

Abstract
Acute promyelocytic leukemia (APL) is characterized by expression of promyelocytic leukemia (PML)/retinoic acid (RA) receptor α (RARα) protein and all-trans-RA-mediated clinical remissions. RA treatment can confer PML/RARα degradation, overcoming dominant-negative effects of this oncogenic protein. The present study uncovered independent retinoid degradation mechanisms, targeting different domains of PML/RARα. RA treatment is known to repress PML/RARα and augment ubiquitin-activating enzyme-E1-like (UBE1L) protein expression in NB4-S1 APL cells. We previously reported RA-induced UBE1L and the IFN-stimulated gene, 15-kDa protein ISG15ylation in APL cells. Whether the ubiquitin-like protein ISG15 directly conjugates with PML/RARα was not explored previously and is examined in this study. Transient transfection experiments with different PML/RARα domains revealed that RA treatment preferentially down-regulated the RARα domain, whereas UBE1L targeted the PML domain for repression. As expected, ubiquitin-specific protease 18 (UBP43/USP18), the ISG15 deconjugase, opposed UBE1L but not RA-dependent PML/RARα degradation. In contrast, the proteasomal inhibitor, N-acetyl-leucinyl-leucinylnorleucinal, inhibited both UBE1L- and RA-mediated PML/RARα degradation. Notably, UBE1L induced ISG15ylation of the PML domain of PML/RARα, causing its repression. These findings confirmed that RA triggers PML/RARα degradation through different domains and distinct mechanisms. Taken together, these findings advance prior work by establishing two pathways converge on the same oncogenic protein to cause its degradation and thereby promote antineoplastic effects. The molecular pharmacologic implications of these findings are discussed.
Introduction
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (FAB, M3; ref. 1). APL is characterized by accumulation of abnormal promyelocytes expressing the oncogenic product of the balanced t(15;17) rearrangement between the promyelocytic leukemia (PML) and retinoic acid (RA) receptor α (RARα) genes (2, 3). PML/RARα is a dominant-negative transcriptional factor (3). All-trans-RA treatment is clinically successful in APL by triggering leukemic cell differentiation (4). RA treatment can cause degradation of PML/RARα protein, thereby reversing its dominant-negative effects (5).
Protein degradation is regulated by the ubiquitin-proteasome pathway (6). Ubiquitin, a 76–amino acid polypeptide, is conjugated to target proteins through isopeptide bonds, resulting in monoubiquitinated and polyubiquitinated substrates (6). Polyubiquitination marks the substrate protein for proteolysis by the proteasome (6). Protein ubiquitination is tightly regulated by a ubiquitin-conjugating multienzyme cascade, including the ubiquitin-activating enzyme (E1), ubiquitin conjugases (E2), ubiquitin ligases (E3), and deubiquitinating (DUB) enzymes, which are deconjugases that remove ubiquitin from substrate proteins (6, 7).
The IFN-stimulated gene, 15-kDa protein (ISG15), is a ubiquitin-like protein (7, 8). Ubiquitin-like proteins conjugate intracellular proteins via an isopeptide linkage (7, 8). ISG15ylation is reversible and regulated by a similar set of enzymes as in the ubiquitin system (7, 8). The ubiquitin-activating enzyme-E1-like protein (UBE1L; E1), ubiquitin-conjugating enzyme 8 (E2), estrogen-responsive finger protein (E3), and ubiquitin-specific protease 18 (UBP43 or USP18) have each been reported as enzymes involved in ISG15ylation (9–12). Biochemical steps involved in ubiquitination have been examined previously, but consequences of ISG15ylation and fate of ISG15ylated substrates are still being determined (6–8).
RA is a differentiation-inducing agent that has been used clinically to treat APL and other diseases, as reviewed (13, 14). Notably, RA treatment can induce complete clinical remissions in APL cases that express PML/RARα (4, 15). Prior work revealed pharmacologic treatment with RA overcomes dominant-negative PML/RARα effects (3). RA response is accompanied by induced PML/RARα degradation through caspase- and proteasome-dependent pathways (5, 16), which inhibits PML/RARα activity. RA causes ubiquitination of RARα and PML/RARα, implicating the RARα domain in mediating this degradation (5, 17). Mutagenesis studies identified the DNA-binding domain and the transcription-activation domain (AF-2) of RARα as involved in proteasomal degradation (17). Involvement of the PML domain in RA-mediated degradation of PML/RARα has been less extensively studied. One study found RA engaged caspase-3 activation, which targeted the PML domain of PML/RARα for repression (16).
Prior work with microarray and biochemical analyses uncovered UBE1L as a direct RA target gene that repressed PML/RARα (18, 19). However, how UBE1L causes this was not discerned. RA treatment of APL cells induced ISG15 and ISG15ylation and whether ISG15 physically conjugates with PML/RARα was not determined (19). The current study sought to examine how retinoid-mediated induction of UBE1L and ISG15ylation targets PML/RARα for repression. Findings reported here establish that, in addition to RA effects on ubiquitination of RARα and PML/RARα, RA engaged a distinct biochemical pathway through induction of UBE1L and ISG15. This caused ISG15ylation of the PML domain of PML/RARα to confer its repression. Taken together, these findings revealed that retinoid treatment engages two distinct pathways that converge on PML/RARα to trigger its degradation and overcome its oncogenic effects in APL.
Materials and Methods
Cells and Reagents
The PML/RARα-expressing NB4-S1 APL cell line was cultured in Advanced RPMI 1640 (Invitrogen) supplemented with 2% fetal bovine serum (Gemini Bio-Products), 4 mmol/L l-glutamine (Invitrogen), 100 units/mL penicillin (Invitrogen), and 100 μg/mL streptomycin (Invitrogen) as described previously (18, 19). The BEAS-2B immortalized human bronchial epithelial cell line was cultured in serum-free LHC-9 medium (Biofluids; refs. 18, 19). The COS-1 monkey kidney fibroblast cell line was cultured in Advanced DMEM (Invitrogen) supplemented with 2% fetal bovine serum, 4 mmol/L l-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (20). The Chinese hamster ovary cells were cultured as in prior work (15). Cell lines were each cultured at 37°C in 5% CO2 using a humidified incubator. RA, N-acetyl-leucinyl-leucinylnorleucinal (ALLN), and DMSO were each purchased (Sigma-Aldrich). Stock RA (10 mmol/L) and ALLN (100 mmol/L) solutions were dissolved in DMSO, stored in liquid nitrogen, and used under subdued light during experiments.
