

Abstract
Mitochondrial DNA (mtDNA) is required for mitochondrial activities because it encodes key proteins for oxidative phosphorylation and the production of cellular ATP. We previously reported the existence of a specific mitochondrial topoisomerase gene, TOP1mt in all vertebrates. The corresponding polypeptide contains an N-terminal mitochondrial targeting sequence and is otherwise highly homologous to the nuclear topoisomerase I (Top1). In the present study, we provide biochemical evidence for the presence of endogenous Top1mt polypeptide in human mitochondria. Using novel antibodies against Top1mt, we detected the corresponding 70-kDa polypeptide in mitochondria but not in nuclear fractions. This polypeptide could be trapped to form covalent complexes with mtDNA when mitochondria from human cells were treated with camptothecin. Mapping of Top1mt sites in the regulatory D-loop region of mtDNA in mitochondria revealed the presence of an asymmetric cluster of Top1mt sites confined to a 150-bp segment downstream from, and adjacent to, the site at which replication is prematurely terminated, generating a ≈ 650-base (7S DNA) product that forms the mitochondrial D-loop. Moreover, we show that inhibition of Top1mt by camptothecin reduces formation of the 7S DNA. The present results suggest novel roles for Top1mt in regulating mtDNA replication.
Somatic cells contain thousands of copies of mitochondrial DNA (mtDNA), which consist of duplex DNA circles encoding genes essential for oxidative phosphorylation and cellular metabolism. MtDNA replication must, therefore, be tightly controlled. Defects in mtDNA result in mitochondrial diseases, including neurological degeneration, ataxia, heart failure, diabetes, aging, and cancers (1-4). MtDNA represents 1 to 5% of the total cellular DNA content (5). Typically, mtDNA consists of 16,569 base pair circles containing intron-less genes. Mitochondrial genes are compactly arranged on both DNA strands (the H- and the L-strands) and code for 2 rRNAs, 13 proteins implicated in oxidative phosphorylation, and 22 tRNAs (6, 7) (www.mitomap.org).
The regulatory region for mtDNA transcription and replication consists of a 1.3 kilobase non-coding region flanked by the three promoters (HSP1, HSP2 and LSP) and the transcription termination region for the H-strand (see Figures 3B and 8). It also includes the origin of replication. In animal mtDNA replication, most nascent strands from the leading, heavy-strand origin (OH) are prematurely terminated, generating a 650-base, 7S DNA product that defines the 3′ boundary of the so-called “displacement loop” (D-loop). Proper formation of the D-loop is critical to the entire replication process and therefore to the integrity of the cell, but the control elements for it have not been identified (8, 9). In cells, mtDNA is packaged in protein-DNA complexes named nucleoids (10) that are attached to the inner mitochondrial membrane by the mtDNA regulatory region (11).
Figure 3.

Experimental flowchart summarizing the experimental design for mapping Top1mt-induced cleavage sites in the mtDNA regulatory region. A. Schematic representation of the PL-PCR protocol. a) Primer extension was used to copy the broken strand (26, 27). b) A universal primer (PL: phosphate linker) was ligated to the end of the extended strand. This linker has a phosphate at the 5′-end (P) and a dideoxynucleoside at the 3′-end (H) of the complementary strand to avoid unwanted ligation. c) PCR with a nested P2 primer and a linker primer (LP) complementary to PL. d) Labeling using a nested primer (P3) 5′-end-labeled with 32P- labeled (asterisk). B. Schematic representation of the mtDNA non-coding regulatory region. The normally arrested D-loop (7S DNA) is represented with its termination at position 16107. Primers used for the PL-PCR are indicated with double arrowheads (see Table 2 for genomic positions and sequence). L: light strand; H: heavy strand; OH: replication origin for heavy strand; LSP: light strand promoter. Dotted horizontal gray line corresponds to the RNA primer from which the D-loop initiates.
Figure 8.

Mapping of the Top1mt sites in the regulatory D-loop region of mtDNA. A. Top1mt sites in organello are clustered (blue circle) downstream from the D-loop. B. Top1mt sites in purified mtDNA are more widely distributed compared to the in organello sites. Primer sets used for the PL-PCR in mitochondria and primer used on mtDNA are indicated with horizontal arrow with double arrowhead. Top1mt sites are indicated by vertical arrowhead pointing down for cleavage in the heavy (H) and light (L) strand, respectively. OH: origin of replication for the H-strand. The red arrow depicts the 7S DNA forming the D-loop and resulting from replication pausing at site 16107. LSP: light strand promoter, which also serves as the RNA primer for H-strand synthesis. HSP1 and HSP2: H-strand promoters 1 and 2. T and P: tRNAs for threonine and proline. TAS: termination-associated sequence implicated in replication elongation arrest and D-loop formation (www.mitomap.org).
The proteins required for mtDNA transcription, replication, repair and nucleoid structure are all encoded in the nuclear genome and imported into mitochondria (7). Just like for any other DNA, mtDNA needs DNA topoisomerases for transcription and replication. Topoisomerases use DNA strand scission and rejoining activities to adjust DNA topology. They are classified into two categories. Type I enzymes transiently break one DNA strand at a time, whereas type II enzymes cleave and religate both strands of the DNA double helix in concert (12, 13). Human cells contain two type IA topoisomerases (Top3α and Top3β), two type IB topoisomerases (Top1 and Top1mt) (14, 15), and two type IIA topoisomerases (Top2α and Top2β) (13, 16). Topoisomerase activities have been identified in mitochondria from many species and tissues (15, 17-19). Because of the minute amount of mitochondrial topoisomerase activities compared with their nuclear counterparts, attempts to identify mitochondrial topoisomerases have been largely unsuccessful until the discovery of a specific mitochondrial DNA topoisomerase gene from human cells—TOP1mt (20).
