

Abstract
Integrin-linked kinase (ILK) has been implicated in the regulation of a range of fundamental biological processes such as cell survival, growth, differentiation, and adhesion. In platelets ILK associates with β1- and β3-containing integrins, which are of paramount importance for the function of platelets. Upon stimulation of platelets this association with the integrins is increased and ILK kinase activity is up-regulated, suggesting that ILK may be important for the coordination of platelet responses. In this study a conditional knockout mouse model was developed to examine the role of ILK in platelets. The ILK-deficient mice showed an increased bleeding time and volume, and despite normal ultrastructure the function of ILK-deficient platelets was decreased significantly. This included reduced aggregation, fibrinogen binding, and thrombus formation under arterial flow conditions. Furthermore, although early collagen stimulated signaling such as PLCγ2 phosphorylation and calcium mobilization were unaffected in ILK-deficient platelets, a selective defect in α-granule, but not dense-granule, secretion was observed. These results indicate that as well as involvement in the control of integrin affinity, ILK is required for α-granule secretion and therefore may play a central role in the regulation of platelet function.
Introduction
Integrin-linked kinase (ILK) was originally identified and cloned in 1996 as the result of a 2-hybrid screen for β1-integrin–interacting proteins1 and is present in many tissues including heart, placenta, liver, kidney, pancreas,1 and platelets.2 Originally recognized for its ability to regulate integrin-mediated signal transduction, ILK has subsequently been shown to be involved in normal cardiac function,3,4 cardiac-hypertrophy,5,6 hepatocyte differentiation,7 dendrite formation,8 bone development,9 and several types of cancer.10–12
The structural characterization of this 59-kDa cytosolic protein implicates ILK in the coordination of cell signaling through its potential to interact directly and indirectly with receptors, cytoso-lic-signaling proteins, and cytoskeletal proteins across its 3 structurally conserved domains.1 The C terminus of ILK contains a serine-threonine kinase domain that binds to β1- and β3-containing integrins, parvin, paxillin (Hic5), kindlin (UNC-112 or MIG2) and PDK1.2,13–15 Adjacent to this is a pleckstrin homology domain that binds to phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3).16 The binding of PtdIns(3,4,5)P3 is thought to regulate kinase activity and facilitates interaction with α-parvin, which supports the formation of focal adhesions.13,17 At the N terminus of this molecule 4 ankyrin repeats bind to both ILK-associated phosphatase (ILKAP) and particularly interesting new cystine-histidine protein (PINCH).18–25 These interactions with integrins, intercellular signaling complexes and matrix proteins link the structural frameworks of the extracellular matrix (ECM) and the cytoskeleton and allow the transmission of bidirectional signals.26
Debate has surrounded whether the cellular role of ILK involves kinase activity. Work in ILK-deficient murine endothelial cells and in HEK-293 cells, using siRNA to silence ILK expression, has demonstrated that ILK has a role in the phosphorylation of protein kinase B (PKB)/Akt at Ser473.27,28 This was confirmed by coimmunoprecipitation, mass spectrometry, and mutational analysis.29 The sequence of the kinase domain, however, is not well conserved between species,18 with sequence differences to other serine/threonine kinases found in the catalytic loop and conserved DXG motif.30 In some systems kinase dead mutations are still able to function or rescue function in ILK-deficient cells,26,31 and in ILK-deficient mouse fibroblasts PKB phosphorylation is unaffected.31 These discrepancies may underscore cell-specific and potentially species-specific differences in the mechanisms through which ILK contributes to cell function.
Platelets play a fundamental role in hemostasis. Upon injury they become exposed to subendothelial matrix proteins, particularly collagens, to which they initially adhere via the GPIb-V-IX complex and plasma von Willebrand factor (VWF). This interaction with collagen is stabilized via direct interactions between platelet collagen receptor glycoprotein VI (GPVI) and integrin α2β1. The binding of collagen to GPVI and receptor clustering results in the tyrosine phosphorylation of the FcR γ-chain and triggers a signaling pathway that leads to PLCγ2 phosphorylation and calcium mobilization.32 At sites of injury platelets are also exposed to thrombin generated due to the activation of the coagulation pathways, resulting in platelet signaling through the protease-activated receptors, PAR1 (PAR3 in mouse), and PAR4.33 Both signaling pathways result in cell spreading, positive feedback signaling, and signaling to other platelets. The latter occurs via secretion of thrombotic factors and inside-out integrin signaling, through which the affinity of integrins α2β1 and αIIbβ3 become increased. The bivalent binding of fibrinogen to integrin αIIbβ3 supports platelet-platelet adhesion and hence thrombus formation. The resulting thrombus is stabilized by the increasing affinity of α2β1 for collagen.34
Platelets depend upon integrins for their role in hemostasis, as demonstrated by conditions where expression is reduced, such as Glanzmann thrombocytopenia.35 Through inside-out and outside-in signaling the integrins mediate a variety of cellular processes36 and are important in transduction pathways linking extracellular adhesive proteins (such as VWF, fibrinogen, and collagen), with intracellular signaling molecules (such as focal adhesion kinase [FAK] and paxillin) and cytoskeletal components (including talin, filamin, and α-actinin37,38).
