerase chain reaction (PCR) and subcloned into the GST vector pGEX 5X-1 (Amersham Biosciences) in frame to generate the constructs GST-cTnI, GST-cTnT, and GST-cTnC, which were sequence verified. GST protein expression and purification were performed as described previously (16).
GST Pulldown Assay, Immunoprecipitations, and Western Blot
Lysates from cultured myocytes were prepared in ice-cold 1×radioimmune precipitation assay (50 mmol/liter Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxy-cholate, 150 mmol/liter NaCl, 1 mmol/liter EDTA, 1 mmol/liter phenylmethylsulfonyl fluoride, 1 mg/ml each of aprotinin, leupeptin, pepstatin, 1 mmol/liter Na3VO4, 1 mmol/liter NaF). For GST pulldown assays, five μg of GST protein was added to 100 μg of cell lysate and incubated overnight at 4 °C. 30 μl of 50% glutathione-agarose slurry was added and incubated for one hour. Glutathione agarose containing bound GST-troponin complexes were collected by centrifugation and washed three times with ice-cold 1× radioimmune precipitation assay buffer, then resuspended in 2× sample buffer (Alliance for Cell Signaling (AfCS) solution protocol 00000437), and boiled for 5 min. For immunoprecipitation assays, phospho-antibodies recognizing either the PKC Ser phosphorylation motif (R/K)X(S)X(R/K) or Thr phosphorylation TXR motif (Cell Signaling) were incubated with lysates overnight at 4 °C. Protein complexes were immunoprecipitated with protein A/G-agarose (Santa Cruz Biotechnology) and were collected by centrifugation and washed three times in ice-cold phosphate-buffered saline. All protein complexes were resolved by SDS-PAGE and transferred to 0.2-micron polyvindyline difluoride membrane (PVDF) membrane (Bio-Rad). Primary antibodies were as follows: protein kinase A catalytic subunit (PKAC) (BD Biosciences), PP2A (Upstate), PKC isoforms α, βII, δ, ε, ζ (Santa Cruz), PKCμ/protein kinase D (PKD) (Abgent), cTnI (Research Diagnostics, Inc), cTnT (CT3), and Tm (CH1) (Developmental Studies Hybridoma Bank, University of Iowa), MyBP-C (a gift from Richard Moss), and desmin (Biomeda). Secondary antibodies conjugated to horseradish peroxidase (Jackson ImmunoLabs) were used for detection. Western blots were developed with enhanced chemiluminescence and exposed to film (Amersham Biosciences). Data obtained were from six different experiments.
Adenoviral Construction, Production, and Expression of PKCζ
Human PKCζ cDNA (17) was purchased from American Type Culture Collection (ATCC MGC-10512). PKCζ cDNA was amplified by PCR and subcloned into pDsRed monomer-C1 (Clontech) in frame to generate the construct DsRed monomer PKCζ WT (wild type). This construct was used as a template to generate constitutively active mutations in the pseudo-substrate (A119E) (13), the PDK1 phosphorylation site (T410E) (14), or the auto-phosphorylation site (T560E) (14) of PKCζ using the QuikChange site-directed mutagenesis kit (Stratagene). All PKCζ constructs were transferred to the AdEasy (18) system for adenovirus production and were sequence verified. Adenoviruses were amplified up to four times in low passage 293 cells (ATCC) then purified using the ViraKit AdenoMini-4 kit (Virapur) according to manufacturer’s recommendations. Viral titers were determined by cytopathic effect assay (19). Cardiac myocytes cultured for two hours were infected with adenoviruses with titers at 108–1011 plaque forming units (pfu) per ml at a multiplicity of infection (MOI) of 100 overnight and cultured for 2 days at 37 °C, 5% CO2. Adenoviral PKCζ construct expression was verified by Western blot with a PKCζ antibody (Santa Cruz).
Confocal Microscopy of Ventricular Myocytes
Adenovirus infection of cultured myocytes with PKCζ constructs were performed as described above. Cells were fixed in 70% ice-cold methanol in 1× phosphate-buffered saline, permeabilized in 0.25% Triton X-100 in phosphate-buffered saline, and blocked with 8% bovine serum albumin fraction V (Roche). A primary mouse monoclonal antibody for α-actinin (Upstate) in 1% bovine serum albumin was used for detection of Z-discs in myocytes. For the secondary antibody, a chicken anti-mouse IgG Fluor 488 (Alexa) in 1% bovine serum albumin was used for green fluorescence. Cells were mounted using Fluoromount-G (SouthernBiotech) with glass coverslips. Image acquisition was performed as described previously (20). Images from at least six different myocytes per treatment from three different myocyte cultures were taken.