Plasmid Constructs
The pCMV-hemagglutinin (HA)-PML/RARα and pSG5-UBE1L expression vectors were described in prior work (18, 19). The coding regions of the long (L) and short (S) forms of PML/RARα, the two predominant isoforms of PML/RARα that are expressed in APL cells, were independently cloned into the pCMV-HA vector using EcoRI and BglII restriction endonuclease sites to engineer pCMV-HA-PML/RARα (S) and pCMV-HA-PML/RARα (L), respectively (3, 15). The pCMV-HA-PML expression vector was engineered using an amplified cloned fragment encoding residues 1 to 390 of PML/RARα. The pSG5-RARα vector was engineered using an amplified cloned fragment coding for amino acids 86 to 462 of RARα. The pcDNA-His-UBP43 plasmid was provided by Dr. Bret Hassel (University of Maryland). The enhanced green fluorescent protein (EGFP) plasmid (pEGFP-N2) and the β-galactosidase reporter plasmid (pCH111) were eachpurchased (Clontech).
Transfection Procedures
Transient transfection of BEAS-2B cells was accomplished using Effectene transfection reagent (Qiagen) and methods described in previous work (21). Briefly, BEAS-2B (1 × 106) cells were plated per treatment group and allowed to attach overnight. Cells were then transfected with Effectene reagent for 24 h. Following this, cells were exposed for 0 to 24 h to different treatments as described in the figure legends. Transient transfection of NB4-S1 APL cells was accomplished using the Nucleofector technology (Amaxa) that combines electroporation with cell-specific optimized solutions to achieve entry of expression vectors into these cells. The EGFP or β-galactosidase reporter plasmids were cotransfected to control for transfection efficiency in transfectants. In selected experiments, single-cell expression assays for transfected EGFP determined the percentages of transfected cells using established methods (20). Coomassie blue staining of gels was also used to confirm comparable loading of protein lysates in each lane. Quantification of signal intensities was accomplished and displayed using previously optimized methods (22).
Immunoblot Analyses
Cells were treated as described in individual figure legends. Whole-cell lysates were prepared using radioimmunoprecipitation assay buffer [150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.4), 1 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L sodium orthovanadate, 1 mmol/L NaF, 1% Triton X-100, 0.1% SDS, and 0.5% sodium deoxycholate] and established techniques (18–20, 23). Protein concentrations in cell lysates were determined by Bradford assays (Bio-Rad); 10 to 90 μg protein/sample were loaded in each lane of 10% SDS-PAGE gels; and immunoblot analyses were done using established techniques (18–20, 22). An anti-RARα polyclonal antibody and anti-actin polyclonal antibody were each purchased (Santa Cruz Biotechnology). An anti-HA monoclonal antibody (Covance Research Products) and an anti-EGFP monoclonal antibody (BD Biosciences) were purchased. The polyclonal antibody recognizing the amino terminus of UBE1L was described previously (18). The monoclonal anti-ISG15 antibody was provided by Dr. Ernest Borden (Cleveland Clinic Foundation). Horseradish peroxidase–linked anti-rabbit and anti-mouse IgG were purchased (Amersham Biosciences). Actin immunoblot analysis served as loading control and EGFP immunoblot analysis served as control for transfection efficiency.
Immunoprecipitation
BEAS-2B cells were plated and transfected as described above. Following treatments, cells were lysed in radioimmunoprecipitation assay buffer, and 1 mg total protein was incubated for 30 min on ice with 20 μL protein A/G plus-agarose beads (Santa Cruz Biotechnology) along with an appropriate anti-mouse (0.25 μg IgG/sample) or anti-rabbit control IgG (0.25 μg IgG/sample; Santa Cruz Biotechnology) in a total volume of 1 mL. The mixture was centrifuged at 3,000 × g for 5 min to clear lysates of proteins nonspecifically bound. The supernatant was incubated overnight with either anti-RARα polyclonal antibody (1:100 dilution) with 20 μL protein A/G plus-agarose beads or 20 μL polyclonal HA-conjugated agarose beads (Santa Cruz Biotechnology) at 4°C to precipitate the immune complexes. The bound proteins were pelleted at 3,000 × g for 5 min and washed with radioimmunoprecipitation assay buffer three times. The obtained pellets were resuspended in 40 μL of 1× SDS sample buffer and then boiled for 5 min before loading onto a 10% SDS-PAGE gel. Immunoblot analyses were done using established techniques (18–20, 22).
Results
Kinetic effects of RA treatment on PML/RARα and UBE1L expression were studied in NB4-S1 APL cells. As shown by immunoblot (Fig. 1A) and densitometric analyses (Fig. 1B), RA repressed PML/RARα protein expression as early as 2 h post-treatment. This decline increased until PML/RARα protein was nearly undetected by 48 h. RA treatment augmented UBE1L expression beginning by 2 h and peaking at 48 h (Fig. 1A). This confirmed and extended previous work showing that RA induced UBE1L expression in NB4-S1 APL cells while increasing both ISG15 expression and conjugation (18, 19). Prior work reported that UBE1L is basally expressed in NB4-S1 APL cells and UBE1L can trigger PML/RARα repression (18, 19). Previous studies also revealed that RA-dependent ubiquitination also can confer PML/RARα degradation (5, 17). RA treatment of NB4-S1 cells augmented both global ubiquitination and ISG15ylation, with ubiquitination preceding ISG15ylation (data not shown). Taken together, these data established that RA activates at least two programs, ubiquitination and ISG15ylation. Each can affect PML/RARα protein stability and cause PML/RARα repression. Experiments were undertaken to understand the mechanistic basis for these pathways causing PML/RARα repression. Expression vectors containing HA tagged to PML/RARα or PML were employed for these studies. Initial transient transfection experiments were conducted in BEAS-2B human bronchial epithelial cells because transfection conditions needed to conduct these experiments were optimized previously in these cells (18, 19).
Figure 1.