Top1mt, like its nuclear counterpart Top1, consists of four domains: an N-terminal localization domain, a core domain, a linker domain, and a C-terminal domain containing the catalytic tyrosine (15, 20, 21). The N-terminal domain of Top1mt is short (50 amino acid residues) and consists primarily of a mitochondrial localization signal. By contrast, the N-terminal domain of (nuclear) Top1 consists of 214 amino acid residues encoding nuclear localization signals and protein-protein interaction motifs (13). The core and C-terminal domains, and to a lesser extent the linker domain are highly similar between Top1mt and Top1 (15). These three domains are sufficient for topoisomerase activities and are encoded by 13 conserved exons. The 13-exon motif is conserved in all vertebrate type IB topoisomerase genes (21). Both Top1 (nuclear) and recombinant Top1mt can be trapped by camptothecin (CPT) (20, 22), a plant alkaloid highly selective for Top1, and from which two derivatives (topotecan and irinotecan) have been derived to treat human cancers (14).
Here we demonstrate the association of Top1mt with mtDNA using novel anti-Top1mt antibodies. We mapped the Top1mt sites induced by CPT in the regulatory D-loop region of mtDNA and show their asymmetrical localization downstream from the D-loop in mitochondria whereas the Top1mt sites exhibited a more diffuse distribution when recombinant Top1mt was incubated with purified mtDNA. We also show that Top1mt inhibition by CPT affects formation of the 7S DNA associated with regulatory D-loops.
Material and Methods
Top1mt antibodies
Top1mt protein was over expressed using pET system (Novagen, Madison, WI) (20). His -tagged Top1mt was purified and injected into rabbits (Spring Valley Laboratories, Woodbine, MD). The anti-serum was affinity-purified using GST-tagged Top1mt.
Preparation of mitochondria
Human breast cancer MCF-7 cells were washed with ice-cold phosphate buffered saline (PBS), scrapped off, and spun down at 500 g for 5 minutes. Mitochondrial preparation was performed as described (23). Briefly, cell pellets were resuspended in CaRSB buffer (10 mM of NaCl, 1.5 mM CaCl2, and 10 mM Tris-HCl, pH 7.5 at 25°C) for 5 minutes. Following osmotic shock, cells were homogenized using a glass Dounce homogenizer (35 strokes). One-sixth volume of stabilizing buffer (2 M sucrose, 35 mM EDTA, and 50 mM Tris-HCl, pH7.5 at 25°C) was added to the cell lysates, and mixed with 2 additional strokes with glass Dounce homogenizer. Cell lysates were centrifuged at 750 g for 5 minutes to remove the nuclei and cell debris. Supernatants were collected and the previous step was repeated one more time. The mitochondria were spun down from supernatant at 10,000 g for 20 minutes and washed with MT buffer (250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 5 mM DTT and 20 mM Hepes-KOH, pH7.4 at 25°C), and re-suspended in MT buffer.
Preparation of mtDNA
Mitochondria in MT buffer were lysed by adding sodium dodecyl sulfate (SDS) to a final concentration of 0.6% and proteinase K (0.1 mg/ml). The remaining steps followed the manufacturer's protocol for the DNeasy tissue kit (Qiagen, Valencia, CA).
Detection of Top1mt cleavage complexes using the ICE Bioassay
The In vivo complex of enzyme (ICE) bioassays were performed as described (24, 25). Mitochondria were lysed with 1% sarkosyl and gently loaded on top of a CsCl gradient. After ultracentrifugation (Beckman SW40 rotor at 30700 rpm for 20 hours at 20°C), the samples were collected from the bottom of the centrifuge tubes, and transferred to the Imobilon P membrane (Millipore, Bedford, MA). Top1mt was detected using standard Western blotting protocol.
Phosphate-Linked Ligation-mediated PCR (PL-PCR)
The PL-PCR was derived from the LM-PCR previously used to map the DNA cleavage sites of nuclear Top1 in human ribosomal DNA (26, 27). To map the cleavage complexes produced by Top1mt in mtDNA, 3 primers were used in each set of experiment (Figure 3 and table 1). The first primer was used to create the substrate for the phosphate linker (step a, Figure 3). The second primer was for PCR amplification (step c, Figure 3), and the third one for labeling the PCR product to generate detectable signals (step d, Figure 3). Briefly, primer 1 was mixed with DNA samples in first strand buffer (40 mM NaCl, 10 mM Tris-HCl, pH 8.9 at 25°C, 5 mM MgSO4, 0.01% Gelatin, 250 μM dNTP) with Vent DNA polymerase (NEB, Beverly, MA) (step a, Figure 3). Incubations cycles were: 95°C for 5 minutes, 60°C for 30 minutes, and 76°C for 10 minutes (step a, Figure 3). The ligase dilution solution (110 mM Tris-HCl, pH 7.5 at 25°C, 17.5 mM MgCl2, 50 mM DTT, 125 μg/ml BSA) and annealed phosphate linker were added, and ligation was carried out with T4 DNA ligase at 17°C overnight (step b, Figure 3). The ligation mixture was precipitated, dissolved in H2O, and was used as PCR template. PCR was carried out with primer 2 and the linker-primer (step c, Figure 3). The PCR conditions were: step 1: one cycle at 95°C for 3 minutes; step 2: twenty cycles consisting of 95°C for 1 minute, 66°C for 2 minutes, and 76°C for 3 minutes; step 3: one cycle at 76°C for 10 minutes. The 32P-labeled primer 3 was added to the PCR solutions and the tubes were further incubated at 95°C for 4 minutes, 70°C for 2 minutes, 76°C for 3 minutes, 95°C for 1 minute, 70°C for 2 minutes, and 76°C for 10 minutes (step d, Figure 3). The final solution was mixed with equal volume of 2x loading buffer, and directly loaded on 6% PAGE/urea.