Data Analysis
All images were analyzed using National Institutes of Health (NIH) ImageJ software. Data are presented as means ±S.E. One-way analysis of variance (ANOVA) was used for multiple comparisons when appropriate. p <0.05 was considered significant.
RESULTS
Association of Protein Kinase C to Cardiac Troponins Is Isoform-specific
To determine the relevant PKC isoforms that may regulate the proteins of cardiac troponin complex, we developed a GST-troponin pulldown assay. First, we tested antibody specificity and verified the presence of various kinases and the protein phosphatase, PP2A. Using lysates from untreated isolated and cultured adult rat ventricular myocytes and rat brain as control, PKA, PP2A, and the PKC isoforms α, βII, δ, ε, ζ and PKD were found in both brain and ventricular myocytes (supplemental Fig. S1). We first tested the ability of GST-cTnI to pull down the catalytic subunit of protein kinase A. As seen in Fig. 1A (left panel), only GST-cTnI bound specifically to endogenous PKA. Endogenous PP2A was pulled down by GST-cTnI and GST-cTnT (Fig. 1A, right panel). The GST-troponin assay was then used to test the hypothesis that PKC associates with cardiac troponins in an isoform-specific manner. Of the conventional PKCs (Fig. 1B, top panel), only PKCα associated with GST-cTnT alone. PKCβII did not associate with any GST protein, though it was detected in myocyte lysates (supplemental Fig. S1). With the novel PKCs, the δ isoform associated with only GST-cTnT. PKCε associated with both GST-cTnI and GST-cTnT (Fig. 1B, middle panels). We found the atypical PKCζ to be specifically associated with only GST-cTnI (Fig. 1B, bottom left). The related kinase PKD was also associated with only GST-cTnI (Fig. 1B, bottom right). No kinases or PP2A were found to associate with GST alone or GST-cTnC.
FIGURE 1. GST-troponin pulldown assays.

A, PKAC pulldown by GST-cTnI (left panel) and PP2A pulldown by GST-cTnI and GST-cTnT (right panel). B, PKC pulldowns. G, GST only; I, GST-cTnI; T, GST-cTnT; C, GST-cTnC.
Localization of PKCζ in Cardiac Myocytes Is Dependent on Its State of Activation
Based on the finding that endogenous PKCζ pulled down with GST-cTnI (Fig. 1B), our initial goal was to characterize the role of PKCζ in cardiac myocytes. Adenoviral constructs of PKCζ were tagged with DsRed monomer (DsRedM), and their localization was examined by confocal microscopy. Expression of the constitutively active mutants along with the wild type (WT) and DsRedM alone are compared in data shown in Fig. 2. PKCζ WT and the T560E mutant were localized in the myofilaments. When either the A119E pseudo-substrate or the PDK1 phosphorylation T410E mutation was introduced, there was increased translocation to the Z-discs of cardiac myocytes. This was confirmed by counter-staining with an antibody against α-actinin, a Z-disc protein. Expression of the DsRed monomer alone was diffused throughout the cell.
FIGURE 2. Localization of PKCζ in cultured adult rat ventricular myocytes.

WT and mutants PKCζ cDNAs were tagged with DsRed monomer (DsRedM) and adenovirally expressed. Fixed cells were counter-stained with an α-actinin antibody (Upstate) and visualized by confocal microscopy. Data are representative of three independent experiments of >30 cells per experiment. Scale bar represents 10 microns.
Thin Filament Protein Phosphorylation by Protein Kinase Cζ
The amino acid sequences of cTnI, cTnT, and Tm were examined, and motifs were found that could serve as substrates for PKC-dependent phosphorylation. To determine whether PKCζ can affect the phosphorylation states of thin filament proteins and to correlate the state of PKCζ activation with thin filament protein phosphorylation, we performed immunoprecipitations (IP) with PKC phospho-motif antibodies in lysates of cardiac myocytes expressing adenoviral PKCζ. IP complexes were resolved by SDS-PAGE and were then blotted for cTnI, cTnT, and Tm. Uninfected (Un) myocyte lysates were used as controls. With cTnI (Fig. 3A, top) and cTnT (3B, top), no Ser phosphorylation was observed. Surprisingly, Thr phosphorylation of cTnI (Fig. 3A, middle panel) and cTnT (3B, middle panel) was decreased with PKCζ activation, especially by the T560E auto-phosphorylation PKCζ mutant. With Tm, Ser phosphorylation was increased by 50% in all constitutively active PKCζ mutants compared with controls (Fig. 3C, top). No Thr phosphorylation of Tm was observed (Fig. 3C, middle panel). Expression of DsRed monomer only or PKCζ WT did not alter Ser or Thr phosphorylation compared with uninfected myocytes. Total cTnI, cTnT, and Tm protein levels were unchanged with PKCζ expression.
