

Abstract
The topology of β-sheets is defined by the pattern of hydrogen-bonded strand pairing. Therefore, predicting hydrogen bonded strand partners is a fundamental step towards predicting β-sheet topology. At the same time, finding the correct partners is very difficult due to long range interactions involved in strand pairing. Additionally, patterns of aminoacids observed in β-sheet formations are very general and therefore difficult to use for computational recognition of specific contacts between strands. In this work, we report a new strand pairing algorithm. To address above mentioned difficulties, our algorithm attempts to mimic elements of the folding process. Namely, in addition to ensuring that the predicted hydrogen bonded strand pairs satisfy basic global consistency constraints, it takes into account hypothetical folding pathways. Consistently with this view, introducing hydrogen bonds between a pair of strands changes the probabilities of forming hydrogen bonds between other pairs of strand. We demonstrate that this approach provides an improvement over previously proposed algorithms. We also compare the performance of this method to that of a global optimization algorithm that poses the problem as integer linear programming optimization problem and solves it using ILOG CPLEX™ package.
Keywords: Biology and genetics, Combinatorial algorithms
I. Introduction
The prediction of protein structure from protein sequence is a long-held goal that would provide invaluable information regarding the function of individual proteins and the evolution of protein families. The increasing amount of sequence and structure data, allowed to decouple the structure prediction problem from the problem of modeling of protein folding process. Indeed, a significant progress has been achieved by bioinformatics approaches such as homology modeling, threading, and assembly from fragments [22]. At the same time, the fundamental problem of how actually a protein acquires its final folded state remains a subject of controversy. Can successes/failures of computational method shade some light on this issue?
It is generally accepted that proteins fold to their global free energy minimum. Through his famous Paradox, Levinthal made an important point that a protein cannot explore all conformational states in the search of the optimal conformation and therefore a protein chain has to fold by following some directed process or a folding pathway [19]. One view that has been gathering a lot of support since at nearly three decades is the concept of hierarchical protein folding [1], [2], [6], [17], [18], [26]. Consequently, many structure prediction algorithms use hierarchical approach in which the structure is assembled in a bottom up fashion (e.g. where smaller locally folded fragments are assembled into larger folded units [4], [11], [20], [29]).
Protein structure is hierarchic: protein primary sequence is organized into secondary structures and the spatial arrangement of these structures defines protein fold. In this work we focus on particular type of secondary structures - β-strands. Here, by a β-strand we understand a continuous segment of aminoacids adopting an extended conformation, and stabilized by hydrogen bonds between such strands. An assembly of β-strands that, trough hydrogen-bonds between pairs of strands, forms a continuous surface in the space is called a β-sheet. The order of hydrogen-bounded β-strands within a β-sheet defines the topology of the β-sheet.
Studies of β-sheets topology indicate that the way strands assemble into larger sheets may be quite complex. While about half of hydrogen bonded pairs of strands are adjacent in the sequence of strands within protein sequence, many are separated by a significant distance.
The problem of predicting the paring between β-strands, despite of many attempts, remains unsolved. Early work by Hubbard [9] has been followed by other studies directed towards understanding and predicting β-sheet topology [10], [21], [28], [31], [32], [34], [35]. In a more recent work, Cheng and Baldi [5] addressed the strand pairing problem using a three-stage approach. In the first stage they compute, for the input protein sequence, the scores (estimated probabilities) of residue pairs as potential partners in a β-strand pairing. This computation is performed by a neural network with input describing a window of size five around each residue and the additional information about the distance between the two residues in the protein sequence. In the second stage, the above pairwise scores are used to define alignment scores for pairs of strands, and for each pair a highest scoring alignment is found with the use of dynamic programming. The alignment scores are used in the third and final stage to run a greedy selection algorithm.
The important novelty of the approach of Cheng and Baldi when compared with previous methods (e.g.Hubbard [9], Zhu and Braun [35] and Steward and Thornton [31]) is that the prediction of residue pairs that are partners in strand pairing is not performed independently for each pair, but instead it takes into account a wider context; to wit, the information about 10 surrounding residues and the distance between them.
Cheng and Baldi reported 59% positive predictive value and 54% sensitivity which is significantly better than what is achieved by a naive algorithm predicting that all pairs of strands that are consecutive in the sequence form hydrogen bonded partners is space. (The performance of such naive algorithm was approximated to be 42% positive predictive value and 50% sensitivity [5].)