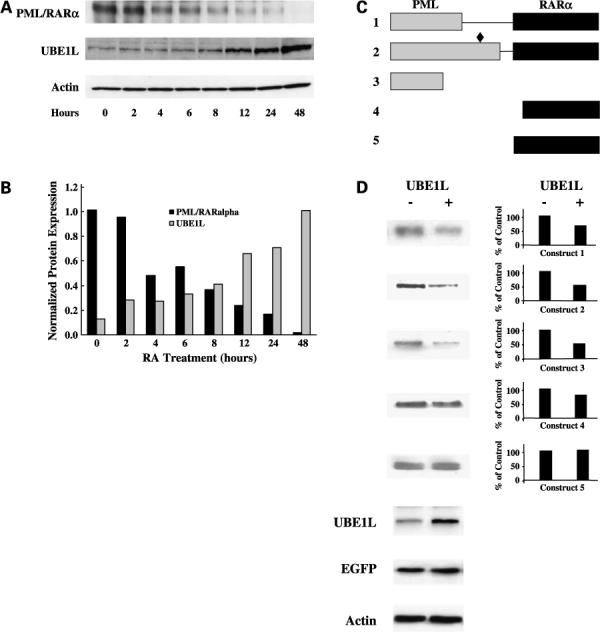
Immunoblot analyses of whole-cell lysates of NB4-S1 APL cells following RA treatment. A, NB4-S1 (2 × 106) cells per experimental group were treated with RA (1 μmol/L) for 0 to 48 h and immunoblot analyses were done to assess effects of RA treatment on expression of PML/RARα and UBE1L. B, scanning densitometric analyses were done for the immunoblots displayed in A. Expression of PML/RARα and UBE1L was each normalized for actin expression, which served as a loading control. This study confirmed induction of UBE1L concurrent with PML/RARα repression following RA treatment of NB4-S1 APL cells. That UBE1L targeted the PML domain of PML/RARα was shown. C, schematic representation of five different PML/RARα expression constructs designated 1 to 5. Gray box, PML domain; black box, RARα domain. Construct 1 depicted PML/RARα (S), which differs from the PML/RARα (L) depicted in construct 2 in that it does not contain the aspartate residue at position 522 (black diamond), the known caspase-3 cleavage site (16). Expression vectors were also engineered to independently contain a portion of the PML domain (construct 3), a portion of the RARα domain (construct 4), or the entire RARα domain (construct 5) of PML/RARα. D, immunoblot analyses for UBE1L and UBE1L-mediated repression of protein products of constructs 1 to 5. Each construct was cotransfected with the UBE1L expression plasmid (+) or with an insertless control vector (−) in BEAS-2B cells followed by immunoblot analyses. Cotransfection of the EGFP expression vector served as a transfection control. Actin expression confirmed similar amounts of protein lysates were loaded. Expression levels of transfected UBE1L are displayed as are the indicated quantified signals. This experiment revealed that UBE1L mediates PML/RARα repression by targeting the PML domain of PML/RARα. PML/RARα (L) construct 2, PML domain construct 3, and RARα domain construct 5 were used for subsequent experiments.
The domain-specific UBE1L effects on PML/RARα expression were determined by cotransfection experiments using constructs independently expressing five different PML/RARα domains (Fig. 1C and D). Previous work reported that RA activates caspase-3, which conferred PML/RARα degradation, and an aspartate residue at amino acid position 522 was identified as the caspase-3 cleavage site in PML/RARα (16). Hence, two PML/RARα constructs were designed, with (Fig. 1C, construct 2) or without (Fig. 1C, construct 1) the aspartate residue at position 522, to discern involvement of caspase-3 in these UBE1L effects. Immunoblot analyses displayed (Fig. 1D) reveal that UBE1L cotransfection repressed both pCMV-HA-PML/RARα (S) (Fig. 1C, construct 1) and pCMV-HA-PML/RARα (L) (Fig. 1C, construct 2), independent of presence of the caspase-3 cleavage site. UBE1L also repressed expression of PML/RARα construct 3 containing a PML domain fragment (Fig. 1C) but had no observed effect on constructs 4 and 5 containing different RARα domains (Fig. 1C). Quantification of signals was displayed in Fig. 1D. Transfection efficiency was assessed by engineered EGFP expression. Engineered UBE1L overexpression did not affect the proportion of EGFP-expressing BEAS-2B cells (113 of 500 = 22.6% of cells for insertless control versus 126 of 505 = 24.95% of cells for UBE1L transfectants). Together, these findings establish UBE1L triggers repression by targeting the PML but not RARα domain of PML/RARα. This effect is independent of the caspase-3 cleavage site.
Immunoblot analyses displayed in Fig. 2 sought to determine whether RA and UBE1L act through similar or different domains of PML/RARα to cause its repression. Transfection assays presented in Fig. 2A confirmed RA (1 μmol/L) treatment for 24 h or UBE1L cotransfection each can repress PML/RARα (L) expression. Because PML/RARα (L) is the PML/RARα isoform present in NB4-S1 APL cells (15), this isoform was the one used in subsequent transfection experiments. RA treatment had no detected effect on expression of the PML domain fragment (Fig. 2B), whereas the RARα domain fragment (Fig. 2C) of PML/RARα was readily down-regulated by RA treatment. This confirmed previous findings that RA targets PML/RARα degradation through its RARα domain (17). In contrast, UBE1L cotransfection repressed the PML domain (Fig. 2B) but had no observed effect on the RARα domain of PML/RARα (Fig. 2C). Quantification of signal intensities is displayed.
Figure 2.
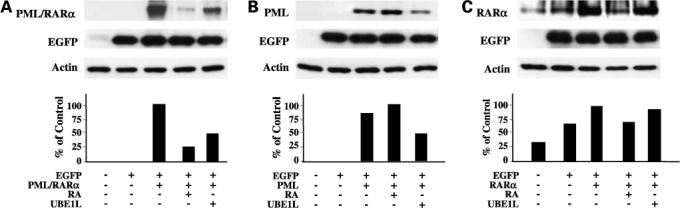
RA and UBE1L affect different PML/RARα domains. BEAS-2B cells were independently transfected with (A) PML/RARα, (B) PML, or (C) RARα expression constructs as described in Materials and Methods. Indicated groups were cotransfected with or without UBE1L and then treated for 24 additional hours with or without RA (1 μmol/L). Immunoblot analyses identified different domains of PML/RARα as being individually targeted by RA and UBE1L for repression of PML/RARα. Bottom, quantification of each of these signals.