FIGURE 3. Thin filament protein phosphorylation by PKCζ.

A, cTnI:PKC phospho-motifs are highlighted (Ser) and underlined (Thr), representative IP-Westerns of cTnI Ser and Thr phosphorylation (left), quantification of Thr phosphorylation of cTnI (right). B, cTnT:PKC phospho-motifs are highlighted (Ser) and underlined (Thr), representative IP-Westerns of cTnT Ser and Thr phosphorylation (left), quantification of cTnT Thr phosphorylation (right). C, Tm:PKC phospho-motifs are highlighted (Ser) and underlined (Thr), representative IP-Westerns of Tm Ser and Thr phosphorylation (left panels), quantification of Ser phosphorylation (right panel). *, p < 0.05 versus Un, **, p < 0.01 versus Un.
Myosin Binding Protein-C Phosphorylation Is Increased by PKCζ Activation
To determine whether thick filament protein phosphorylation is altered by PKCζ activation, MyBP-C was assessed. As seen in Fig. 4A, MyBP-C contains a number of PKC phospho-serine and phospho-threonine motifs. When myocytes were infected with the different constitutively active PKCζ constructs, the levels of Ser (Fig. 4B, top panel) and Thr (4B, middle panel) phosphorylation of MyBP-C were significantly increased with only the PKCζ pseudo-substrate A119E mutant. There were increased Ser and Thr phosphorylations of MyBP-C observed in the wild type, T410E, and T560E PKCζ constructs, but they did not reach significance. Total MyBP-C protein levels were unchanged by adenoviral PKCζ expression (Fig. 4B, bottom panel).
FIGURE 4. MyBP-C phosphorylation by PKCζ.

A, PKC phospho-motifs are highlighted (Ser) and underlined in MyBP-C. B, representative IP-Westerns of MyBP-C phosphorylation: PKC phospho-serine IP-Western (top panel); PKC phospho-threonine IP-Western (middle panel); lysate only (bottom panel). C, quantification of PKC Ser (top panel) and Thr phosphorylation (bottom panel). *, p < 0.05 versus Un; **, p < 0.01 versus Un.
PKCζ Phosphorylation of Desmin
To determine whether the translocation of PKCζ to the Z-discs results in changes in phosphorylation of Z-disc proteins, we examined the Z-disc/intermediate filament protein desmin. As seen in Fig. 5A, desmin contains a number of phospho-PKC Ser and Thr motifs. Without changing total desmin protein levels (Fig. 5B, bottom panel), expression of the pseudo-substrate A119E and the PDK1 phosphorylation T410E constitutively active mutants of PKCζ increased both Ser (5B, top panel) by up to 50% (5C, top panel) and Thr phosphorylation (5B, middle panel) by up to 60% (5C, bottom panel). No significant changes in Ser or Thr phosphorylation by PKCζ WT or the auto-phosphorylation mutant were observed. Taken together, translocation of the constitutively active PKCζ mutants A119E and T410E to the Z-discs (Fig. 2) results in increased desmin protein phosphorylation.
FIGURE 5. Desmin phosphorylation by PKCζ.

A, desmin amino acid sequence with PKC phospho-motifs highlighted (Ser) and underlined (Thr). B, representative IP-Westerns of desmin phosphorylation: PKC phospho-serine IP-Western (top panel), PKC phospho-threonine IP-Western (middle panel), lysate only (bottom panel). C, quantification of PKC Ser (top panel) and Thr (bottom panel) phosphorylation. *, p < 0.05 versus Un, **, p < 0.01 versus Un.
Evidence for PKCζ Involvement in the Pak1/PP2A Pathway
To explain the apparent threonine dephosphorylation of cTnI and cTnT by expression of activated PKCζ (Fig. 3), we tested the hypothesis that PKCζ is involved in the Pak1/PP2A pathway (20) by demonstrating that PKCζ exists in a complex with Pak1 and PP2A. Using myocyte lysates as well as brain as a control, IPs were first performed with a PKCζ antibody(Fig. 6A). Western blotting was performed with the protein complexes resolved by SDS-PAGE and found to have Pak1 (left panel) and PP2A (middle panel). Conversely, IPs in myocyte lysates with an antibody that recognizes Pak1 (Fig. 6B) resulted in identification of an association with PKCζ (left panel) and PP2A (middle panel). Finally, IPs with a PP2A antibody (Fig. 6C) in myocyte lysates were found to have PKCζ (left panel) and Pak1 (middle panel) associated with PP2A. Together with the Thr dephosphorylation of cTnI and cTnT reported in Fig. 3, these results indicate that PKCζ is involved in the Pak1/PP2A pathway leading to Thr dephosphorylation of cTnI and cTnT.