The third stage of algorithm of Cheng and Baldi is a very simple greedy algorithm, which raises a question: Would a more elaborate approach increase the quality of prediction even further? In particular, would a more sophisticated optimization method (e.g., as discussed by Berman and Jeong in [3]) improve on these earlier results. To address this question, we designed a new optimization algorithm. The objective of this algorithm is very similar to the approach of Cheng and Baldi, but rather than having a two-stage greedy selection heuristic, it poses the problem as integer linear programming optimization problem and solves it using ILOG CPLEX™ package.
We also consider a second approach based on the ideas borrowed form principles of hierarchical folding. In her classic 1977 paper, Richardson proposed a set of folding rules where consecutive β-strands grow into larger hydrogen-bonded structures in successive steps, and blocks of strands obtained in this way coalesce, providing they are consecutive in the chain [25]. Richardson showed, by manual inspection, that 37 known strand topologies can be constructed using these rules.
Subsequently, Przytycka et al. [24] proposed a modified set of folding rules for all -proteins where the folding rules were motivated by the prevalent supersecondary structures. The concept of folding rules stems from the assumption of the hierarchic nature of protein folding. Namely, first one or more pairs of neighboring strands are brought together to form super-secondary structures such as hairpins. The formation of these substructures brings to relative spatial proximity pairs of strands that are distant in sequence, increasing the probability of contacts between them. At each stage of this hierarchical process, a compact substructure is formed. It has been hypothesized, that such procedures are related to actual folding pathways. Obviously such folding rules remain hypothetical and simplistic. However, the fact that that majority of fold families (>80%) can be completely folded using rules proposed in [24] indicates that such approach can be helpful in prediction of β-sheet topology in general, and the pairing of β-strands in particular.
In a more recent paper, Maity et al. proposed the view in which previously formed, so called, foldons guide and stabilize subsequent foldons to progressively build the native protein [20]. A subsequent paper proposed predetermined pathway optional error (PPOE) folding model which puts together cooperative formation of native-like foldon units and the sequential stabilization process together generate predetermined stepwise pathways with an allowance for optional missfolding errors [16]. Compact substructures generated by folding rules can be naturally seen as such stabilized foldons.
The assumption that proteins fold through such stepwise process provides also the cornerstone of protein folding simulations in the LINUS program [30] as well as in the more recent zipping and assembly model [23]. In both cases, the energy contacts of neighboring residues is computed first and only after enforcing stable contacts detected in this way, further contacts are estimated. Thus the energy of those subsequent contacts is dependent on the contacts made in the previous step.
How can one bring the ideas behind models of hierarchical folding into strand paring prediction? Scores from a crystal structure typically do not indicate kinetic pathways but rather estimate contact probabilities in a folded structure. Since folding rules of Przytycka et al. ensure that at each step the partially formed substructures are compact, they provide a way of organizing strands into putative foldons without performing folding simulations. In the current work, we consider only one type of a initial foldon, motivated by the hairpin supersecondary structure. This initial compact substructure can be subsequently extended, via a narrow set of folding rules, to form a larger compact unit. In future, we plan to extend this approach to more complex folding steps. Here, we take an advantage of the fact that the scoring function developed by Chang and Baldi, due to the specific machine learning procedure applied by these authors, is very successful in recognizing hairpins (and in general contacts between strands that are consecutive is sequences). This allows for discovering putative initial foldons which can then be propagated with our folding rules. The idea of stabilization and propagation of subsequent foldons implies that strands that brought together into spatial proximity as a result of previously made contacts between other strands, have increased probability of making a contact. In our simple approach, this is achieved by dynamically increasing the scores of pairs of strands that are brought to common spatial neighborhood by formation of a compact substructure.
Both, the linear programming algorithm and greedy folding rule promoting algorithm, provided noticeable improvement over the previous approach. Importantly, a more significant improvement was obtained with the approach that promotes folding rules. This is remarkable, since in the case of integer linear program we are heuristically solving a NP-complete problem using about 100 times more time than folding rules promotion algorithm (almost entire time of the latter algorithm is consumed by the dynamic programming that computes optimal pairing/alignment for each pair of strands).
While the improvement, taken in absolute numbers, is not drastic (about 2.7% in sensitivity and 1% in positive predictive), one has to keep in mind that the problem is quite hard and the improvement of Cheng and Baldi over a naive algorithm was only 4−5 times larger. In another perspective, without any new predictor or data source we decreased the number of false positives by 10% while increasing the number of true positives.
II. Methods
We assume that we are given a protein sequence together with the secondary structure annotation. That is we assume that, for each input protein sequence, we know where each strands starts and ends. Our goal is to find, for each strand, its hydrogen-bounded strand partners.