UBE1L targeting of the PML domain of PML/RARα revealed that UBE1L likely activated PML/RARα repression through a mechanism distinct from that induced by RA treatment and which affected the RARα domain. To investigate this further, UBP43, the ISG15 deconjugase for UBE1L-dependent ISG15ylation, was cotransfected into BEAS-2B cells with expression constructs independently containing PML/RARα, the PML domain fragment or the RARα domain fragment with or without UBE1L transfection and with or without RA treatment. Immunoblot data displayed in Fig. 3 confirmed as expected that UBP43 cotransfection inhibited UBE1L-mediated repression of PML/RARα (Fig. 3A) and the PML domain (Fig. 3B) of PML/RARα. UBP43 did not oppose RA-mediated repression of PML/RARα (Fig. 3A) or the RARα domain (Fig. 3C) of PML/RARα. Because UBE1L induction can augment ISG15ylation and UBP43 opposes UBE1L effects on PML/RARα stability, this raised the possibility that PML/RARα itself undergoes ISG15ylation through direct interaction with the PML domain. Experiments were undertaken to address this by first determining how proteasomal inhibition would independently affect actions of RA and UBE1L on PML/RARα or its domains.
Figure 3.
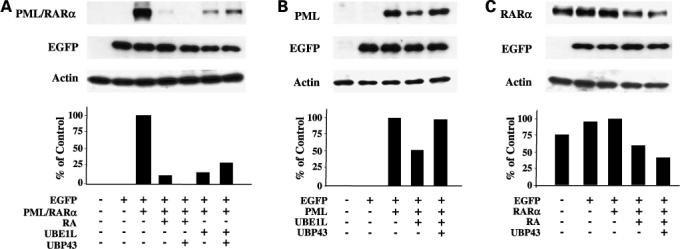
Independent effects of UBP43 on RA- and UBE1L-mediated repression of PML/RARα and its domains. BEAS-2B cells were independently transfected with (A) PML/RARα, (B) PML, or (C) RARα domain containing expression vectors. Indicated groups were either cotransfected with UBE1L or treated for 24 h with vehicle (DMSO) or RA (1 μmol/L). Cells were also cotransfected with or without a UBP43 expression vector as indicated. Immunoblot analyses revealed that UBP43 preferentially opposes UBE1L- but not RA-mediated repression of the indicated PML/RARα species. Bottom, quantification of each of these signals.
Preferential effects of UBP43 in opposing UBE1L-mediated but not RA-dependent PML/RARα repression led to exploring involvement of the proteasome in conferring PML/RARα repression through each of these pathways. Experiments employed as a pharmacologic tool the reversible proteasome inhibitor, ALLN, to investigate proteasomal involvement in these RA and UBE1L effects on PML/RARα. Immunoblot analyses revealed that ALLN stabilized PML (Fig. 4A) and RARα (Fig. 4B) domain expression, indicating that protein products of these domains are each regulated by the proteasome. ALLN treatment antagonized UBE1L-mediated repression of the PML domain (Fig. 4A) and RA-mediated repression of the RARα domain (Fig. 4B) of PML/RARα. These findings strongly implicated proteasomal involvement in regulating stability of these domains of PML/RARα by both UBE1L transfection and RA treatment. These findings are consistent with the view that proteins that are degraded by the proteasome are stabilized by proteasome inhibitors (23).
Figure 4.
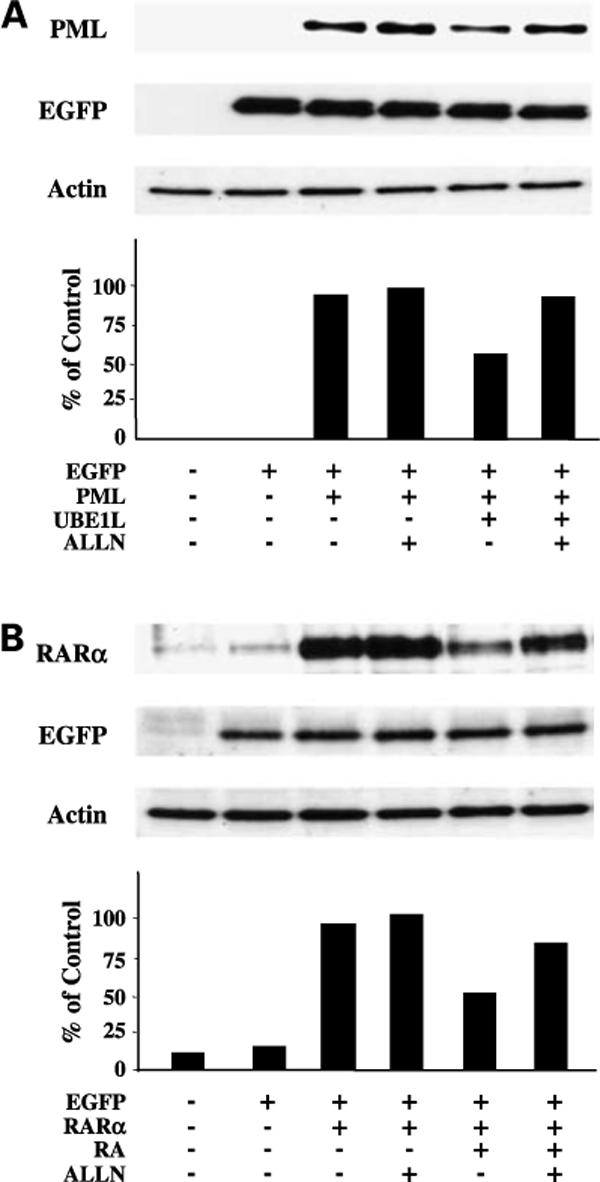
Effects of proteasome inhibition by ALLN on RA- and UBE1L-mediated repression of PML/RARα domains, respectively. BEAS-2B cells were independently transfected with (A) PML or (B) RARα domain containing expression constructs. Indicated groups were also cotransfected with or without UBE1L. Following transfection, cells in the ALLN treatment groups were incubated with ALLN (100 μmol/L) for 90 additional minutes. RA (1 μmol/L) was then added to the respective RA treatment groups and incubated for 6 additional h. Remaining groups were incubated with the vehicle (DMSO) during this same treatment period. Immunoblot analyses revealed that ALLN inhibited RA-dependent repression of the RARα domain and UBE1L-dependent repression of the PML domain of PML/RARα. Bottom, quantification of each of these signals.