Table 1.
| Primer | Name and sequence (from 5′ to 3′) | Length | Tm |
|---|---|---|---|
| HP1.1 | TTTCAGTGTATTGCTTTGAGGAGG | 24mer | 59.3°C |
| HP1.2 | GAGGAGGTAAGCTACATAAACTGTGGG | 27mer | 65.0°C |
| HP1.3 | GTAAGCTACATAAACTGTGGGGGGTGTC | 28mer | 66.6°C |
| HP2.1 | GTTATGATGTCTGTGTGGAAAGCG | 24mer | 61.0°C |
| HP2.2 | ATGTCTGTGTGGAAAGCGGCTGTG | 24mer | 64.4°C |
| HP2.3 | GTGGAAAGCGGCTGTGCAGACATTC | 25mer | 66.3°C |
| HP3.1 | CGGTAAATGGCTTTATGTGCTATG | 24mer | 59.3°C |
| HP3.2 | GGGTGGGTAGGTTTGTTGGTATCCTAG | 27mer | 66.5°C |
| HP3.3 | GGGTAGGTTTGTTGGTATCCTAGTGGGTG | 29mer | 68.1°C |
| LP2.1 | CTGTTCTTTCATGGGGAAGCAG | 22mer | 60.3°C |
| LP2.2 | GAAGCAGATTTGGGTACCACCCAAG | 25mer | 64.6°C |
| LP2.3 | GGGTACCACCCAAGTATTGACTCACCC | 27mer | 68.0°C |
| LP3.1 | GACTCCTAGCCGCAGACCTC | 20mer | 59.6°C |
| LP3.2 | CTGAATCGGAGGACAACCAGTAAGC | 25mer | 64.6°C |
| LP3.3 | GAATCGGAGGACAACCAGTAAGCTACCC | 28mer | 68.0°C |
| HM15643 | GTTTATGACTGTATGGTGTATGTCAG | 26mer | 60.1°C |
| HM15637 | GACTGTATGGTGTATGTCAGATAACACAG | 29mer | 63.9°C |
| Phosphate linker (PL) | p-GAATTCAGATCTCCCGGGTCACCGC | ||
| Complementary linker | GATCTGAATTC-dd (2′, 3′ dideoxy C): | ||
| Linker primer (LP) | GCGGTGACCCGGGAGATCTGAATTC |
In vitro Top1mt cleavage assay
Double-stranded mtDNA fragments and oligonucleotides were 3′-end-labeled with 32P. Labeled DNA (50 fmol/reaction) was incubated with 50 ng of recombinant Top1mt (20) with or without drug at 37°C for 10 minutes in 10 μl reaction buffer (10 mM Tris-HCl, pH 8.5, 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/ml BSA, final concentrations). Maxam Gilbert loading buffer was added to the reaction mixtures. Aliquots were separated in denaturing polyacrylamide gels (7 M urea) in 1X TBE. Imaging and quantitation were performed using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
Mapping the 3′ end of 7S DNA associated with the mtDNA D-loop
A modified version of PL-PCR was used. DNA samples were directly ligated with annealed phosphate linker by T4 DNA ligase at 17°C overnight. After ligation, the protocol followed steps c and d of PL-PCR (see Figure 3). The primers used for PCR were LP and HM15643 for mouse, and LP and HP3.2 for human mitochondria (see Table 1). Labeling and sequencing were performed after extension of the 32P-5′-end-labeled HM15637 and HP3.3 nested primer for mouse and human mitochondria, respectively (see Table 1).
Generation of the Top1mt-/- MEF cell lines
The Top1mt-/- MEF cells were generated from Top1mt-/- mice, which were constructed by deletion of the last two exons of TOP1mt gene (21).
Results
Top1mt antibodies demonstrate the presence of Top1mt in mitochondria
In spite of the high homology between Top1mt and Top1 (nuclear) (20), Western blotting of mitochondrial lysates with available polyclonal and monoclonal antibodies against Top1 (nuclear) failed to detect a band with the expected size for Top1mt (Figure 1 and additional data not shown) even when the mitochondrial lysates showed high signal for Cox4 (a well-establish mitochondrial polypeptide; data not shown). Thus, we generated anti-Top1mt polyclonal antibodies using recombinant full-length Top1mt to immunize rabbits. Those antibodies recognize a band of ≈70 kDa in mitochondrial lysates (Figure 1, lane M). That size is in agreement with the predicted molecular weight of Top1mt (69.9 kDa for the 601 amino acid polypeptide analyzed with MacVector™). Our Top1mt antibodies also recognized nuclear Top1 (Figure 1, lane N). Those experiments indicate the presence of Top1mt in mitochondria. The lack of signal for Top1 (nuclear) in the mitochondrial fractions also demonstrates the absence of nuclear contamination in our mitochondrial preparations.
Figure 1.