We start by introducing the common notions used in the description of the three algorithms discussed in this paper:
strand: interval of residue indexes predicted to form a β-strand; we visualize a strand as a sequence of boxes where each box represents an amino-acid. The number in the box corresponds to the index of the amino-acid in the protein sequence.
contact: adjacency (hydrogen bonding) of two strands, as in Fig. 1; each contact is represented by a sequence of adjacent pairs of residues. For each pair of strands, we store only contacts that are optimal for this pair.
side of a strand: Each strand has two sides denoted here by upper and lower side respectively.
side of a contact: Each contact has two sides denoted also by upper and lower side respectively.
parallel contact: a contact where the indexes of residua of contacting strands have the same monotonicity (both are increasing or both are decreasing)
anti-parallel contact: a contact where the indexes of the residua of contacting strands have opposite monotonicity.
score of a contact: sum of scores for all pairs of residues adjacent in the contact. For the original Chang-Baldi algorithm and our Integer Linear Programming Algorithm, the scores for pairs of residues are directly computed by Cheng-Baldi neural network. For our greedy path promoting algorithm, the initial scores are also the neural network scores, but they can be increased (this is done by multiplication of a given score by a scalar) if a folding rule applies.
Fig. 1.
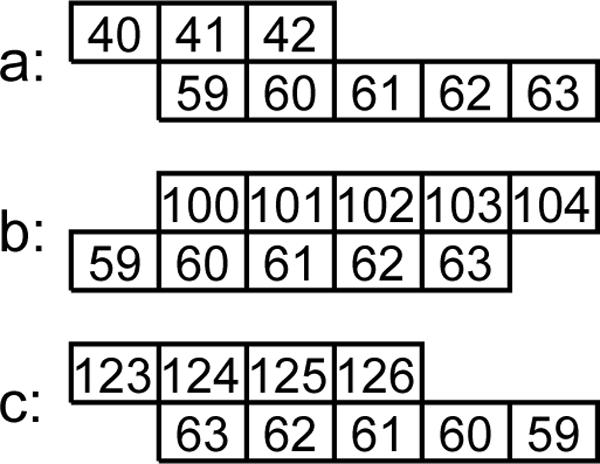
Conflicting and non-conflicting contacts. Each box represents an aminoacid (the number in a box corresponds to the index of the corresponding aminoacid in the protein chain) and rows of boxes represent strands. The contacts b and c are in conflict as they are not overlap-free, while contacts a and c are in conflict as they are not direction-consistent.
Thus a contact, c, is characterized by following parameters: upper strand, lower strand, parallel (or not), the offset (relative shift of the strands). The score of c, E(c) was computed using dynamic programming (we allowed a single gap of length 1 in the alignment).
A solution returned by a strand paring algorithm is a collection of contacts that satisfies the following (minimal) constraints:
uniqueness: a single pair of strands may form at most one contact;
sidedness: contacts of a strand are on one of the two sides of that strand;
overlap-free: each residue can participate in at most one contact on the same side;
direction-consistent: contacts on the same side of a strand are either all parallel, or all anti-parallel.
In Fig. 1, contacts b and c are in conflict as not overlap-free, while contacts a and c are in conflictas not direction-consistent.
While these constraints are necessary, they allow for many impossible combinations of contacts. After some experimentation we added the constraint that a solution is cycle-free (as did Cheng and Baldi [5]). In the data set, among all 916 protein chains and ca. 9000 strands there were only 80 cycles. At the same time, without prohibition of all cycles, our program was returning solutions with many cycles, ca. 99% of them wrong.
Lastly, we disallowed contacts with score below 0.06 from further consideration. This caused the number of predicted contacts (true and false positives in Table I) to roughly coincide with the number of actual contacts (true positives and false negatives).
TABLE I.
Comparison of results of three tested algorithms on a set of 916 protein chains. Note that the discriminating power of the potential function quickly decreases as the separation grows and the statistical quality measures are largely determined by contacts separated by up to three other strands.