UBE1L targets PML/RARα for repression through its PML domain, implicating ISG15 conjugation of PML/RARα as responsible for this effect. Figures 5 and 6 present the conjugation assays conducted to examine this possibility. Several cell lines (BEAS-2B, COS-1, and Chinese hamster ovary) were examined as models for these conjugation assays (Figs. 5 and 6; data not shown), and the BEAS-2B cell line was found as optimal for these studies. This cell line is of human origin and is known to contain the necessary components for activation of ISG15ylation (24). Prior work established that RA treatment induced UBE1L and ISG15 expression in these cells, thus showing that these key components of the ISG15 conjugation pathway are not only expressed but also regulated in these cells (24). BEAS-2B cells can be readily transfected, and previous work revealed that UBE1L overexpression efficiently repressed transfected PML/RARα expression in these cells (18).
Figure 5.
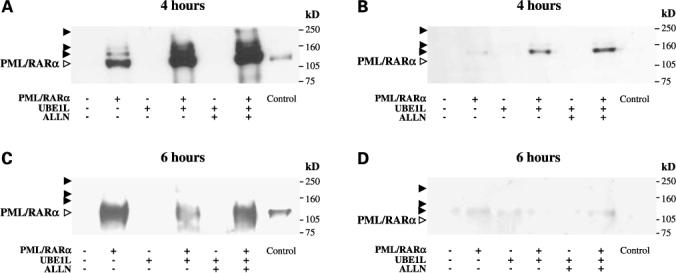
Effects of UBE1L on ISG15ylation of PML/RARα. BEAS-2B cells were individually transfected with PML/RARα containing expression constructs. Indicated groups were cotransfected with or without UBE1L. Following transfection, respective groups were treated with either ALLN (100 μmol/L) or vehicle (DMSO) for 4 (A and B) or 6 (C and D) additional hours. Immunoprecipitation followed by immunoblot analyses were done as described in Materials and Methods. The filters were probed with either a monoclonal anti-HA antibody to detect PML/RARα (A and C) or a monoclonal anti-ISG15 antibody to detect ISG15ylated forms of PML/RARα (B and D). Open arrows, positions of unconjugated PML/RARα proteins; closed arrows, positions of post-translational modification products of PML/RARα; Control, whole-cell lysates of COS-1 cells transfected with PML/RARα serve as a signal for unconjugated PML/RARα protein. These experiments established that UBE1L confers ISG15 conjugation to PML/RARα with subsequent repression of this protein.
Figure 6.
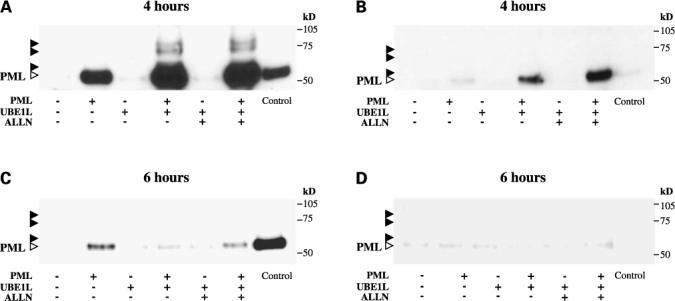
Effects of UBE1L on ISG15ylation of the PML domain of PML/RARα. BEAS-2B cells were individually transfected with the PML domain fragment of PML/RARα containing expression construct. Indicated groups were cotransfected with or without UBE1L. Following transfection, respective groups were treated with either ALLN (100 μmol/L) or vehicle (DMSO) for 4 (A and B) or 6 (C and D) additional hours. Immunoprecipitation followed by immunoblot analyses were done as described in Materials and Methods. The filters were probed with either a monoclonal anti-HA antibody to detect PML (A and C) or a monoclonal anti-ISG15 antibody to detect ISG15ylated forms of PML (B and D). Open arrows, positions of unconjugated PML protein; closed arrows, positions of post-translational modification products; Control, whole-cell lysates of COS-1 cells transfected with PML serve as a signal for the unconjugated form of PML. These experiments established that UBE1L confers ISG15 conjugation to the PML domain of PML/RARα with subsequent repression of this protein.
UBE1L was independently cotransfected with either PML/RARα (Fig. 5), the PML domain of PML/RARα (Fig. 6), or the RARα domain of PML/RARα (data not shown). These studies sought to learn whether ISG15ylation of these species occurred and whether these putative ISG15ylated targeted proteins were repressed. Separate studies (data not shown) determined kinetics of PML/RARα and PML repression following UBE1L cotransfection over a period of 24 h. These experiments revealed that repression of PML/RARα and PML was undetected until 4 h but increased from 6 h post-transfection through 24 h. ISG15 conjugation assays for PML/RARα and the PML domain fragment were done at 4 h post-cotransfection with UBE1L because these species did not undergo appreciable repression at this time point (data not shown). To determine whether these ISG15ylated species undergo repression, a 6-h time point was selected because PML/RARα and PML domain fragments undergo repression by this time point. Following cotransfection with or without UBE1L and treatment with or without ALLN, PML/RARα (Fig. 5) or the PML domain fragment (Fig. 6) was pulled down by immunoprecipitation either after 4 or 6 h and immunoblotted to determine presence and consequence of ISG15 conjugation. Appearance of proteins with higher molecular weights (closed arrows) than transfected PML/RARα (Fig. 5A and C) or PML (Fig. 6A and C) provides evidence for post-translational conjugation. Reactivity of these species with a monoclonal anti-ISG15 antibody confirmed that ISG15 conjugation occurred (Figs. 5 and 6).