FIGURE 6. IP of PKCζ, Pak1, and PP2A in brain (B) and myocyte (M) lysates.
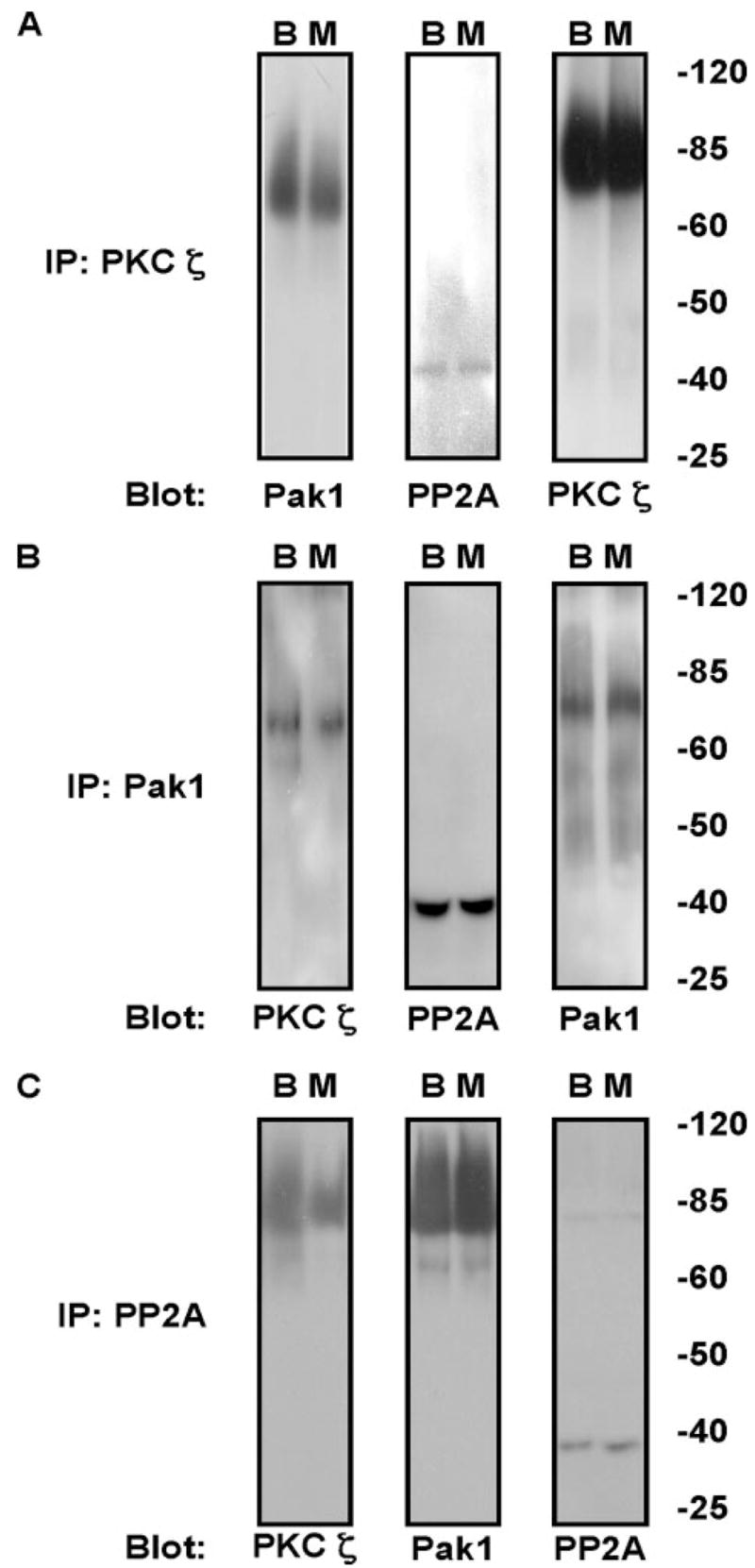
A, IP with an antibody to PKCζ and blotted for Pak1, PP2A, and PKCζ. B, Pak1 immunoprecipitation followed by blotting for PKCζ, PP2A, and Pak1. C, immunoprecipitation with PP2A antibody followed by PKCζ, Pak1, and PP2A antibodies.