In this study a conditional ILK-deficient mouse model was used to examine the role of ILK in the regulation of platelet function, signaling, and thrombus formation under arterial flow conditions. The data indicate an important role for this molecule in the control of platelet biology.
Methods
Reagents
Collagen-related peptide (CRP), a GPVI-selective agonist was synthesized by Pepceuticals LTD (Nottingham, United Kingdom) and chemically cross-linked.39 Horm-Chemie collagen (collagen fibers from equine tendons) was purchased from Nycomed (Munich, Germany) and thrombin from Sigma (Poole, United Kingdom). Antibodies for immunoblotting: anti-ILK (C-19, sc-7516), anti-α2 (N-19, sc-6586), anti-αIIb (C-20, sc-6602), anti-β1 (N-20, sc-6622), anti-β3 (C-20, sc-6602), anti-PINCH1 (N-19, sc-47 914), anti-talin (C-20, sc-7534), and anti-PLCγ2 (Q-20, sc-407) were purchased from Santa Cruz Biotechnology (Autogenbioclear; Calne, United Kingdom). Anti–P-selectin (RB40.34) was supplied by BD Biosciences, anti-phosphotyrosine (4G10) by Upstate (Millipore, Watford, United Kingdom), and anti-GAPDH (6C5), anti-kindlin polyclonal (ab36957), and anti-parvin alpha (ab11336) by AbCam (Cambridge, United Kingdom). For flow cytometry phycoerythrin (PE)–labeled CD62P (P-selectin) and fluorescein isothiocyanate (FITC) anti-mouse CD41 (integrin αIIb) were purchased from BD Biosciences. FITC-labeled fibrinogen was produced by incubating fibrinogen (2.5 mg/5 mL in phosphate-buffered saline [PBS]) in the dark with FITC (1 mg/mL in acetonitrile) in the presence of reaction buffer (PBS with 0.1% SDS, 1% triethylamine at pH 9) in a 1:1:1 ratio. The solution was gently mixed at 65°C in the dark for 30 minutes, and FITC-labeled fibrinogen then purified by ultrafiltration. Reagents used for the platelet factor 4 assay include: anti-rabbit (whole molecule)–peroxidase (Sigma A6154), rabbit anti–human platelet factor 4 (AYNRHPF4; Accurate Chemical & Scientific, Westbury, NY) and 3,3′,5,5′-tetramethyl-benzidine (TMB) was from Sigma. The chemiluminescent Western blotting substrate was supplied by Pierce (Perbio, Cramlington, United Kingdom) and x-ray film by GE Healthcare (Little Chalfont, United Kingdom). Densitometry was performed using a BIO-RAD GS-710 calibrated imaging densitometer, and Quantity One analysis software. Fluo-4AM was from Invitrogen (Fisher Scientific, Loughborough, United Kingdom).
Generation of mice
LoxP-flanked ILK-transgenic mice3,9,27 were crossed with mice containing a Cre transgene under control of the Mx1 promoter. Founders and breeding stock were backcrossed onto the C57BL6 genetic background and litters were genotyped by polymerase chain reaction (PCR) to select ILK(lox/lox):MxCre31 mice and littermate ILK(+/+):MxCre (control [Ctrl]) mice. All mice were given an intraperitoneal injection of 300 μg Poly(I)-Poly(C) (pIpC) 8 days prior to experiments.