Immunoblot analyses in Fig. 5A revealed that PML/RARα was pulled down (open arrows) along with expected higher-molecular-weight species (closed arrows) 4 h after cotransfection with or without UBE1L. This indicated PML/RARα undergoes post-translational conjugation even under basal physiologic conditions. Cell lysates of COS-1 cells transfected with PML/RARα served as positive control (Fig. 5A, lane 7) for determining position of unconjugated PML/RARα protein (open arrows) compared with the three PML/RARα-conjugated species (closed arrows). UBE1L cotransfection (Fig. 5A, lane 4) enhanced the conjugation of PML/RARα and treatment with ALLN (Fig. 5A, lane 6) led to further stabilization of these PML/RARα-conjugated species at this 4-h time point, consistent with the fate of these conjugation proteins as being affected by the proteasome (23). The immunoblot filter displayed in Fig. 5A was stripped and reprobed with an anti-ISG15 antibody (Fig. 5B) to confirm ISG15 immunoreactivity of higher-molecular-weight species (Fig. 5B, lanes 2, 4, and 6). This immunoreactivity established that a physical complex exists between PML/RARα and ISG15. Cell lysate of COS-1 cells transfected with PML/RARα (Fig. 5B, lane 7) did not display immunoreactivity to ISG15 and served as a negative control for this conjugation assay. Figure 5C and D revealed that conjugation of PML/RARα to ISG15 leads to its repression following cotransfection with UBE1L for 6 h. The decrease in PML/RARα signal in the UBE1L cotransfection experiment (Fig. 5C, lane 4) is consistent with this species undergoing degradation, which is inhibited by ALLN treatment (Fig. 5C, lane 6). The lack of immunoreactivity of PML/RARα to ISG15 provides further evidence that PML/RARα conjugation to ISG15 leads to its repression (Fig. 5D).
Figure 6A and B provides evidence for a physical association between ISG15 and the PML domain of PML/RARα. The immunoprecipitated PML domain of PML/RARα is depicted in Fig. 6A (open arrows, lanes 2, 4, and 6), and UBE1L cotransfection for 4 h in Fig. 6A (lane 4) conferred its post-translational conjugation as evidenced by presence of higher-molecular-weight species (closed arrows), which were stabilized by ALLN treatment (Fig. 6A, lane 6). Cell lysates of COS-1 cells transfected with the PML domain of PML/RARα were used as a positive control (Fig. 6A, lane 7) for distinguishing between the position of unconjugated PML immunoreactive protein (open arrows) and three identified higher-molecular-weight PML conjugation products (closed arrows). The immunoblot filter in Fig. 6A was stripped and reprobed with the anti-ISG15 antibody (Fig. 6B) to confirm that UBE1L caused ISG15ylation of the PML domain of PML/RARα (Fig. 6B, lane 4). The depicted ISG15-conjugated species were stabilized by ALLN treatment (Fig. 6B, lane 6), consistent with prior work indicating that ISG15ylated substrates are affected by the proteasome (23). Cell lysates of COS-1 cells transfected with the PML domain of PML/RARα (Fig. 6D, lane 7) did not show immunoreactivity to ISG15 and thus served as a negative control for this conjugation assay. Figure 6C and D show that ISG15ylation of the PML domain of PML/RARα leads to its repression following cotransfection with UBE1L for 6 h. The decrease in PML following UBE1L cotransfection (Fig. 6C, lane 4) indicated that it undergoes repression, which is inhibited by treatment with ALLN (Fig. 6C, lane 6). The loss of immunoreactivity of PML to ISG15 provided evidence for ISG15 conjugation to PML leading to PML repression (Fig. 6D).
Additional experiments were done to confirm that UBE1L directly repressed PML/RARα expression in APL cells. Using the Nucleofector technology, cotransfection in NB4-S1 cells of PML/RARα and UBE1L expression vectors determined (relative to insertless controls) that UBE1L triggers repression of PML/RARα (see Supplementary Fig. S1).4 This established relevancy of these UBE1L transfection experiments in the APL cell context.
Discussion
Targeting expression of the oncogenic PML/RARα fusion protein through its pharmacologic repression has been a feature of APL therapy (1–5). RA treatment induces clinical remissions in APL patients by causing leukemic cell differentiation; this has been associated with triggering of PML/RARα degradation (4, 5, 16–19). It is important to unravel those mechanisms responsible for this because an improved understanding of induced PML/RARα degradation would reveal the interplay of post-translational programs that finely regulate stability and turnover of this oncogenic protein. This knowledge should uncover novel molecular pharmacologic targets. The present study has investigated how RA causes PML/RARα degradation.
RA treatment of APL cells led to PML/RARα repression along with induction of UBE1L expression as reported in our prior work (18, 19). Ubiquitination is known to trigger PML/RARα degradation and UBE1L, the ISG15-activating enzyme, has also been independently shown to cause PML/RARα repression (17–19). These findings raised the prospect that ISG15ylation, like ubiquitination, is involved in regulating PML/RARα degradation and implicated a level of cooperativity that links these related but biochemically distinct post-translational programs (6–8). Because RA is able to engage both of these pathways in APL cells, it would be intriguing to learn through future work whether activation of one affects the other as suggested by prior work (25). Likewise, it will be important to discern precisely how RA regulates these events.
Prior work revealed that UBE1L is a RA-targeted gene (18, 19, 26). The present study independently confirmed that RA treatment caused UBE1L induction in APL cells and this was associated with a repression of PML/RARα protein, indicating a link between these events. Indeed, findings from transient transfection experiments established that UBE1L has domain-specific effects on PML/RARα by targeting the PML but not the RARα domain for repression. Moreover, UBP43, the ISG15 deconjugase, and ALLN, the proteasome inhibitor, were each able to oppose these UBE1L effects. UBE1L caused a physical association between ISG15 and PML, resulting in PML repression. Findings revealed that the target domain for UBE1L effects is located in the PML region of PML/RARα, as stability of transfected RARα domain was unaffected by UBE1L. These findings are of interest as they advance prior work by uncovering a direct link between retinoid response, UBE1L and ISG15 induction, and PML/RARα repression. Also, the fate of the identified ISG15ylated proteins was shown to be regulated by the proteasome.