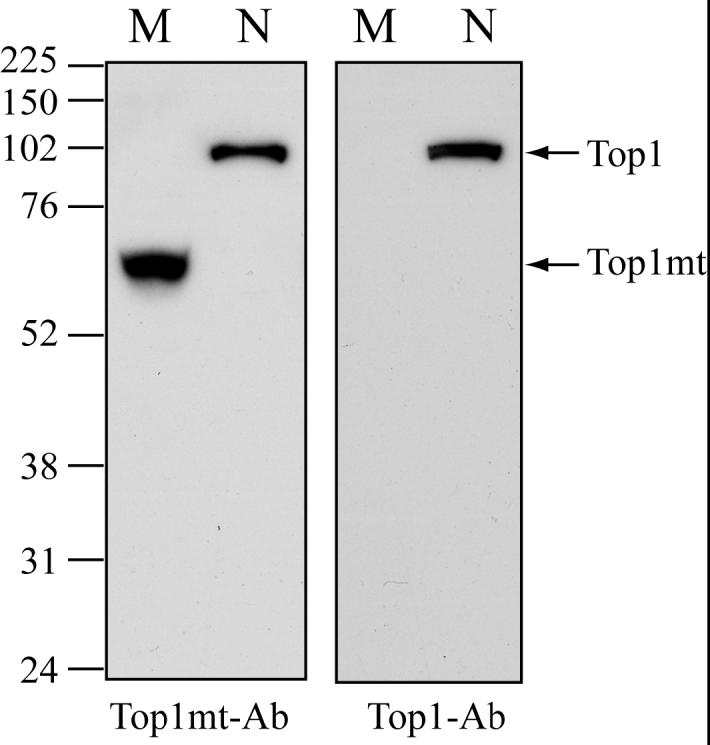
Detection of Top1mt in mitochondria by immunoblotting. Left panel: Western blotting with Top1mt antibodies (Top1mt-Ab); Right panel: the same membrane was striped and reprobed with monoclonal Top1-Ab (Top1-Ab; C21); Lanes M: mitochondrial fraction; Lanes N: nuclear fraction. Migration position of protein marker (kDa) is indicated at left.
Trapping of Top1mt by camptothecin in mitochondria
To reveal Top1mt interaction with mtDNA, we took advantage of the fact that Top1mt cleavage complexes can be trapped specifically by CPT (20) and that Top1 cleavage complexes can be visualized using Top1 antibodies following isolation of DNA by cesium gradient centrifugation (24, 25). Having generated the antibodies that recognize Top1mt (see above), we were able to look for the presence of Top1mt cleavage complexes in mitochondria treated with CPT. Figure 2 shows that treatment of cells with a relatively high dose of CPT (5 μM for 1 hour) that are known to produce high levels of cleavage complexes with nuclear Top1 (26-28) failed to produce detectable Top1mt-DNA complexes (Figure 2A). However, treatment of isolated mitochondria with CPT for half hour generated Top1mt-mtDNA complexes (Figure 2). Topotecan, a clinical CPT derivative, was as effective as CPT (data not shown). We also tested the non-camptothecin indenoisoquinoline Top1 inhibitor, MJ-III-65 (NSC 706744) (29). MJ-III-65 also produced Top1mt-mtDNA cleavage complexes, but was less potent than CPT (Figure 2B). Low levels Top1mt-mtDNA cleavage complexes were also consistently detected in the absence of drug at 37°C (the condition used for drug treatments) (Figure 2B, bottom left), whereas no detectable Top1 cleavage complexes were visible when mitochondria were kept at 0°C (Figure 2B, top left). These results suggest that Top1mt-mtDNA cleavage complexes may form spontaneously. When mitochondria from different human cell lines, HCT116 colorectal carcinoma and Jurkat leukemia T cells were used, CPT also trapped Top1mt-mtDNA cleavage complexes (data not shown). As the above immunoassays demonstrated the presence of Top1mt mtDNA-complexes, which presumably corresponded to cleavage complexes, we designed experiments to map Top1mt cleavage sites in mtDNA.
Figure 2.

Induction of Top1mt-DNA complexes by CPT and the indenoisoquinoline (MJ-III-65, NSC 706744) in mitochondria from human breast cancer MCF-7 cells. A. Detection of Top1mt-DNA complexes in cells vs. mitochondria. Control: untreated cells; CPT in cells: cells were treated with 5 μM CPT for 1 hour at 37°C before mitochondria were purified in the absence of CPT; CPT in mito: mitochondria isolated from untreated cells were treated with 5 μM CPT for 30 minutes at 37°C. B. Induction of Top1mt-DNA complexes in isolated mitochondria. Mitochondria were treated with the indicated drug concentrations for 30 minutes at 37°C as indicated. Two control untreated samples are included: 0: sample without drug for 30 minutes at 37°C; 0*: untreated sample kept on ice for 30 minutes while the other samples were incubated at 37°C.
In organello cleavage sites of Top1mt in mtDNA
As Top1 forms cleavage complexes it binds covalently to the DNA 3′-end and generates a free hydroxyl group at the 5′-end of the DNA break (20) (see scheme at the top of Figure 3). To map the cleavage sites induced by Top1mt in mtDNA, we modified the ligation-mediated polymerase chain reaction (LM-PCR) approach (26, 27) by adding a phosphate group at the 5′-end of the linker (Figure 3b), so that the linker could be ligated to the 3′-OH group of the newly extended DNA strand (Figure 3a). To further increase the signal over noise ratio, we employed a dideoxynucleotide (ddNTP) at the 3′ end of the complementary strand of the linker (Figure 3b; noted as “H”). We refer to this protocol as PL-PCR for phosphate linker, ligation mediated PCR. Cycles of PCR were performed using a nested P2 primer and a primer (LP) complementary to the primer linker (PL) (Figure 3c). The broken DNA strands were labeled by primer extension with a third primer (P3; Figure 3d) labeled at the 5′-end with 32P.