Preparation and aggregation of washed platelets
Blood was obtained from KO (ILK(lox/lox):MxCre) and control (ILK(+/+):MxCre) litter-matched mice via cardiac puncture following termination. Blood (1 mL) was drawn into a syringe containing acidic citrate dextrose (100 μL) (ACD; 120 mM sodium citrate, 110 mM glucose, 80 mM citric acid) as anticoagulant. Platelets were prepared from whole blood by differential centrifugation in the presence of prostacyclin (0.1 μg/ mL) and resuspended in modified Tyrode-HEPES buffer (134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12mM NaHCO3, 20 mM HEPES, 5 mM glucose, 1 mM MgCl2, pH 7.3) to a density of 8 × 108 cells/mL and rested for 30 minutes at 30°C prior to experiments. Platelets (90 μL) were stimulated with collagen at 37°C with continuous stirring (1200 rpm) in an optical aggregometer for 90 seconds. For aggregation experiments in platelet-rich plasma (PRP), blood was taken into citrate (3.13% [wt/vol]). PRP was collected and control and KO samples matched for platelet number (at a density no less than 2 × 108) and stimulated with collagen at 37°C with continuous stirring (1200 rpm) in an optical aggregometer. All protocols involving the use of animals were approved by the University of Reading Local Ethical Review Panel and are authorized by a United Kingdom Home Office License.
Immunoblotting and immunoprecipitation
Immunoprecipitation, SDS-PAGE, and immunoblotting onto polyvinylidine difluoride membrane were all performed using standard techniques.40 Densitometry was performed using GAPDH levels to normalize for protein loading.
Flow cytometry
Surface integrin αIIbβ3 levels were assessed with anti-αIIb (CD61) antibody and fibrinogen binding was measured using FITC-labeled fibrinogen. Surface exposure of P-selectin was observed as a measure of α-granule secretion using a PE-conjugated anti-CD62P (P-selectin) antibody. Flow cytometry was carried out on a FACScan system and analyzed using CellQuest software (Becton Dickinson, Oxford, United Kingdom).
Dense granule secretion
Platelets (8 × 108 cells/mL) were loaded with [3H] 5-HT (37 kBq/mL) by incubation in PRP for 1 hour at 30°C. [3H] 5-HT secretion following activation was measured by scintillation spectrometry.41
α-granule secretion: assay for platelet factor 4 (PF4)
Forty microliters of washed platelets at a density of 4 × 108 were stimulated with collagen (10 μg/mL) or thrombin (5 units/mL) for 90 seconds and stopped with 5 μL of 6% glutaraldehyde, then immediately centrifuged. Ten microliters of supernatant of lysed (maximum), resting and stimulated platelets were added in triplicate to 96-well plates containing 100 μL of binding buffer (50 mM 3-[cyclohexylamino]-1-propanesulfonic acid [CAPS] at pH 11.5). After incubating for 2 hours the plate was washed twice with wash buffer (0.165 mM Na2HPO4, 0.035 mM NaH2PO4, 1.5 mM NaCl) then blocked with 5% dried skimmed milk in TBS for 30 minutes. One hundred microliters of antiplatelet factor 4 antibody (10 ng/mL) was added to the wells for 1 hour and after 2 washes, 1:500 dilution of anti-rabbit peroxidase was added for 1 hour. Plates were washed 3 times and developer (3,3′,5,5′-tetramethyl-benzidine [TMB] in 40% methanol and 0.015% H2O2) was added, color was allowed to develop for 15 minutes before 50 μL HCl (1 M) was added, and the plate was read at 450 nm on a Molecular Devices Emax precision microplate reader.
Intracellular calcium mobilization analysis
Fluo-4AM was added to murine PRP, prepared from whole blood by centrifugation, at a final concentration of 10 μM and incubated for 45 minutes at 30°C. Platelets were centrifuged and resuspended in Tyrode as normal and rested for 30 minutes. Fifty microliters of washed platelets were added to 96-well black-walled, clear-bottom microtiter plates (Greiner Bio-One, Frickenhausen, Germany). The plate was loaded into a Flexstation (Molecular Devices, Wokingham, United Kingdom) and allowed to equilibrate to temperature (37°C). CRP was then added to the plate to give 10 μg/mL final concentration (sufficient to stimulate full aggregation). Settings were: excitation 488 nm, emission 525 nm, reading sensitivity “normal,” and photomultiplier tube (PMT) sensitivity “high.” Run time was 180 seconds, with a fluorescence reading taken every 1.4 seconds. Analysis was performed using Softmax Pro software (Molecular Devices).