DISCUSSION
Our data provide the following novel results: 1) The demonstration that there are PKC isoform-specific associations with individual cardiac troponins and 2) the definition for diverse roles for PKCζ in controlling sarcomeric protein phosphorylation and dephosphorylation in cardiac myocytes. To our knowledge, our data are the first to report endogenous PKCα association with cTnT and PKCζ association with cTnI.
Identification of the relevant kinases and phosphatases that modulate troponin phosphorylation has been attempted with varying results (8, 9, 21). Jideama et al. (9) using recombinant PKC isoforms α, δ, ε, and ζ to phosphorylate purified troponin complex demonstrated that α and δ isoforms preferentially phosphorylated cTnI over cTnT, whereas PKCζ preferentially phosphorylated cTnT. PKCε phosphorylated both cTnI and cTnT (9). It has been reported that in cardiac-targeted PKCε over-expressing mice, PKCε associates in a complex with cTnI and cTnT (21). To better identify the relevant signaling molecules, we developed a GST-troponin pulldown assay with the idea that an endogenous kinase or phosphatase can be identified by its affinity to its substrate. We found that endogenous PKA pulled down by GST-cTnI in myocyte lysates (Fig. 1B, left panel), consistent with PKA phosphorylation of cTnI (22). Protein phosphatase 2A has been shown to dephosphorylate cTnI (23), and this was confirmed by GST-cTnI pulldown of PP2A. In addition, GST-cTnT also pulled down PP2A, suggesting PP2A also dephosphorylates cTnT (Fig. 1B, right panel). Of the PKC isoforms examined, GST-cTnT associated with PKCs α, δ, and ε (Fig. 1C). GST-cTnI associated with PKCε, PKD, and PKCζ (Fig. 1C). With respect to PKCε, these data agree with the previous findings in the studies of cTnI and cTnT phosphorylation associated with transgenic mice over-expressing PKCε (21). PKD pulldown by GST-cTnI is consistent with results reported by Haworth et al. (24), where PKD association with cTnI was identified by yeast two-hybrid. Of particular interest is that neither GST-cTnI nor GST-cTnT pulled down PKCβII (Fig. 1C), though it was found in myocytes (1A). Increased cTnI phosphorylation has been shown in constitutively active PKCβII transgenic mice (6) as well as in vitro (25). More recently, p90 ribosomal S6 kinase has been shown to also phosphorylate cTnI by H2O2-induced PKCβII activation (26). Though PKCβII can phosphorylate cTnI in vitro (25), we believe PKCβII works upstream of p90 ribosomal S6 kinase to phosphorylate cTnI in cultured myocytes and would explain our findings.
Our demonstration of robust interaction of PKCζ with cTnI prompted further investigation of the signaling associated with this atypical isoform, which has not been well characterized in myocardium. Yet because the exact extrinsic signaling pathways leading to PKCζ activation are unknown in cardiac myocytes, we therefore generated three constitutively active mutants of PKCζ because it is neither Ca2+ nor DAG sensitive (3). When either PKCζ WT or the T560E auto-phosphorylation mutant was expressed in cardiac myocytes, they were localized diffusely in the myofilaments (Fig. 2). Upon introduction of the A119E or T410E mutation, there was an increase in localization to the Z-discs, as confirmed by co-localization with the Z-disc protein, α-actinin (Fig. 2). This is consistent in part with a report of Kang and Walker (27) who observed PKCs δ and ε translocation to the Z-discs upon phobol ester treatment. In addition, disruption of the Z-disc protein network perturbed PKC signaling in the myofilaments by a modest reduction in the cardiac actin capping protein CapZ (28). Taking into account all these data, we conclude that the constitutively active mutants of PKCζ, A119E (pseudo-substrate) and T410E (PDK1 phosphorylation) are most representative of PKCζ activation. Though the T560E auto-phosphorylation mutant did not translocate to the Z-discs, but was still activated, we think this is a conformational change of PKCζ that may act as a signal for other pathways, which alter myofilament protein phosphorylation (see below).
In our studies of phosphorylation of thin filament proteins, active PKCζ induced phosphorylation of Thr but did not induce Ser phosphorylation in either cTnI or cTnT (Fig. 3, A and B). Previous studies identified PKC phosphorylation sites in cTnI and cTnT (29). In cTnI, Ser-43 and Ser-45 and Thr-144 are PKC phosphorylated as well as the nominal PKA sites Ser-23 and Ser-24, notably by PKCδ (9). Kobayashi et al. (30) observed that PKCε also phosphorylated recombinant cTnI in vitro at the PKA sites. In cTnT, Ser-201 and Thr-197, Thr-206, and Thr-287 were identified and found to be phosphorylated by PKCs α, δ, ε, and ζ (9). We think the differences between our data reported here and previous studies is that they were performed in vitro and not in cardiac myocytes.