Figures 4 and 5 show representative experiments for Top1mt-induced cleavage sites in isolated mitochondria. We selected five primer sets (P1, P2 and P3 for each set; see Tables I and II) to cover the regulatory D-loop region (Figure 3B). PL-PCR experiments showed a cluster of three sites consistently induced by CPT (Figs 4 & 5). Sequencing demonstrated their location at H15826, H15973 (Figure 4) and L15932 (Figure 5). Cleavage at those sites could be reversed by heat, a characteristic of Top1 cleavage complexes (28, 29). These experiments demonstrated the presence of Top1mt cleavage complexes in mtDNA, and their selective location in the regulatory region of mtDNA.
Figure 4.

Mapping of Top1mt sites on the Heavy-strand of the mtDNA regulatory region. A. PL-PCR result. The arrowheads indicate the Top1mt sites and numbers correspond to their genomic location in mtDNA. B. Identification by sequencing of the corresponding DNA fragment obtained by PL-PCR for site 15826. The sequences of chromatograms show the mtDNA-linker junctions in the corresponding DNA fragments. The double-stranded sequences under each chromatogram show the corresponding mtDNA sequences. The bold face letters indicate the sequence portions presented in the chromatograms, the italic letters mean the portions missed from the chromatograms. The numbers indicate the positions in mtDNA. Bold face numbers linked to the italic sequences are the sites where Top1mt was covalently linked to mtDNA. LS, light-strand mtDNA; HS, heavy-strand mtDNA. C. Same for site 15973.
Figure 5.

Mapping of Top1mt sites on the Light-strand of the mtDNA regulatory region. A. PL-PCR result. The arrowhead indicates the Top1mt site. B. Identification by sequencing of the corresponding DNA fragment obtained by PL-PCR. The sequence of chromatogram shows the mtDNA-linker junction in the corresponding DNA fragment. The double-stranded sequence under chromatogram shows the corresponding mtDNA sequence. The bold face letters indicate the sequence portion presented in the chromatogram, the italic letters mean the portion missed from the chromatogram. The numbers indicate the positions in mtDNA. Bold face number linked to the italic sequence is the site where Top1mt covalently linked to mtDNA. LS, light-strand mtDNA; HS, heavy-strand mtDNA.
Table 2.
Oligonucleotides for mapping Top1mt sites (see Figure 6)
| L-15826 | 5′-TACTATACTTCACAACAATCCTAATC |
| H-15826 | 5′-GATTAGGATTGTTGTGAAGTATAGTA |
| L-15932 | 5′-GTAAACCGGAGATGAAAACCTTTTTC |
| H-15932 | 5′-GAAAAAGGTTTTCATCTCCGGTTTAC |
| L-15973 | 5′-AGAAAAAGTCTTTAACTCCACCATTA |
| H-15973 | 5′-TAATGGTGGAGTTAAAGACTTTTTCT |
DNA cleavage sites induced by recombinant Top1mt in DNA oligonucleotides corresponding to the sequences cleaved in mitochondria
To determine whether Top1mt-induced cleavage at the three sites of the mtDNA regulatory region were determined by the local DNA sequence, three 26-mer duplex oligonucleotides corresponding to the Top1mt sites shown in Figures 4 and 5 were designed (H-15826, L-15932, and H-15973; Table 2). The potential cleavage sites previously observed in mtDNA were positioned in the center of the oligonucleotides to generate 13-mers cleavage products. The oligonucleotides H-15826, L-15932, and H-15973 were 3′-end-labeled with α-32P-cordycepin and annealed to their corresponding complimentary oligonucleotides, as Top1 requires duplex DNA for cleavage. Figure 6 shows that Top1mt cleaved the oligonucleotides at the same sites as in mitochondria. Cleavage efficiency was best for the H-15826 site, and was relatively weak for the H15973 site. The difference in cleavage intensity may reflect an impact of mtDNA structure in vivo for optimum Top1mt activity.
Figure 6.

Top1mt sites in DNA oligonucleotides. Purified Top1mt was reacted with the indicated 32P-3′-end-labeled oligonucleotides for 10 minutes at 37°C. The oligonucleotides sequences are listed in Table 2. Arrowheads point to the Top1mt cleavage sites.
Cleavage sites induced by recombinant Top1mt in mtDNA in vitro
To further determine the effect of local mtDNA structure on Top1mt activity, cleavage assays were carried out with long mtDNA fragments corresponding to the regions mapped in mitochondria. Those DNA fragments were obtained by PCR (see Figure 8B and Table 3) and were incubated with recombinant Top1mt. When the fragment encompassing to the H15826 and H15973 region was used, different patterns of cleavage sites were observed in vitro and in mitochondria (Figure 7). The H15973 site observed in mitochondria was not observed in vitro, which is consistent with the weak cleavage observed with the oligonucleotide centered by that site (see Figure 6). A new site, H15829 was observed in mtDNA in vitro. Additional experiments were performed to scan both mtDNA strands in the region from 15741 to 16569 in vitro (see Figure 8B). We identified Top1mt sites that were spread along the isolated mtDNA but which had not been detectable in the mitochondria experiments (Figure 8B). The differences in Top1mt cleavage pattern in organello and with isolated mtDNA suggest that mtDNA packaging regulate the cleavage sites induced by Top1mt in intact mitochondria.
Table 3.
Primers for mapping Top1mt sites, in vitro (see Figure 7)
| Bam15741 | GCGGATCCTCATTCTAACCTGAATC |
| Bam16249 | GCGGATCCAAAGCCACCCCTCACC |
| Bam1611R | GCGGAACCGTACAATATTCATGGTGG |
| Bam-end | GCGGATCCATCGTGATGTCTTATTTAAG |
Figure 7.