Thrombus formation in whole blood under arterial flow conditions
Platelets in whole blood were loaded with the lipophilic fluorescent dye 3,3′-dihexyloxacarbocyanine iodide (DiOC6; 0.5 ng/mL final) in 50% ethanol (0.05% [vol/vol] final) in the dark at room temperature for 30 minutes prior to perfusion. This had no effect on platelet aggregation (not shown), consistent with other reports.42 Whole blood was perfused through a collagen-coated (100 μg/mL) glass microcapillary using a syringe pump at a shear rate of 1000 s−1. Twenty microliters of Tyrode-HEPES buffer was then perfused though the system to remove unattached cells. The capillary was transferred to a Leica DMIRE2 inverted confocal microscope, and images were collected at room temperature through the thrombi at 2-μm intervals in the Z-plane (using N PLANL 20 × 0.4 objective lens with 0-2 mm correction) within 5 randomly selected fields of view. The resulting Z-stack data were analyzed using TCS SP2 software (Leica, Milton Keynes, United Kingdom) to calculate thrombus depth. t tests were performed using Microsoft Excel after confirming normality using SPSS software. Results are presented as mean values plus or minus standard error of the mean. P values of less than .05 were considered significant.
Tail-bleeding assay
Mice were anesthetized using ketamine (80 mg/kg) and xylazine (5 mg/kg) administered via the intraperitoneal route and placed on a heated mat. One millimeter of tail tip was removed using a scalpel blade and the tail tip bathed in sterile saline at 37°C. The time to cessation of bleeding was measured up to 10 minutes, after which the assay was terminated.
Transmission electron microscopy
Washed platelets were dually fixed with glutaraldehyde and osmic acid adapted from a method described by White.43 In summary, an equal volume of 0.1% glutaraldehyde in White saline was added to washed platelets in Tyrode buffer for 15 minutes at room temperature (RT), after which platelets were centrifuged at 800g for 5 minutes then resuspended in 0.3% (vol/vol) glutaraldehyde for 45 minutes at RT. Platelets were then washed in White saline (0.7% NaCl [wt/vol], 0.0375% KCl [wt/vol], 0.05% MgSO4 [wt/vol], 0.075% Ca[NO3]2 4H2O [wt/vol], 0.055% NaHCO3 [wt/vol], 0.01% Na2HPO4 · 7H2O [wt/vol], 0.003% KH2PO4 [wt/vol], trace Phenol red) and resuspended in 1% (wt/vol) osmium for 1 hour. Samples were then dehydrated by a graded series of acetone and embedded in EPON resin made as per the manufacturer's instructions (Agar; Stansted, United Kingdom). Sections of approximately 0.7 μm were produced and floated onto a formvar film strengthened with carbon, then stained with lead citrate for 15 minutes, washed in water, and observed on a Philips CM20 analytical TEM at ambient temperature, 80 kV and 25 K true magnification. The imaging medium was Kodak Electron image film, SO-163.
Results
Production of conditional ILK-deficient mice
The association of ILK with the integrins α2β1 and αIIbβ3 suggest that ILK may have a role in platelet function.32 The systemic deletion of the ILK gene is embryonically lethal in mice at 3 to 5 days' gestation.31 Therefore, a conditional deletion model based on the CRE-Lox system44 was used to enable the deletion of the ILK gene. A double transgenic mouse colony was established by crossing a mouse in which LoxP sites flank the ILK gene, with a mouse in which the Cre-recombinase gene was placed under the control of the Mx1 promoter. Upon intraperitoneal injection with Poly(I)-Poly(C) (pIpC), an antiviral-type response causes interferon-mediated activation of the Mx1 promoter and subsequent production of Cre-recombinase.44 This results in the site-specific recombination of LoxP sites and consequent removal of the ILK gene. Platelets have a rapid turnover and immunoblot analysis of platelet whole cell lysate indicated that 8 days after pIpC injection ILK protein was undetectable (Figure 1A). When the ILK-deficient (ILK(lox/lox):MxCre) and control (ILK(+/+):MxCre) litter mates were injected with pIpC, the mice showed no noticeable deterioration in health over the 8-day period.
Figure 1.