Transfection experiments established that RA targeted PML/RARα for degradation through the RARα domain. RA effects were opposed by ALLN treatment, but not by UBP43 transfection, underscoring that this pathway was distinct from that engaged by UBE1L activation. Prior work has shown that the DNA binding and transactivation AF-2 domains located in the RARα portion of PML/RARα protein are involved in ubiquitin-proteasomal degradation (17). Additionally, the RARα portion of PML/RARα was found to contain the 26S proteasome regulatory subunit-1 that binds to the 19S proteasome leading to PML/RARα degradation. Yet, the specific lysines regulating this ubiquitination have not yet been identified (17).
The conjugation assay data displayed in Fig. 5A showed the presence of protein species with molecular weights higher than unconjugated PML/RARα protein when cotransfected for 4 h with UBE1L. Similarly, the conjugation assay data presented in Fig. 6A revealed higher-molecular-weight products of the PML domain fragment after 4 h of UBE1L cotransfection. These data indicated PML/RARα and the PML domain undergo multiple post-translational modifications. However, only one of the observed conjugation products in both proteins displayed immunoreactivity with the anti-ISG15 antibody (Figs. 5B and 6B). Notably, transfection for 6 h with UBE1L resulted in repression of both PML/RARα (Fig. 5C and D) and PML (Fig. 6C and D). These findings indicated that PML/RARα undergoes mono-ISG15ylation through the PML domain and ISG15ylated PML/RARα is targeted for repression.
These observations provided a mechanistic basis for repression of PML/RARα by UBE1L in that the ISG15 complexed PML/RARα is readily repressed and only transiently detected in cells. The possibility of ISG15 conjugating to multiple sites in PML/RARα cannot be excluded. Prior work has shown that PML/RARα can conjugate to ubiquitin and SUMO-1 (17, 27). It is possible that the remaining higher-molecular-weight species represented other conjugation products, and these are not detected by the anti-ISG15 antibody. The PML domain of PML/RARα is capable of conjugating to SUMO-1, a member of the ubiquitin-like protein family, whereas the RARα domain of PML/RARα undergoes ubiquitin conjugation (17, 24, 28). That stability of a single oncogenic protein, PML/RARα, is regulated by multiple post-translational modifications raises the possibility of cross-talk between these programs. Determining the nature of this is a future direction of this work, as these findings are relevant to other oncogenic proteins whose stabilities are subjected to similar post-translational modifications.
We reported previously that RA does not augment expression of the classic E1 enzyme in APL cells (18). UBE1L does not activate ubiquitin, thereby ruling out this direct level of cross-talk (6–8). Although ubiquitination and ISG15ylation are distinct with respect to their activating enzymes, E1 and UBE1L, respectively, the conjugase and the ligase enzyme components of these multienzyme cascades display redundancy and can participate in aspects of these programs (7–11). RA might affect these regulatory enzymes to modulate their actions. The specific conjugase and ligase recruited by RA treatment to engage these systems in APL cells remain to be determined. Identification of these enzymes alone would still not explain how RA activates ubiquitination without directly augmenting E1 expression.
Ubiquitin and ISG15 are conjugated to specific lysines in a protein substrate. A comprehensive analysis is indicated in future work using biochemical and site-directed mutagenesis assays to discern which of the lysines in PML/RARα are targeted for these modifications. This knowledge would elucidate how these programs cross-talk and perhaps affect each other. A similar approach has proven useful to uncover the sites of ubiquitination within cyclin D1 (22). Protein sequence analysis revealed that the PML domain alone contains 11 [PML/RARα (S)] or 21 [PML/RARα (L)] lysines, with the RARα domain containing 27 additional lysines. A systematic analysis of each of these lysines should enhance an understanding of how PML/RARα is regulated by retinoids. Prior studies have already uncovered cross-talk between retinoid and IFN signaling pathways as likely responsible for some aspects of the post-translational modifications of PML/RARα (29–31). It would be intriguing to delineate which of the lysines present in PML/RARα are modified before or after RA treatment by ubiquitin, ISG15 and SUMO, and whether any of these sites are shared between them. The physiologic relevance for studying this and for examining differential modifications engaged by UBE1L transfection or RA treatment comes from the experiment conducted in NB4-S1 APL cells where UBE1L overexpression was accomplished and found to preferentially repress PML/RARα expression (see Supplementary Fig. S1).4
In summary, the current study established a direct link between PML/RARα ISG15ylation and stability of PML/RARα protein. These findings provided a mechanistic basis for how UBE1L confers PML/RARα repression. This study also uncovered a pharmacologic agent, RA, which can activate multiple pathways that independently regulate stability and expression of a single oncogenic protein, PML/RARα. Taken together, these findings highlighted the usefulness of retinoids as tools to probe biochemical pathways that regulate the stability of PML/RARα and potentially other oncogenic proteins.
Acknowledgments
We thank Dr. Ernest Borden (Cleveland Clinic Foundation) and Dr. Bret Hassel (University of Maryland) for providing the monoclonal anti-ISG15 antibody and the pcDNA-His-UBP43 plasmid, respectively.
Grant support: NIH, National Cancer Institute grants R01-CA087546, R01-CA062275, and R01-CA111422 (E. Dmitrovsky); Samuel Waxman Foundation Cancer Research Award (E. Dmitrovsky); American Cancer Society Institutional Grant (S.J. Freemantle); and NIH grant T32-CA00959 (S. Blumen).