| separation | true positive | false negative | false positive | true negative | sensitivity | ppv | corr. coef. |
|---|---|---|---|---|---|---|---|
| greedy — Cheng & Baldi's version | |||||||
| ALL | 5032 | 3140 | 3370 | 61563 | 0.599 | 0.616 | 0.557 |
| 0 | 3748 | 363 | 2136 | 3577 | 0.637 | 0.912 | 0.541 |
| 1 | 521 | 485 | 484 | 7418 | 0.518 | 0.518 | 0.457 |
| 2 | 407 | 523 | 355 | 6710 | 0.534 | 0.438 | 0.423 |
| 3 | 169 | 359 | 161 | 6412 | 0.512 | 0.320 | 0.368 |
| 4 | 100 | 276 | 89 | 5788 | 0.529 | 0.266 | 0.348 |
| 5 | 38 | 241 | 58 | 5130 | 0.396 | 0.136 | 0.209 |
| 6 | 29 | 195 | 32 | 4482 | 0.475 | 0.129 | 0.230 |
| 7 | 11 | 157 | 10 | 3891 | 0.524 | 0.065 | 0.175 |
| 8+ |
9 |
541 |
45 |
18155 |
0.167 |
0.016 |
0.044 |
| ILP optimizer | |||||||
| ALL | 5092 | 3080 | 3253 | 61603 | 0.610 | 0.623 | 0.568 |
| 0 | 3781 | 330 | 2084 | 3621 | 0.645 | 0.920 | 0.558 |
| 1 | 538 | 468 | 552 | 7342 | 0.494 | 0.535 | 0.449 |
| 2 | 427 | 503 | 317 | 6741 | 0.574 | 0.459 | 0.457 |
| 3 | 167 | 361 | 119 | 6447 | 0.584 | 0.316 | 0.398 |
| 4 | 94 | 282 | 72 | 5798 | 0.566 | 0.250 | 0.352 |
| 5 | 36 | 243 | 39 | 5143 | 0.480 | 0.129 | 0.230 |
| 6 | 30 | 194 | 10 | 4498 | 0.750 | 0.134 | 0.306 |
| 7 | 14 | 154 | 13 | 3883 | 0.519 | 0.083 | 0.196 |
| 8+ |
5 |
545 |
47 |
18130 |
0.096 |
0.009 |
0.021 |
| greedy — with pathway-based promotion | |||||||
| ALL | 5089 | 3083 | 3035 | 61821 | 0.626 | 0.623 | 0.577 |
| 0 | 3715 | 396 | 1733 | 3972 | 0.682 | 0.904 | 0.596 |
| 1 | 594 | 412 | 619 | 7275 | 0.490 | 0.590 | 0.473 |
| 2 | 472 | 458 | 385 | 6673 | 0.551 | 0.508 | 0.469 |
| 3 | 142 | 386 | 122 | 6444 | 0.538 | 0.269 | 0.347 |
| 4 | 81 | 295 | 68 | 5802 | 0.544 | 0.215 | 0.318 |
| 5 | 37 | 242 | 41 | 5141 | 0.474 | 0.133 | 0.231 |
| 6 | 30 | 194 | 10 | 4498 | 0.750 | 0.134 | 0.306 |
| 7 | 11 | 157 | 14 | 3882 | 0.440 | 0.065 | 0.158 |
| 8+ | 7 | 543 | 43 | 18134 | 0.140 | 0.013 | 0.034 |
A. ILP formulation
We can view the strand pairing problem as an optimization problem which identifies a solution with the maximum sum of contact scores, where the score are designed to approximate the energy function. As shown in [5], this problem cannot be solved in polynomial time in the worst case. However, in almost all instances in the test set, an ILP solver found provably optimal solutions.
While there are many ILP methods used for protein structure prediction (e.g., see [14], [15], [33]) none of them operated in our particular framework, instead, they were used in the context of all-atom model, threading etc.
A contact is characterized by these parameters: upper strand, lower strand, parallel (or not), the offset (relative shift of the strands). The score E (c) of a contact c was computed using dynamic programming (we allowed a single gap of length 1 in the alignment). We kept only the contacts with the optimal offset values.
For every possible contact c we introduced a variable xc, and for every pair of strands i, j a variable yi,j. The value of xc indicates if contact c is in the solution (xc = 1) or not (xc = 0). Similarly, yi,j = 1 means that strands i and j were paired, i.e.that we selected a contact that binds these two strands together.
To formulate our ILP we introduce two classes of 0−1 vectors: Ci,j such that Cc = 1 if and only if contact c binds strand i with strand j, and (S) such that γ(S)i,j = 1 if and only if . We also set conflict(c, d) to be true if there is a conflict between contact c and contact d.
We wish to solve the following ILP:
This set of constraints is often too large as an input to ILP solver: when the number of strands reaches 20, the number of cycle-free constraints reaches 106 and for the largest protein domains, with more than 40 strands, it exceeds 1012.
To avoid that problem, we start with a single cycle-free constraint with S = {1, . . . , n} and run a row generation loop: we submit ILP, we obtain a solution, and if it contains a cycle of strands we add a cycle-free constraint for its set of nodes. When the number of repetitions is too large (as it happened in ca. 15% of the cases) we give up and return the solution of the greedy algorithm described below.