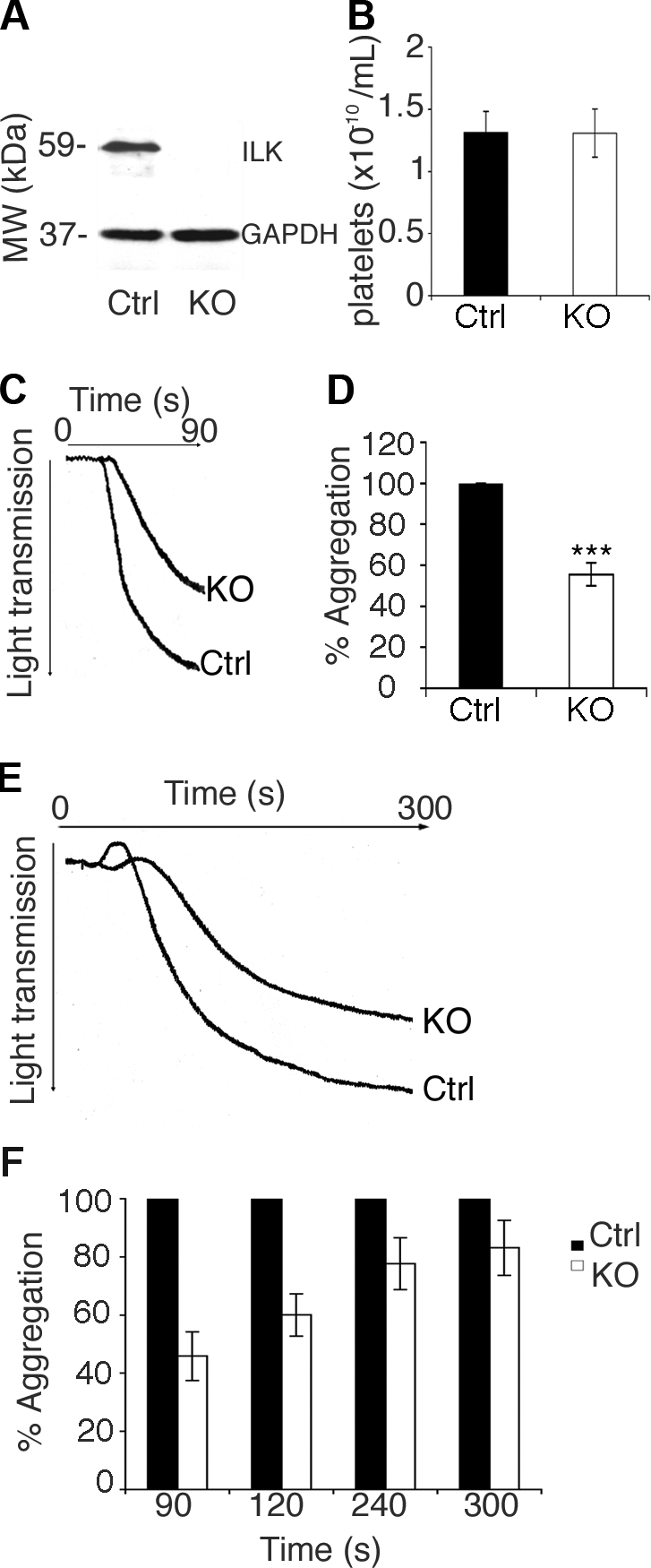
Aggregation is attenuated in ILK-deficient platelets. The levels of ILK protein in control (Ctrl) ILK(+/+):MxCre and KO mice ILK(lox/lox):MxCre at 8 days after pIpC injection were assessed by immunoblotting, and a GAPDH reprobe was used to check for equal loading (A). Platelet counts in whole blood from control (Ctrl) and KO are plotted (B; n = 4, P = .98). Washed platelets from ILK KO and control (Ctrl) mice were stimulated with collagen at a final concentration of 100 μg/mL. A representative aggregation trace is shown (C), and the percentage aggregation is plotted from 3 replicate experiments (D; n = 3, mean ± SEM; ***P < .005). Platelets in PRP from ILK KO and control (Ctrl) mice were matched for platelet number and stimulated with collagen at a final concentration of 100 μg/mL and monitored for 5 minutes. A representative aggregation trace is shown (E), and the percentage aggregation (where the control = 100%) is plotted from 3 replicate experiments (F; n = 3, mean ± SEM; all **P < .05).