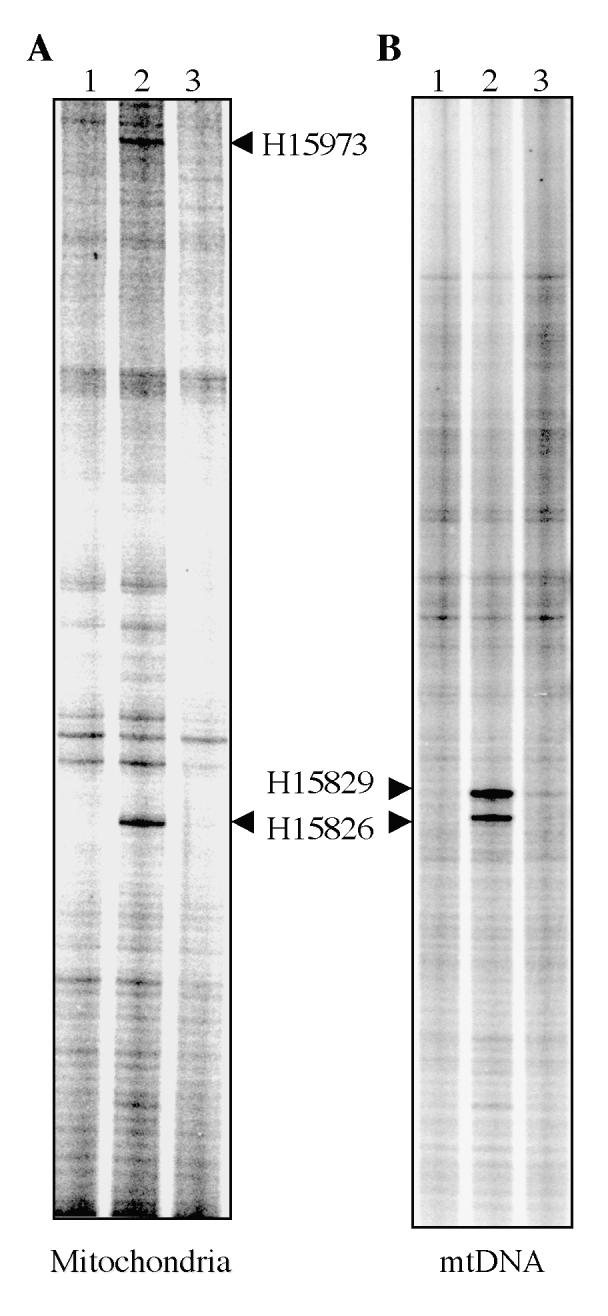
Comparison of Top1mt sites induced by CPT in mitochondria and with purified mtDNA and recombinant Top1mt. A. Top1mt cleavage sites in mitochondria. B. Top1mt cleavage sites in vitro. Arrowheads point to the Top1mt sites. Lanes 1: DNA alone; lanes 2: + CPT (50 μM); lane 3: + CPT with heat reversal (65°C for 5 minutes).
Regulation of 7S DNA by Top1mt
The selective distribution of Top1mt sites ahead of the D-loop region in mtDNA suggested the possibility that Top1mt might play a role in regulating the premature termination of mtDNA replication that gives rise to the D-loop. Although the premature termination has been mapped to highly specific sites (8, 9), the molecular factors that control premature termination remain unknown. Using our PLPCR assay (see steps b-d in Figure 3), we mapped the normal mouse mtDNA premature termination site at H15426 (9) (Figure 9).
Figure 9.
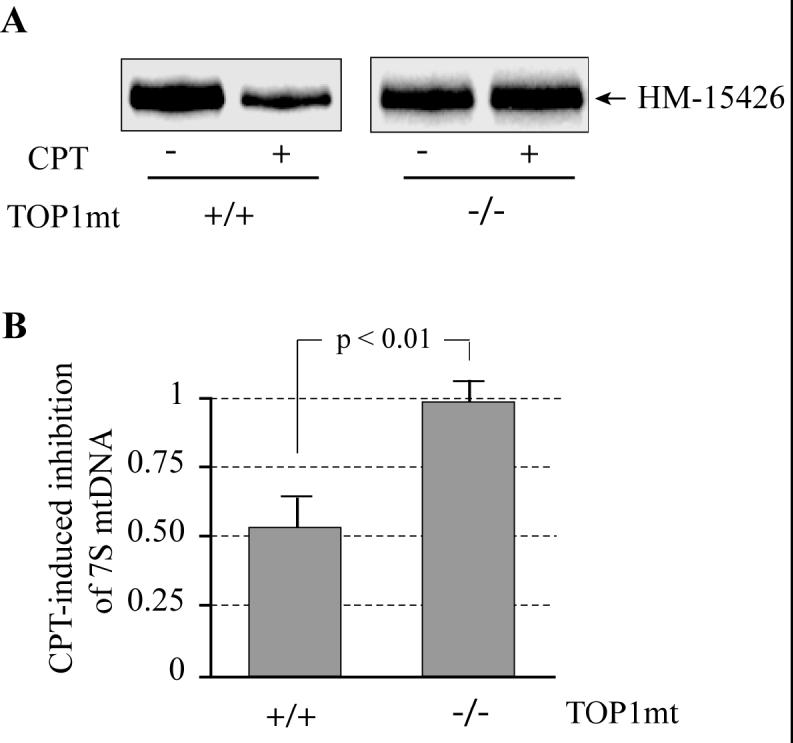
Inhibition of Top1mt by camptothecin (CPT) reduces the formation of 7S DNA. A. Representative experiments in normal (TOP1mt +/+) and Top1mt-deficient (TOP1mt -/-) MEF mitochondria treated with CPT (as indicated; 50 μM at 37°C for 30 min). B. Top1mt-dependent inhibition of 7S DNA by CPT (50 μM at 37°C for 30 min). Mean ± SD (at least 3 independent experiments).