Data reported here support previous findings that Thr phosphorylation of cTnI and cTnT is significant (8, 11, 25) and demonstrate that these sites can be altered by PKCζ activation (Fig. 3). Our laboratory previously implicated Thr-206 of cTnT, located in the region where cTnT interacts with cTnI and cTnC, to be a particularly critical and functionally relevant PKC site (12). In studies using the in vitro motility assay, we have also previously reported that cTnI phosphorylation at Thr-144, found in the inhibitory region (1), leads to decreased Ca2+ sensitivity (11). However, Wang et al. (25) have indicted that phosphorylation of cTnI at Thr-144 by PKCβII in vitro leads to an increase in Ca2+ sensitivity. The significance of Thr-144 in cTnI has been further implicated by Malhotra et al. (8), who reported persistent Thr phosphorylation of cTnI with elevated PKCζ activity in myocytes treated with high glucose.
In view of findings reported in Figs. 3 and 6, we propose a novel aspect of regulation of troponin phosphorylation involving an induction by active PKCζ of dephosphorylation of cTnI at Thr-144 and cTnT at Thr-206 through a PKCζ-Pak1/PP2A pathway. There are a number of examples where the activation of upstream kinases can lead to modulation of protein phosphatase activity, which in turn can lead to either a change in total protein phosphorylation or changes in the state of phosphorylation of specific cardiac regulatory proteins. We observed that expression of the auto-phosphorylation PKCζ mutant (T560E) led to Thr dephosphorylation of cTnI (Fig. 3A) and cTnT (3B). In Fig. 6, we have demonstrated PKCζ exists as a complex with Pak1 and PP2A in cardiac myocytes. Recently a Pak1/PKCζ pathway has been found in non-muscle cells to regulate myosin II-B phosphorylation and filament assembly (31). Studies from our laboratory have demonstrated that activation of PP2A by constitutively activated Pak1 lead to total dephosphorylation of cTnI and myosin-binding protein C (20). Moreover, activation of p38-MAPK leads to dephosphorylation of α-tropomyosin and cTnI by PP2C, but does not dephosphorylate cTnT or myosin light chain-2 (32).
We also examined PKCζ phosphorylation of Tm, MyBP-C, and desmin. Tropomyosin exists as an α-helical dimer arranged head-to-tail along the actin filament. Upon Ca2+ binding to cTnC, the inhibitory effect of cTnI is relieved, allowing cTnT to interact with Tm and overall allow myosin cross-bridges to react with actin (reviewed in Ref. 1)). Tm phosphorylation at Ser-283 has been implicated to increase the MgATPase activity of myosin and to promote the head-to-tail interactions of Tm along the actin filament (33). In our examination of the Tm sequence, we identified a PKC phospho-serine motif at Ser-215 (Fig. 3C, top). Activation of PKCζ led to increased Ser phosphorylation of Tm (Fig. 3C, bottom left). Ser-215 may represent a novel PKC site in Tm. It is also possible that the head-to-tail interactions of Tm create a unique phosphorylation motif at Ser-283.
Our data are the first to demonstrate PKC phosphorylation of MyBP-C in cardiac myocytes. Expression of the constitutively active PKCζ A119E pseudo-substrate mutant most profoundly increased Ser and Thr phosphorylation of MyBP-C and to a lesser extent the T410E and T560E mutants (Fig. 4, B and C). MyBP-C is a large 150-kDa-protein thick filament protein attached to myosin at the head-to-neck region. Phosphorylation of MyBP-C by PKA along with cTnI has been associated with decreased Ca2+ sensitivity of force (reviewed in Ref. 1). Mohamed et al. (34) identified the PKA and PKC phosphorylation sites in a loop region near the N terminus of MyBP-C (Ser-286, Ser-295, and Ser-315 of rat MyBP-C). Our analysis of this region has revealed a PKC phospho-serine motif at Ser-315 (Fig. 4A), which is also a PKA phosphorylation site. We believe that Ser phosphorylation of MyBP-C by PKCζ mainly occurs in the loop region at Ser-315, supporting the idea that this area is a major regulatory region for MyBP-C (34). Our data are the first to demonstrate Thr phosphorylation of MyBP-C by PKCζ (Fig. 4, B and C). We identified eleven putative PKC phospho-threonine sites in rat MyBP-C and only one, the loop region that is unique to rats (Fig. 4A). We cannot exclude the possibility that PKC may phosphorylate other regions of MyBP-C because it has other PKC phospho-motifs (Fig. 4A).