Footnotes
Supplementary material for this article is available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1.Frankel SR, Miller WH, Jr., Dmitrovsky E. Retinoic acid and its rearranged receptor in the etiology and treatment of acute promyelocytic leukemia. Oncology. 1992;6:74–8. [PubMed] [Google Scholar]
- 2.Xiao YH, Miller WH, Jr., Warrell RP, Dmitrovsky E, Zelenetz AD. Pulsed-field gel electrophoresis analysis of retinoic acid receptor-α and promyelocytic leukemia rearrangements. Detection of the t(15;17) translocation in the diagnosis of acute promyelocytic leukemia. Am J Pathol. 1993;143:1301–11. [PMC free article] [PubMed] [Google Scholar]
- 3.Kakizuka A, Miller WH, Jr., Umesono K, et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RARα with a novel putative transcription factor, PML. Cell. 1991;66:663–74. doi: 10.1016/0092-8674(91)90112-c. [DOI] [PubMed] [Google Scholar]
- 4.Warrell RP, Jr., Frankel SR, Miller WH, Jr., et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med. 1991;324:1385–93. doi: 10.1056/NEJM199105163242002. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida H, Kitamura K, Tanaka K, et al. Accelerated degradation of PML-retinoic acid receptor α (PML-RARA) oncoprotein by all-trans-retinoic acid in acute promyelocytic leukemia: possible role of the proteasome pathway. Cancer Res. 1996;56:2945–8. [PubMed] [Google Scholar]
- 6.Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol. 2005;23:4776–89. doi: 10.1200/JCO.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 7.Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol. 2005;6:599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 8.Staub O. Ubiquitylation and isgylation: overlapping enzymatic cascades do the job. Sci STKE. 2004;245:pe43. doi: 10.1126/stke.2452004pe43. [DOI] [PubMed] [Google Scholar]
- 9.Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20:362–71. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao C, Beaudenon SL, Kelley ML, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-α/h-induced ubiquitin-like protein. Proc Natl Acad Sci U S A. 2004;101:7578–82. doi: 10.1073/pnas.0402528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zou W, Zhang DE. The interferon-inducible ubiquitin-protein isopep-tide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem. 2006;281:3989–94. doi: 10.1074/jbc.M510787200. [DOI] [PubMed] [Google Scholar]
- 12.Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277:9976–81. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 13.Nason-Burchenal K, Dmitrovsky E. The retinoids: cancer therapy and prevention mechanisms. In: Nau H, Blaner WS, editors. Handbook of experimental pharmacology. Vol. 139. Springer-Verlag; Heidelberg: 1999. pp. 301–22. [Google Scholar]
- 14.Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22:7305–15. doi: 10.1038/sj.onc.1206936. [DOI] [PubMed] [Google Scholar]
- 15.Miller WH, Jr., Kakizuka A, Frankel SR, et al. Reverse transcription polymerase chain reaction for the rearranged retinoic acid receptor clarifies diagnosis and detects minimal residual disease in acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1992;89:2694–8. doi: 10.1073/pnas.89.7.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nervi C, Ferrara FF, Fanelli M, et al. Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARα fusion protein. Blood. 1998;92:2244–51. [PubMed] [Google Scholar]
- 17.Zhu J, Gianni M, Kopf E, et al. Retinoic acid induces proteasome-dependent degradation of retinoic acid receptor (RAR) and oncogenic RAR fusion proteins. Proc Natl Acad Sci U S A. 1999;96:14807–12. doi: 10.1073/pnas.96.26.14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitareewan S, Pitha-Rowe I, Sekula D, et al. UBE1L is a retinoid target that triggers PML/RARα degradation and apoptosis in acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:3806–11. doi: 10.1073/pnas.052011299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitha-Rowe I, Hassel BA, Dmitrovsky E. Involvement of UBE1L in ISG15 conjugation during retinoid-induced differentiation of acute promyelocytic leukemia. J Biol Chem. 2004;279:18178–87. doi: 10.1074/jbc.M309259200. [DOI] [PubMed] [Google Scholar]
- 20.Freemantle SJ, Portland HB, Ewings K, et al. Characterization and tissue-specific expression of human GSK-3-binding proteins FRAT1 and FRAT2. Gene. 2002;291:17–27. doi: 10.1016/s0378-1119(02)00594-2. [DOI] [PubMed] [Google Scholar]
- 21.Dragnev KH, Pitha-Rowe I, Ma Y, et al. Specific chemopreventive agents trigger proteasomal degradation of G1 cyclins: implications for combination therapy. Clin Cancer Res. 2004;10:2570–7. doi: 10.1158/1078-0432.ccr-03-0271. [DOI] [PubMed] [Google Scholar]
- 22.Feng Q, Sekula D, Muller R, Freemantle SJ, Dmitrovsky E. Uncovering residues that regulate cyclin D1 proteasomal degradation. Oncogene. 2007 Feb 19; doi: 10.1038/sj.onc.1210309. Epub. [DOI] [PubMed] [Google Scholar]
- 23.Liu M, Li XL, Hassel BA. Proteasomes modulate conjugation to the ubiquitin-like protein, ISG15. J Biol Chem. 2003;278:1594–602. doi: 10.1074/jbc.M208123200. [DOI] [PubMed] [Google Scholar]
- 24.Pitha-Rowe I, Petty WJ, Feng Q, et al. Microarray analyses uncover UBE1L as a candidate target gene for lung cancer chemoprevention. Cancer Res. 2004;64:8109–15. doi: 10.1158/0008-5472.CAN-03-3938. [DOI] [PubMed] [Google Scholar]
- 25.Desai SD, Haas AL, Wood LM, et al. Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res. 2006;66:921–8. doi: 10.1158/0008-5472.CAN-05-1123. [DOI] [PubMed] [Google Scholar]
- 26.Pitha-Rowe I, Petty WJ, Kitareewan S, Dmitrovsky E. Retinoid target genes in acute promyelocytic leukemia. Leukemia. 2003;17:1723–30. doi: 10.1038/sj.leu.2403065. [DOI] [PubMed] [Google Scholar]
- 27.Lallemand-Breitenbach V, Zhu J, Puvion F, et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor α degradation. J Exp Med. 2001;193:1361–71. doi: 10.1084/jem.193.12.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamitani T, Nguyen HP, Kito K, Fukuda-Kamitani T, Yeh ET. Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J Biol Chem. 1998;273:3117–20. doi: 10.1074/jbc.273.6.3117. [DOI] [PubMed] [Google Scholar]
- 29.Nason-Burchenal K, Gandini D, Botto M, et al. Interferon augments PML and PML/RAR α expression in normal myeloid and acute promyelocytic cells and cooperates with all-trans retinoic acid to induce maturation of a retinoid-resistant promyelocytic cell line. Blood. 1996;88:3926–36. [PubMed] [Google Scholar]
- 30.Dao CT, Luo JK, Zhang DE. Retinoic acid-induced protein ISGylation is dependent on interferon signal transduction. Blood Cells Mol Dis. 2006;36:406–13. doi: 10.1016/j.bcmd.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Malakhova OA, Kim KI, Luo JK, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25:2358–67. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]