B. Greedy algorithm with pathway-based promotion
The greedy algorithm constructs a set of contact, by increasing the solution set one contact at the time, always choosing the new contact with the maximum possible score. On one hand, the initial choices may limit subsequent choices and thus prevent the algorithm from finding a solution with the maximum score. On the other hand, the greedy algorithm is much more flexible in checking the consistency requirements, as they do not have to be formulated in the form of linear inequalities. Additionally, such stepwise approach allows for dynamic modification of scores and promoting strand paring consistent with our folding rules.
In the preliminary stage of the algorithm, for each pair of strands we pre-select the best parallel and the best anti-parallel contact, and we order them according to their score. We consider candidates starting with the one with the largest score, and we never consider a candidate again.
We represent contacts with unordered pairs of strands, which means that we do not declare which strand is the upper one and which one is lower. This allows us to avoid, for example, the following anomaly: we greedily choose contacts for pairs (1,2) and (3,4), and decide that, say, strands 1 and 3 are upper ones. Then we cannot choose contact (1,4): if in the latter strand 1 is upper, we have conflict with (1,2), and if strand 4 is lower, we have a conflict with (3,4).
Such representation makes it less obvious how to verify the constraints of sidedness, overlap-free and direction-consistent. (Verifying the constraints of uniqueness, cycle-free constraint, as well as metric consistency described below is straightforward.) The crucial observation is that given a set of contacts we can efficiently test if there exists a consistent assignment of sides. To check for the existence of a consistent assignment we construct the following consistency graph:
The nodes of the consistency graphs are (strand) contacts
There is an edge between two nodes if and only if the corresponding contacts share a strand (e.g.(i, j) and (k, j)) and either (a) one is parallel and one is anti-parallel, or (b) they share a residue of the common strand.
Figure Fig. 2 (a), shows the consistency graph for the set of contacts from figure Fig. 1. Note that two contacts that are connected by an edge in the consistency graph cannot be assigned to the same side of the common strand. Consequently, there exists consistent assignment of sides to these contacts if and only if the corresponding consistency graph is two-colorable. In particular, this criterion tells us that there does not exist an consistent assignment of sides to the tree contacts from figure Fig. 1. Figure Fig. 2 (b), shows consistency graph for a different hypothetical set of contacts and Fig. 2 (c) shows a consistent assignment of sides to these contacts.
Fig. 2.

Examples of consistency graphs. Recall that in the construction of the graph, the contacts are treated as unordered pairs of strands and they don't have assigned sides. (a) The consistency graph for the set of contacts from Figure 1; (b) A consistency graph for a different hypothetical set of contacts; (c) β-sheet corresponding to the connected component of the consistency graph (b)
Since two-colorability of a graph is an easy problem, thus one can test efficiently if there exist a consistent assignment of sides to a given set of contacts.
Connected components of the consistency graph have an important interpretation. Namely, such components correspond to β-sheets. The strands of such β-sheet can be mapped onto a grid in such a way that strands form rows and paired partners are adjacent in common columns (Fig. 2). Such a layout provides a very crude approximation of the β-sheet geometry (in 3D the surface of a β-sheet is actually curved) but still it allows for a conservative estimate of the minimal length of coils that join the strands in the components. If such a coil is actually shorter, we disallow the candidate. As before, we disallow a candidate if it would create a cycle.
Up to this point, the algorithm does not differ from that of Cheng and Baldi in a significant way. (Their notion of consistency as exhibited by their program is a bit different than the one described in the paper, but in the evaluation it was indistinguishable).
The new element introduced in our algorithm is that after selecting a consecutive contact, say between strands i and i+1, we increase the score of contacts between strand pairs (i, i + 2), (i − 1, i + 1), (i − 1, i + 2) by a multiplicative factor (here factor two was used) and change their position within the ordering to reflect that.
This rule is explicitly promoting a folding pathway. It is actually a part of a more general rule in [24], but it restricts it here to the cases of the relatively small separation between strands and thus, as discussed later, the most reliable scores.
As mentioned before, there are biophysical reasons for which the probability of hydrogen bonding between strands i and i + 2 (Fig. 3) is increased under assumption that i is already hydrogen bonded. Namely, strand i + 2 would stabilize the conformation already acquired by strands i and i + 1. The higher probability of bonding between strands i − 1 and i + 2 upon hydrogen bonding between i and i + 1 is in turn justified by the loss of entropy of subchain separating strands i − 1 and i + 2 resulted from the hairpin formation. This rule can be extended to strands i − 2 and i + 3 but with the current scoring schema it had no effect on the results (see Discussion section).
Fig. 3.