Figure 9 shows that CPT reduced the formation of 7S DNA by approximately 50% [54 ± 10% in mitochondria from wild-type (+/+) MEF cells treated with CPT]. Similar experiments performed in mitochondria from Top1mt (-/-) MEF cells showed a lack of effect of CPT. Those results indicate that blocking Top1mt activity reduces D-loop formation in mitochondria and suggest the possibility that Top1mt acts as a DNA swivel to allow replication fork progression (12-14).
Discussion
The present study provides the first direct evidence for the presence of native Top1mt in mitochondria (see Figure 1), for its association with mtDNA (see Figures 2, 4, 5 and 7), for the non-random formation of Top1mt cleavage complexes in mtDNA in organello (Figures 4-5, 7-8), and for a role of Top1mt in 7S DNA formation associated with mtDNA D-loop.
Although Top1 activity is readily detectable from mitochondria from various sources (15), the biochemical isolation of the enzyme from mitochondria remained elusive until recently. Proteomic analyses published this year confirmed the presence of Top1mt in mitochondrial nucleoids (30). The difficulty in purifying mitochondrial topoisomerases is probably related to the small amount of enzymes present in mitochondria and to their limited and reversible association with mitochondrial nucleoids (30). The present study provides four lines of evidence demonstrating the presence of Top1mt in mitochondria. First, immunoblotting of mitochondrial lysates with our Top1mt antibodies revealed the presence of an immunoreactive band migrating with the expected size range for Top1mt (Figure 1). Second, immunoblotting of mtDNA isolated by cesium chloride gradient from mitochondria treated with CPT showed Top1mt tightly bond to mtDNA (Figure 2). Third, CPT-induced cleavage sites in mtDNA were demonstrated in mitochondria, and those sites coincided with cleavage sites induced by recombinant Top1mt in pure mtDNA (Figures 2-8). And fourth, treatment of mitochondria with CPT reduced the levels of 7S DNA, which was dependent upon the presence of Top1mt (Figure 9).
The need to generate specific anti-Top1mt antibodies to detect protein-DNA complexes was prompted by our finding that the existing antibodies against Top1 (nuclear) failed to cross-react against Top1mt. The lack of detectable cross-reactivity of the anti-Top1 polyclonal antibodies against Top1mt was surprising as the identity scores for the core, linker and c-terminal domains of Top1 and Top1mt polypeptides are high (73%, 53% and 75%, respectively) (15, 20). Thus, it is possible that the titer of the available Top1-polyclonal antibodies tested was too low to detect the normally weak Top1mt signal. The C21 monoclonal antibody, raised against the C-terminal peptide of nuclear Top1, also failed to detect Top1mt, which might reflect the existence of different epitope of Top1mt in this region because five out of fifteen amino acids are different (K/R)-F-A-W-A-(I/L)-(D/A)-M-A-(D/G)-E-D-(Y/F)-E-F (Top1/Top1mt). It is also clear that our rabbit polyclonal antibodies generated against recombinant Top1mt are cross-reactive against (nuclear) Top1 (see Figure 1). Nevertheless, having generated antibodies that could react with Top1mt enabled us to demonstrate the induction of Top1-DNA complexes by CPT and the indenoisoquinoline Top1 inhibitor, MJ-III-65 (14, 29).
The induction of Top1mt-DNA complexes by CPT and MJ-III-65 (NSC706744) (14) demonstrates the functional association of Top1mt with mtDNA in organello. Indeed, Top1 must be engaged in its normal nicking-closing cycle to be trapped by drugs in cleavage complexes (14, 22). We detected the formation of cleavage complexes in mtDNA by immunoblotting mtDNA isolated from mitochondria with the Top1mt-antibodies (see Figure 2). We also mapped the Top1mt cleavage complexes as cleavage sites in mtDNA using a variation of the LM-PCR assay we had previously used for mapping Top1 cleavage sites in rDNA (26, 27). We refer to the assay as PL-PCR (see Figure 3). Mapping of Top1mt sites in the regulatory 1.3 kbp region spanning the regulatory region of mtDNA in organello revealed only three detectable sites clustered within a 150 bp region downstream from the D-loop (summarized in Figure 8A). By contrast, mapping of Top1mt sites in a 0.7 kbp overlapping segment in purified mtDNA revealed 9 sites distributed throughout both strands of the mtDNA including in the D-loop region (see Figure 8B). Two of the in organello sites were exactly at the same location in purified mtDNA (H15826 and L15932). Only one site (H15973) was detectable in organello but not in purified mtDNA. However, that site (H15973) was detectable, albeit only weakly in an oligonucleotide in the presence of recombinant Top1mt (Figure 6). The sequestration of the Top1mt sites next to the end of the D-loop in the mitochondrial regulatory region suggest that the mitochondrial nucleoid structure (30) directs the accessibility of mtDNA to Top1mt.
The D-loop is a segment of the mitochondrial genome defined by the presence of a displacement loop, which has been attributed to the pausing of DNA polymerase γ around position 16100 (8) (see Figure 8). Using the modified PL-PCR technique (see Material and Methods) we mapped the 3′-end of human 7S DNA at position 16107 in breast cancer MCF-7 cells, which agrees with previous data reporting position 16106 (8). However, in mouse cells we found a prominent 7S DNA 3′ end at position 15426 instead of multiple sites around that same region (8). This difference may reflect either the difference of methods or the difference of the mtDNA structure in various mitochondrial preparations. Our findings that 7S DNA was reduced by CPT in a Top1mt-dependent manner (see Figure 9) along with the clustering of Top1mt sites downstream from the 3′-end of the D-loop show that Top1mt may play a role in D-loop homeostasis. The effect of CPT on the D-loop could be due to replication or/and transcription alterations and other DNA-related functions in response to the poisoning of Top1mt.