We determined the phosphorylation of the intermediate filament/Z-disc protein desmin by PKCζ to correlate its translocation to the Z-discs upon its activation. Both the A119E and T410E PKCζ mutants significantly increased desmin Ser and Thr phosphorylation (Fig. 5, B and C) and also displayed increased translocation to the Z-discs (Fig. 2). Previous studies have determined the PKA (Ser-35 and Ser-50) and PKC (Ser-12, Ser-38, and Ser-56) phosphorylation sites, with Ser-29 phosphorylated by both PKA and PKC to be toward the N terminus in the non α-helical head domain of chicken desmin (35). In our examination of the rat desmin sequence, Ser-45 and Ser-60 are corresponding PKA sites, and Ser-13, Ser-48, and Ser-68 are corresponding PKC sites (Fig. 5A). We believe that most of the Ser/Thr phosphorylation of desmin by activated PKCζ occurs in the non α-helical head domain. We found four PKC phospho-threonine sites in desmin and only one is in the head domain (Thr-50) (Fig. 5A). We therefore think that PKC Thr phosphorylation is most likely at Thr-50 of desmin.
Roles for PKCζ in the heart are beginning to emerge. PKCζ has been shown to elevate atrial natriuretic factor at the transcriptional level (13). In a diabetic model, only PKCζ is not inhibited by the angiotensin receptor antagonist losartan, which corresponds with maintained Thr phosphorylation of cTnI (8). Recently Pak1 has been shown to interact with PKCζ to phosphorylate myosin II-B in non-muscle cells (31), and our data in part support this idea in cardiac myocytes (Fig. 6). However, we think that PKCζ activation leads to dephosphorylation of cTnI and cTnT (Fig. 3, A and B) because of its interaction with Pak1 and PP2A. Whatever the case, our data emphasize the importance for a complete understanding of PKCζ as a regulator of cardiac function via myofilament protein phosphorylation.
Footnotes
This work was supported by an American Heart Association pre-doctoral fellowship and National Institutes of Health Training Grants T32 HL07692-17 (to S. C. W.) and R01 HL64035 and PO1 HL62426 (to R. J. S.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
The abbreviations used are: PKC, protein kinase C; cTnI, cardiac troponin I; cTnT, cardiac troponin T; cTnC, cardiac troponin C; Tm, tropomyosin; MyBP-C, myosin-binding protein C; PKA, protein kinase A; PKD, protein kinase D; Pak1, p21-activated kinase 1; PP2A, protein phosphatase 2A; WT, wild type; GST, glutathione S-transferase; Un, uninfected; IP, immunoprecipitation; DsRedM, DsRed monomer; DAG, diacylglycerol; PDK1, 3′-phospho-inositide-dependent kinase-1.
References
- 1.Solaro RJ. Handbook of Physiology. Oxford University Press; New York, NY: 2001. pp. 264–300. [Google Scholar]
- 2.Solaro RJ, Montgomery DM, Wang L, Burkart EM, Ke Y, Vahebi S, Buttrick P. J Nucl Cardiol. 2002;9:523–533. doi: 10.1067/mnc.2002.127626. [DOI] [PubMed] [Google Scholar]
- 3.Newton AC. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 4.Goldspink PH, Montgomery DE, Walker LA, Urboniene D, McKinney RD, Geenen DL, Solaro RJ, Buttrick PM. Circ Res. 2004;95:424–432. doi: 10.1161/01.RES.0000138299.85648.92. [DOI] [PubMed] [Google Scholar]
- 5.Huang L, Wolska BM, Montgomery DE, Burkart EM, Buttrick PM, Solaro RJ. Am J Physiol Cell Physiol. 2001;280:C1114–C1120. doi: 10.1152/ajpcell.2001.280.5.C1114. [DOI] [PubMed] [Google Scholar]
- 6.Takeishi Y, Chu G, Kirkpatrick DM, Li Z, Wakasaki H, Kranias EG, King GL, Walsh RA. J Clin Investig. 1998;102:72–78. doi: 10.1172/JCI2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, Walsh RA. Circ Res. 2000;86:1218–1223. doi: 10.1161/01.res.86.12.1218. [DOI] [PubMed] [Google Scholar]
- 8.Malhotra A, Kang BP, Cheung S, Opawumi D, Meggs LG. Diabetes. 2001;50:1918–1926. doi: 10.2337/diabetes.50.8.1918. [DOI] [PubMed] [Google Scholar]