Characterization of ILK-deficient platelets
Blood from pIpC-injected control and ILK KO mice contained an equal number of platelets (Figure 1B). Platelet size was assessed by flow cytometry, no difference was detected between control and KO platelets (average diameter, 2.09 and 2.07 μm, respectively), although a small difference was noted from the non–pIpC-injected WT mice tested (average diameter, 2.12 μm). Due to their fundamental role in the functional responses of platelets, the levels of key adhesion receptors α2, β1, β3, αIIb integrin subunits and GPVI levels were verified in the control and KO mouse platelets along with key integrin and ILK-binding partners. When ILK-deficient platelets were compared with the control mice, no significant difference was observed in levels of α2, β3, αIIb integrin subunits, GPVI levels, or the levels of integrin-associated proteins kindlin and talin (Table 1). A modest reduction in the levels of β1 integrin subunit was observed (13.28% ± 2.02%, P ≤ .05) and the expression levels of the ILK-binding partners PINCH1 and α-parvin were reduced in ILK-deficient platelets (by 66.8% ± 10.3% and 38.6% ± 8.9%, respectively, P ≤ .05; Table 1). Previous reports show that initially pIpC-injected mice suffer from thrombocytopenia, although platelet numbers are recovered by around day 6.45 The experiments described in this paper were carried out at 8 days after pIpC injection, and equivalent platelet numbers in KO and control littermates were observed (Figure 1B).
Table 1.
Characterization of the ILK-deficient platelets
| Protein | Level detected in the KO as a percentage of Ctrl |
|---|---|
| α2 | 99.15 ± 7.62 |
| αIIb | 106.03 ± 16.25 |
| β1 | 86.72 ± 2.02* |
| β3 | 94.65 ± 4.38 |
| GPVI | 103.15 ± 8.20 |
| PINCH | 34.21 ± 10.30* |
| α-parvin | 61.36 ± 8.92* |
| Kindlin | 105 ± 5.54 |
| Talin | 86.54 ± 6.38 |
Relative levels of key adhesion receptors in the KO platelets compared to control were determined by immunoblotting. Densitometric data are expressed as a percentage of the control following adjustment for equivalent protein levels using GAPDH (35 kDa).
Mean plus or minus SEM, n = 3-8; *P ≤ .05.
Aggregation is attenuated in ILK-deficient platelets
The effect of ILK deficiency on platelet function was assessed. Aggregation in response to collagen was measured in ILK-deficient platelets and compared with sibling-matched control mouse platelets (Ctrl). In washed platelets aggregation was reduced substantially in the ILK-deficient platelets in comparison to controls at both low (10 μg/mL, not shown) and high concentrations (100 μg/mL) of collagen over 90 seconds (46% ± 5.6% reduction, Figure 1C,D). This defect could not be rescued by the presence of plasma components since aggregation responses in PRP were also reduced substantially (54% ± 8.3% reduction; Figure 1E,F) at 90 seconds and continued to be reduced over 5 minutes (16.9% ± 9.4% reduction; Figure 1E,F).
ILK is required for the up-regulation of integrin αIIbβ3 function
ILK is known to associate with the cytoplasmic tail of the β1- and β3-containing integrins. To determine whether the loss of ILK would affect the surface exposure of integrins, surface α2β1 and αIIbβ3 levels were measured by flow cytometry. In ILK-deficient platelets α2β1 surface exposure was unaffected compared with control mice (data not shown). Surface αIIbβ3 was reduced compared with control platelets in resting conditions (a 20% decrease; Figure 2A). Upon activation the surface exposure of αIIbβ3 was increased but to a lesser extent than the control mice (Figure 2B). This resulted in a 35% decrease in the amount of surface αIIbβ3, indicating that the level of recruitment of αIIbβ3 to the surface is reduced in the absence of ILK.
Figure 2.

ILK is required for the up-regulation of integrin αIIbβ3 function. The exposure of integrin αIIbβ3 on the platelet surface was measured by flow cytometry in resting platelets (A; **P ≤ .01), and thrombin-stimulated platelets (B; ***P ≤ .01). Fibrinogen binding was assessed in the ILK-deficient and control (Ctrl) platelets using FITC-labeled fibrinogen and flow cytometry. Platelets were stimulated with either collagen (C; *P ≤ .05) or thrombin (D; ***P ≤ .005) (average reduction of 60% in the KO compared with the WT). Data for fibrinogen binding upon thrombin activation was normalized to account for the reduction in surface receptors (E). All data expressed as mean plus or minus SEM, n = 4.