Acknowledgements
We wish to thank Dr. Yung-Chi Cheng, Yale University, New Haven, CT, for the kind gift of monoclonal C21 antibodies against Top1 and for providing information regarding the epitope to which the antibody was made. We also wish Dr. Keli Agama and Dr. Smitha Antony with their help in performing the ICE-bioassays, as well as Dr. Kurt Kohn and the members of the LMP for stimulating discussions.
Funding information: These studies were supported by the Center for Cancer Research (CCR) of the National Cancer Institute, National Institutes of Health.
Abbreviations:
- CPT:
camptothecin
- mtDNA:
mitochondrial DNA
- Top:
topoisomerase
- Top1:
topoisomerase I
- Top1mt:
mitochondrial topoisomerase I
References
- (1).Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- (2).Kang D, Hamasaki N. Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Curr Med Chem. 2005;12:429–441. doi: 10.2174/0929867053363081. [DOI] [PubMed] [Google Scholar]
- (3).Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–232. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- (4).Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen XJ, Butow RA. The organization and inheritance of the mitochondrial genome. Nat Rev Genet. 2005;6:815–825. doi: 10.1038/nrg1708. [DOI] [PubMed] [Google Scholar]
- (6).Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- (7).Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24:813–825. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
- (8).Doda JN, Wright CT, Clayton DA. Elongation of displacement-loop strands in human and mouse mitochondrial DNA is arrested near specific template sequences. Proc Natl Acad Sci U S A. 1981;78:6116–6120. doi: 10.1073/pnas.78.10.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fish J, Raule N, Attardi G. Discovery of a major D-loop replication origin reveals two modes of human mtDNA synthesis. Science. 2004;306:2098–2101. doi: 10.1126/science.1102077. [DOI] [PubMed] [Google Scholar]
- (10).Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell. 2003;14:1583–1596. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Albring M, Griffith J, Attardi G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc Natl Acad Sci U S A. 1977;74:1348–1352. doi: 10.1073/pnas.74.4.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- (13).Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- (14).Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- (15).Zhang H, Meng LH, Pommier Y. Mitochondrial topoisomerases and alternative splicing of the human TOP1mt gene. Biochimie. 2007;89:474–481. doi: 10.1016/j.biochi.2006.11.002. [DOI] [PubMed] [Google Scholar]
- (16).Corbett KD, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu Rev Biophys Biomol Struct. 2004;33:95–118. doi: 10.1146/annurev.biophys.33.110502.140357. [DOI] [PubMed] [Google Scholar]
- (17).Castora FJ, Lazarus GM. Isolation of a mitochondrial DNA topoisomerase from human leukemia cells. Biochem. Biophys. Res. Commun. 1984;121:77–86. doi: 10.1016/0006-291x(84)90690-9. [DOI] [PubMed] [Google Scholar]
- (18).Lin JH, Castora FJ. Response of purified mitochondrial DNA topoisomerase I from bovine liver to camptothecin and m-AMSA. Arch Biochem Biophys. 1995;324:293–299. doi: 10.1006/abbi.1995.0042. [DOI] [PubMed] [Google Scholar]
- (19).Kosovsky MJ, Soslau G. Mitochondrial DNA topoisomerase I from human platelets. Biochim Biophys Acta. 1991;1078:56–62. doi: 10.1016/0167-4838(91)90092-e. [DOI] [PubMed] [Google Scholar]
- (20).Zhang H, Barcelo JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y. Human mitochondrial topoisomerase I. Proc Natl Acad Sci U S A. 2001;98:10608–10613. doi: 10.1073/pnas.191321998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang H, Meng LH, Zimonjic DB, Popescu NC, Pommier Y. Thirteen-exon-motif signature for vertebrate nuclear and mitochondrial type IB topoisomerases. Nucleic Acids Res. 2004;32:2087–2092. doi: 10.1093/nar/gkh525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- (23).Bogenhagen D, Clayton DA. The number of mitochondrial deoxyribonucleic acid genomes in mouse L and human HeLa cells. Quantitative isolation of mitochondrial deoxyribonucleic acid. J Biol Chem. 1974;249:7991–7995. [PubMed] [Google Scholar]
- (24).Subramanian D, Kraut E, Staubus A, Young DC, Muller MT. Analysis of topoisomerase I/DNA complexes in patients administered topotecan. Cancer Res. 1995;55:2097–2103. [PubMed] [Google Scholar]
- (25).Pourquier P, Takebayashi Y, Urasaki Y, Gioffre C, Kohlhagen G, Pommier Y. Induction of topoisomerase I cleavage complexes by 1-β-D-arabinofuranosylcytosine (Ara-C) in vitro and in ara-C-treated cells. Proc. Natl. Acad. Sci. U.S.A. 2000;97:1885–1890. doi: 10.1073/pnas.97.4.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pondarre C, Strumberg D, Fujimori A, Torres-Leon R, Pommier Y. In vivo sequencing of camptothecin-induced topoisomerase I cleavage sites in human colon carcinoma cells. Nucleic Acids Res. 1997;25:4111–4116. doi: 10.1093/nar/25.20.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hsiang YH, Liu LF. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988;48:1722–1726. [PubMed] [Google Scholar]
- (29).Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential induction of topoisomerase I-DNA cleavage complexes by the indenoisoquinoline MJ-III-65 (NSC 706744) and camptothecin: base sequence analysis and activity against camptothecin-resistant topoisomerases I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- (30).Bogenhagen DF, Rousseau D, Burke S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2008;283:3665–3675. doi: 10.1074/jbc.M708444200. [DOI] [PubMed] [Google Scholar]