Pathway promoting rules. Three configurations that upon formation of contact between strands i and i + 1 promote contacts between (a) strands i − 1 and i + 1 (b) strands i and i + 2 (c) strands i − 1 and i + 1.
III. Results
We used the data set of Cheng and Baldi (see [5], page 176) that consists of 916 protein chains that contain up to 45 β-strands.
We also used the output of their program that given a sequence of amino acids (residues) returns (a) a sequence of secondary structure identifications (α-helix, β-strand, coil) and (b) for every pair of residues classified as β-strand it provides a pseudo-probability that these two residues face each other in a pairing of two β-strands. To evaluate the result we used their file of DSSP identifications of correct secondary structure identifications and correct pairing of β-strand residues.
We defined the population of possible answers in two ways: pairs of β-strands as identified by predict_beta_fasta.sh and as identified by DSSP [12]. Given a pair of predicted strands, we defined the pairing to be true positive(correctly predicted) if for at least one residue of one strand there was a residue in the other strand that was in a contact described by DSSP (predicted by the evaluated program). These two definitions yielded different numbers, but they registered roughly the same differences between various programs, so our conclusions do not seem to depend on this somewhat arbitrary definition.
We compare three programs: the three-stage program of Cheng and Baldi, ILP optimizer and our greedy algorithm with pathway based promotion. The differences in the quality of predictions are very consistent when we use various measures. We use T and F to indicate the number of true and false predictions and ⊕ and ⊖ to indicate positive and negative predictions. In particular, denotes the set of true positives, while denotes the set of false positives. To evaluate the set of prediction, we use the correlation coefficient, as well as positive predictive value/sensitivity pairs.
The correlation coefficient was 0.555 for Cheng and Baldi's, 0.567 for ILP optimizer and 0.577 for the greedy with pathway based promotion.
IV. Discussion and conclusions
We considered two new methods of predicting β-sheet pairing partners using the machine learned scores for inter-residue contacts from [5]. In the first method, we computed optimal set of pairs by solving an instance of integer linear program while the second method was based on ideas borrowed from hierarchical views on protein folding.
The fact that the ILP optimizer provided an improvement over the previous approach indicates that more sophisticated optimization approach can be helpful. On the other hand, imposing pairing preference according to our folding rules provided a more significant improvement, despite the fact that this procedure frequently leads to a solution with suboptimal total score (as measured by the sum of Cheng-Baldi neural network of scores of contacting pairs of residues). Thus the optimal score does not always lead to the correct structure. The fact that stabilization of one portion of a protein's structure contributes to the formation of subsequent contacts [8], [27] suggests that enforcing such folding cooperatively by dynamic change is the scoring function may improve the results of a greedy approach to strand pairing prediction.
Such folding cooperativity has been explored by Dill et al. in their hydrophobic zipper hypothesis: hydrophobic contacts act as constraints that bring other contacts into spatial proximity, which then further constrain and zip up the next contacts, etc. [7] and, in the zipping and assembly model [23]. The assumption that proteins fold through such stepwise process is also a cornerstone of protein folding simulations in the LINUS program [30].
The proposed folding-rule promoting strand pairing algorithm can be seen as a generalization of the hydrophobic zipper hypothesis, where the cooperativity of folding is modeled on the secondary structure level rather than on the residue level. The folding rules are designed so that each stage forms another foldon-like substructure. Here we implemented only a very basic set of folding rules where subsequent folding steps follow a formation of a hairpin-like structure. In the future, more complete set of rules based on the work of Richardson [25] and Przytycka [24] et al. could be added.
Effectively, currently implemented folding rules apply only to strands that, while not being neighbors in protein sequence, are separated by relatively small number of other strands. However, in a recent work, Kamat and Lesk [13] demonstrated that a vast majority of contacts between secondary structures in general and between strands in particular are between secondary structures which a separated in the sequence by at most a few other secondary structures. Thus, for most proteins, the correct predictions of contacts between those strands determine all or nearly all contacts. Therefore correct prediction of the contacts between pairs of strands that are not separated by very large sequence distance is extremely important for the strand pairing prediction.
In this work we demonstrated that a simple, model based algorithm, may perform better than a heavy duty integer linear programming. Our method of enforcing folding rules by simply increasing scores by a multiplicative factor is arguably naive. However having only one additional parameter decreases the possibility of overtraining. Our results suggest that the future line of research should include developing a scoring function that would allow to explore the cooperativivity of the folding process more fully.
Fig. 4.