The up-regulation in affinity of αIIbβ3 was monitored by flow cytometry to measure the binding of FITC-labeled fibrinogen to the platelet surface. Following stimulation with collagen or thrombin, a large increase in fibrinogen binding to control platelets was observed. ILK-deficient platelets, however, displayed only a small increase in fibrinogen binding (Figure 2C,E). The large reduction in fibrinogen binding in the ILK-deficient platelets was still apparent when data were normalized to account for the difference in receptor numbers (Figure 2E), indicating that ILK is involved in the up-regulation of the affinity of αIIbβ3. A small increase in fibrinogen binding upon activation was, however, observed in the ILK-deficient platelets, indicating that either low-level integrin affinity up-regulation is possible in the absence of ILK or that residual ILK expression is maintained below the level of detection. Similar data obtained in response to both collagen and thrombin stimulation shows that ILK is involved in features of platelet activation common to both agonists.
Granule secretion is defective in ILK-deficient mice
The aggregation of washed platelets is dependent upon platelet granule secretion, which supplies fibrinogen that supports aggregation. Therefore the observed reduction in aggregation may be explained by a reduction in either integrin affinity or secretion, or a combination of both of these. After platelet stimulation with collagen the secretion of α-granules was assessed by monitoring the increase in surface P-selectin by flow cytometry. In control platelets collagen stimulated a dramatic increase in surface P-selectin, consistent with previous reports.46 ILK-deficient platelets, however, displayed substantially reduced surface P-selectin exposure following stimulation compared with controls (Figure 3A), while immunoblot analysis demonstrated that total P-selectin levels are unaltered in the ILK-deficient platelets (Figure 3B). To establish whether this was due to a defect in membrane assemblage into the α-granules or a secretion defect, the release of platelet factor 4 (PF4) was assessed. PF4 secretion was reduced in the ILK-deficient platelets when stimulated with collagen and thrombin (Figure 3C), consistent with an α-granule secretion defect. Furthermore, TEM analysis illustrated that the ILK-deficient platelets have normal ultrastructure, including the formation of α-granules (Figure 3D,E) that appear of normal size and number (control mean = 3.1 ± 0.57 α-granules/platelet, KO mean = 3.3 ± 0.40 α-granules/platelet). This reduction in α-granule secretion is consistent with the observed reduced recruitment of αIIbβ3 to the platelet surface in the ILK-deficient platelets (Figure 2A,B) and indicates the presence of a major defect in secretion in the ILK-deficient platelets (Figure 3B).
Figure 3.

α-granule but not dense granule secretion is defective in ILK-deficient mice. The exposure of P-selectin at the platelet surface was measured by flow cytometry in resting (basal) and stimulated (collagen 100 μg/mL) platelets (A; mean ± SEM, n = 4; ***P ≤ .005). Western analysis of total cellular P-selectin (140 kDa) is shown in panel B. Densitometric analysis shows the protein levels of P-selectin are unchanged in the absence of ILK (n = 3, KO = 85.77% ± 5.54%). Equivalent levels of protein loading were demonstrated by reprobing blots for GAPDH (B, bottom panel) (35 kDa). Release of platelet factor 4 (PF4) was measured in washed platelets stimulated with collagen (10 μg/mL) and thrombin (5 units/mL; C; mean ± SEM, n = 4; *P ≤ .05). Washed platelets loaded with [3H] 5-HT were stimulated with collagen (10 μg/mL) (n = 3, P = .64) and thrombin (5 units/mL; P = .69; D) as a measure of dense granule secretion. TEM images of control (Ctrl; E) and KO (F) washed mouse platelets show normal formation of mitochondria (letter M), dense granules (letter D), α-granule (letter A), and glycerol stores (letter G) in the KO platelets.
Dense granule secretion was examined using radiolabeled 5-hydroxytryptamine (5-HT). The analysis of collagen (100 μg/ mL (not shown) and 10 μg/mL [Figure 3D]) and thrombin (5 units/mL [Figure 3D]) mediated release of 5-HT revealed no difference between control and ILK-deficient platelets (Figure 3D), indicating that dense granule secretion is unaffected by ILK depletion. Thus, the observed reduction in aggregation is not due to reduced secretion of dense granule components such as adenosine diphosphate (ADP) that would normally perpetuate activation.
Thrombus formation is diminished in ILK-deficient blood
The effect of ILK depletion on thrombus formation under arterial flow conditions was assessed in an in vitro system. Whole blood labeled with DiOC6 was perfused through a collagen-coated capillary at an arterial shear rate of 1000 s−1 and the resulting thrombi assessed by confocal microscopy. In the ILK-deficient samples the number of thrombi formed was found not to be altered (Figure 4C), indicating that the level of initial adhesion is unaffected by the absence of ILK in platelets (Figure 4A,C). A decrease was observed, however, in the depth of thrombi formed in the ILK-deficient blood compared with the control samples (Figure 4B). This suggests that the assembly of large thrombi by platelet-platelet interactions was reduced (Figure 4A,B). This is consistent with the expected consequences of reduced α-granule secretion (Figure 3A) and reduced collagen-stimulated αIIbβ3 affinity up-regulation in ILK-deficient platelets (Figure 2C,D).