- 9.Jideama NM, Noland TA, Jr, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, Blumberg PM, Hannun YA, Kuo JF. J Biol Chem. 1996;271:23277–23283. doi: 10.1074/jbc.271.38.23277. [DOI] [PubMed] [Google Scholar]
- 10.Noland TA, Jr, Kuo JF. J Biol Chem. 1991;266:4974–4978. [PubMed] [Google Scholar]
- 11.Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. J Biol Chem. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- 12.Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. J Biol Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 13.Decock JB, Gillespie-Brown J, Parker PJ, Sugden PH, Fuller SJ. FEBS Lett. 1994;356:275–278. doi: 10.1016/0014-5793(94)01283-0. [DOI] [PubMed] [Google Scholar]
- 14.Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Biochemistry. 2001;40:249–255. doi: 10.1021/bi0018234. [DOI] [PubMed] [Google Scholar]
- 15.Wolska BM, Solaro RJ. Am J Physiol. 1996;271:H1250–H1255. doi: 10.1152/ajpheart.1996.271.3.H1250. [DOI] [PubMed] [Google Scholar]
- 16.Engel PL, Kobayashi T, Biesiadecki B, Davis J, Tikunova S, Wu S, Solaro RJ. J Biol Chem. 2007;282:183–193. doi: 10.1074/jbc.M512337200. [DOI] [PubMed] [Google Scholar]
- 17.Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, Nishizuka Y. J Biol Chem. 1988;263:6927–6932. [PubMed] [Google Scholar]
- 18.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Carroll SJ, Hall AR, Myers CJ, Braithwaite AW, Dix BR. BioTechniques. 2000;28:408–410. 412. doi: 10.2144/00283bm03. [DOI] [PubMed] [Google Scholar]
- 20.Ke Y, Wang L, Pyle WG, de Tombe PP, Solaro RJ. Circ Res. 2004;94:194–200. doi: 10.1161/01.RES.0000111522.02730.56. [DOI] [PubMed] [Google Scholar]
- 21.Edmondson RD, Vondriska TM, Biederman KJ, Zhang J, Jones RC, Zheng Y, Allen DL, Xiu JX, Cardwell EM, Pisano MR, Ping P. Mol Cell Proteomics. 2002;1:421–433. doi: 10.1074/mcp.m100036-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Solaro RJ, Moir AJ, Perry SV. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 23.Mumby MC, Russell KL, Garrard LJ, Green DD. J Biol Chem. 1987;262:6257–6265. [PubMed] [Google Scholar]
- 24.Haworth RS, Cuello F, Herron TJ, Franzen G, Kentish JC, Gautel M, Avkiran M. Circ Res. 2004;95:1091–1099. doi: 10.1161/01.RES.0000149299.34793.3c. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Grant JE, Doede CM, Sadayappan S, Robbins J, Walker JW. J Mol Cell Cardiol. 2006;41:823–833. doi: 10.1016/j.yjmcc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 26.Itoh S, Ding B, Bains CP, Wang N, Takeishi Y, Jalili T, King GL, Walsh RA, Yan C, Abe J. J Biol Chem. 2005;280:24135–24142. doi: 10.1074/jbc.M413015200. [DOI] [PubMed] [Google Scholar]
- 27.Kang M, Walker JW. J Mol Cell Cardiol. 2005;38:753–764. doi: 10.1016/j.yjmcc.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 28.Pyle WG, Hart MC, Cooper JA, Sumandea MP, de Tombe PP, Solaro RJ. Circ Res. 2002;90:1299–1306. doi: 10.1161/01.res.0000024389.03152.22. [DOI] [PubMed] [Google Scholar]
- 29.Noland TA, Jr, Raynor RL, Kuo JF. J Biol Chem. 1989;264:20778–20785. [PubMed] [Google Scholar]
- 30.Kobayashi T, Yang X, Walker LA, Van Breemen RB, Solaro RJ. J Mol Cell Cardiol. 2005;38:213–218. doi: 10.1016/j.yjmcc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 31.Even-Faitelson L, Ravid S. Mol Biol Cell. 2006;17:2869–2881. doi: 10.1091/mbc.E05-11-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vahebi S, Ota A, Li M, Warren CM, de Tombe PP, Wang Y, Solaro RJ. Circ Res. 2007;100:408–415. doi: 10.1161/01.RES.0000258116.60404.ad. [DOI] [PubMed] [Google Scholar]
- 33.Heeley DH. Eur J Biochem. 1994;221:129–137. doi: 10.1111/j.1432-1033.1994.tb18721.x. [DOI] [PubMed] [Google Scholar]
- 34.Mohamed AS, Dignam JD, Schlender KK. Arch Biochem Biophys. 1998;358:313–319. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 35.Kitamura S, Ando S, Shibata M, Tanabe K, Sato C, Inagaki M. J Biol Chem. 1989;264:5674–5678. [PubMed] [Google Scholar]