The table of pairwise scores for 2C-Methyl-D-erythritol-2,4-cyclodiphosphate Synthase (PDB id: 1iv1, chain a). The entries in the table correspond to color-coded scores: purple codes correspond to scores in the interval 2/3 to 1, and each subsequent color-code (purple-blue, blue, blue-green, red, red-orange, orange, orange-yellow and yellow etc.) codes an interval decreased by 2/3 factor (and white for the remaining values down to zero). Black background codes the true contacts, purple ovals are the contacts found by Cheng & Baldi, and the pink ovals are the contacts found by our version of greedy. After contact 2−3 was selected, contact 1−4 (between strand 1 and strand 7) was promoted over 1−2; once we got contacts 1−4−3−2, contact 1−2 was blocked by cycle-free rule; moreover 1−5 was blocked by 5−6 and 5−7, thus 1−7 became the best available contact for 1 — as well as for 7.
Acknowledgments
The authors thank George D. Rose (JHU), Bonnie Berger (MIT) and Arthur M. Lesk (PSU) for an insightful discussions. We also thank Jailing Cheng for help in using their program. This work was supported in part by the intramural research program, National Institutes of Health, National Library of Medicine.
References
- 1.Baldwin RL, Rose GD. Is protein folding hierarchic? I. II. Folding intermediates and transition states . Trends in Biochemical Sciences. 1999;24(2):77–83. doi: 10.1016/s0968-0004(98)01345-0. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin RL, Rose GD. Is protein folding hierarchic? I. Local structure and peptide folding. Trends in Biochemical Sciences. 1999;134(3):26–33. doi: 10.1016/s0968-0004(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 3.Berman P, Jeong J. Consistent sets of secondary structures in proteins. Algorithmica (Online First) 2007 [Google Scholar]
- 4.Bystroff C, Baker D. Prediction of local structure in proteins using a library of sequence-structure motifs . Journal of Molecular Biology. 1998;281(3):565–577. doi: 10.1006/jmbi.1998.1943. [DOI] [PubMed] [Google Scholar]
- 5.Cheng J, Baldi P. Three-stage prediction of protein beta-sheets by neural networks, alignments and graph algorithms. Bioinformatics. 2005;21(suppl 1):i75–84. doi: 10.1093/bioinformatics/bti1004. [DOI] [PubMed] [Google Scholar]
- 6.Crippen GM. The tree structural organization of proteins. Journal of Molecular Biology. 1978;126:315–332. doi: 10.1016/0022-2836(78)90043-8. [DOI] [PubMed] [Google Scholar]
- 7.Dill KA, Fiebig KM, Chan HS. Cooperativity in Protein-Folding Kinetics. Proceedings of the National Academy of Sciences. 1993;90(5):1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginsburg Ann, Carroll William R. Some specific ion effects on the conformation and thermal stability of ribonuclease. Biochemistry. 1965;4(10):2159–2174. [Google Scholar]
- 9.Hubbard TJ, Park J. Fold recognition and ab initio structure predictions using hidden markov models and β-strand pair potentials. Proteins: Structure, Function, and Genetics. 1995;23(3):398–402. doi: 10.1002/prot.340230313. [DOI] [PubMed] [Google Scholar]
- 10.Huthinson EG, Sessions RB, Thornton JM, Woolfson DN. Determinants of strand register in antiparallel β-sheets of proteins. Protein Science. 1998;7(11):2287–2300. doi: 10.1002/pro.5560071106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inbar Y, Benyamini H, Nussinov R, Wolfson HJ. Protein structure prediction via combinatorial assembly of sub-structural units. Bioinformatics. 2003;19(suppl 1):i158–168. doi: 10.1093/bioinformatics/btg1020. [DOI] [PubMed] [Google Scholar]
- 12.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 13.Kamat AP, Lesk AM. Contact patterns between helices and strands of sheet define protein folding patterns. Proteins: Structure, Function, and Bioinformatics. 2007;66(4):869–876. doi: 10.1002/prot.21241. [DOI] [PubMed] [Google Scholar]
- 14.Kingford CL, Chazelle B, Singh M. Solving and analyzing side-chain positioning problems using linear and integer programming. Bioinformatics. 2004;21(7):1028–1036. doi: 10.1093/bioinformatics/bti144. [DOI] [PubMed] [Google Scholar]
- 15.Klepeis JL, Floudas CA. Astro-fold: A combinatorial and global optimization framework for ab initio prediction of three-dimensional structures of proteins from the amino acid sequence. Biophysical Journal. 2003 October;85:2119–2146. doi: 10.1016/s0006-3495(03)74640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishna Mallela M.G., Walter Englander S. A unified mechanism for protein folding: Predetermined pathways with optional errors. Protein Sci. 2007;16(3):449–464. doi: 10.1110/ps.062655907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kryshtafovych A, Venclovas C, Fidelis K, Moult J. Protein folding: From the levinthal paradox to structure prediction . Journal of Molecular Biology. 1999;293(2):283–293. doi: 10.1006/jmbi.1999.3006. [DOI] [PubMed] [Google Scholar]
- 18.Lesk AM, Rose GD. Folding Units in Globular Proteins. PNAS. 1981;78(7):4304–4308. doi: 10.1073/pnas.78.7.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levinthal C. Are there pathways for protein folding? . Journal de Chimie Physique et de Physico-Chimie Biologique. 1968;65:44. [Google Scholar]
- 20.Maity Haripada, Maity Mita, Krishna Mallela M. G., Mayne Leland, Walter Englander S. Protein folding: The stepwise assembly of foldon units. Proceedings of the National Academy of Sciences. 2005;102(13):4741–4746. doi: 10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menke M, King J, Berger B, Cowen L. Wrap-and-pack: A new paradigm for beta structural motif recognition with application to recognizing beta trefoils. Journal of Computational Biology. 2005;12(6):777–795. doi: 10.1089/cmb.2005.12.777. [DOI] [PubMed] [Google Scholar]
- 22.Moult J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Current Opinion in Structural Biology. 2005;15(3):285–289. doi: 10.1016/j.sbi.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Banu Ozkan S, Albert Wu G, Chodera John D., Dill Ken A. Protein folding by zipping and assembly. Proceedings of the National Academy of Sciences. 2007;104(29):11987–11992. doi: 10.1073/pnas.0703700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Przytycka TM, Srinivasan R, Rose GD. Recursive domains in proteins. Protein Science. 2002;11(2):409–417. doi: 10.1110/ps.24701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richardson JS. beta-Sheet topology and the relatedness of proteins . Nature. 1977;268(5620):495–500. doi: 10.1038/268495a0. [DOI] [PubMed] [Google Scholar]
- 26.Rose GD. Hierarchic organization of domains in globular proteins. Journal of Molecular Biology. 1979;134(3):447–470. doi: 10.1016/0022-2836(79)90363-2. [DOI] [PubMed] [Google Scholar]
- 27.Rose George D., Fleming Patrick J., Banavar Jayanth R., Maritan Amos. A backbone-based theory of protein folding. Proceedings of the National Academy of Sciences. 2006;103(45):16623–16633. doi: 10.1073/pnas.0606843103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruczinski I, Kooperberg C, Bonneau R, Baker D. Distributions of beta sheets in proteins with application to structure prediction . Proteins: Structure, Function, and Genetics. 2002;48(1):85–97. doi: 10.1002/prot.10123. [DOI] [PubMed] [Google Scholar]
- 29.Srinivasan R, Rose GD. LINUS: A hierarchic procedure to predict the fold of a protein. Proteins: Structure, Function, and Genetics. 1995;22(2):81–99. doi: 10.1002/prot.340220202. [DOI] [PubMed] [Google Scholar]
- 30.Srinivasan Rajgopal, George Rose D. Ab initio prediction of protein structure using LINUS. Proteins: Structure, Function, and Genetics. 2002;47(4):489–495. doi: 10.1002/prot.10103. [DOI] [PubMed] [Google Scholar]
- 31.Steward RE, Thornton JM. Prediction of strand pairing in antiparallel and parallel β-sheets using information theory . Proteins: Structure, Function, and Genetics. 2002;48(2):178–191. doi: 10.1002/prot.10152. [DOI] [PubMed] [Google Scholar]
- 32.Woolfson DN, Evans PA, Hutchinson EG, Thornton JM. On the conformation of proteins: The handedness of the connection between parallel β-strands. Journal of Molecular Biology. 1977;110:269–283. doi: 10.1016/s0022-2836(77)80072-7. [DOI] [PubMed] [Google Scholar]
- 33.Xu J, Li M, Kim D, Xu Y. Raptor: Optimal protein threading by linear programming. Journal of Bioinformatics and Computational Biology. 2003;1(1):85–117. doi: 10.1142/s0219720003000186. [DOI] [PubMed] [Google Scholar]
- 34.Zhang C, Kim S.-Hou. The anatomy of protein [beta]-sheet topology . Journal of Molecular Biology. 2002;299(4):1075–1089. doi: 10.1006/jmbi.2000.3678. [DOI] [PubMed] [Google Scholar]
- 35.Zhu H, Braun W. Sequence specificity, statistical potentials, and three-dimensional structure prediction with self-correcting distance geometry calculations of beta-sheet formation in proteins. Protein Science. 1999;8(2):326–342. doi: 10.1110/ps.8.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]