Figure 4.

Thrombus formation is diminished in ILK-deficient blood. Whole blood from KO and control (Ctrl) mice was labeled with DiOC6 and perfused through a collagen-coated capillary at a shear rate of 1000 s−1. Representative topological and composite (stacked) views of the thrombi formed (A) show more large thrombi are formed in the control (Ctrl) compared with the KO, though the KO sample still shows formation of many small plaques. (B) The average maximum depth of thrombi formed in a field of view (mean ± SEM, n = 10; *P ≤ .05). The number of thrombi formed (C) was not significantly altered in the control and KO samples (mean ± SEM, n = 10; P = .34).
Mice with ILK-deficient platelets show extended bleeding
To examine the effect of ILK depletion in platelets on hemostasis, tail bleeding assays were performed. Bleeding time and volume was prolonged moderately in ILK-deficient mice in comparison to the litter-mate controls, with almost all of the KO mice bleeding beyond the 10-minute duration of the assay (Figure 5).
Figure 5.

Bleeding time and volume is increased in the ILK KO mice. The time to cessation of bleeding following transection of the tail tip of ILK-deficient and litter-matched control (Ctrl) was measured over 10 minutes. Data represent individual mice (n = 14) with the mean value indicated. *P < .05 (A). The volume of blood lost was assessed and is represented for individuals. *P < .05 (B).
Early agonist-induced signaling is unaffected by the absence of ILK
PLCγ2 tyrosine phosphorylation and calcium release precede both aggregation and α-granule secretion in collagen-stimulated (GPVI) signaling. To determine whether agonist induced signaling is initiated normally in ILK-deficient platelets, PLCγ2 tyrosine phosphorylation and calcium mobilization levels were examined. PLCγ2 was immunoprecipitated from resting and collagen-stimulated (100 μg/mL for 90 seconds) platelets and immunoblotted for tyrosine phosphorylation and total PLCγ2 levels. PLCγ2 tyrosine phosphorylation (Figure 6A,B) was unaltered in the ILK-deficient platelets compared with the controls. Calcium mobilization was observed by loading resting platelets with Fluo4-AM and measuring the change in fluorescence upon stimulation with CRP. Calcium mobilization (Figure 6C) was unaltered in the ILK-deficient platelets compared with controls. This demonstrated that early GPVI stimulated signaling leading to calcium mobilization was unaffected by ILK depletion (Figure 6).
Figure 6.

Early agonist induced signaling is unaffected by the absence of ILK. PLCγ2 immunoprecipitated from ILK-deficient and control (Ctrl) washed platelets was prepared from resting and collagen-stimulated (100 μg/mL 90 seconds) samples. Samples were immunoblotted for tyrosine phosphorylation, stripped, and reprobed for PLCγ2 loading levels, and densitometric analysis was carried out (A; mean ± SEM, n = 3; P = .44). A representative blot of PLCγ2 and tyrosine phosphorylation (4G10) levels is shown (B). Calcium mobilization in Fluo-4AM– labeled control (Ctrl) and ILK-deficient platelets was monitored in a Flexstation II, and change in fluorescence was expressed as the percentage increase in calcium from resting levels (mean ± SEM, n = 3; P = .16; C).
Discussion
Previous work has shown that ILK physically interacts with β1 and β3 integrin subunits in platelets and that these interactions are increased upon platelet activation.2,14,32 Since these integrins play a major role in platelet function, adhering platelets to sites of damage and facilitating platelet-platelet interactions, it has been proposed that this association may be important to platelet function.2,14 Furthermore, it has been shown that ILK isolated from platelets exhibits kinase activity that is increased transiently upon collagen and thrombin stimulation.2
To allow analysis of the role of ILK in platelet function and signaling, a conditional ILK knockout mouse model was established using the Cre-LoxP system. By day 8 following Cre-induction, ILK expression was reduced to undetectable levels in platelets (Figure 1A). Equivalent platelet numbers were observed in control and ILK KO mice, and levels of receptors were relatively unchanged in the platelets, including the integrin subunits α2, αIIb and β3, GPVI (Table 1), and P-selectin (Figure 2D). Although a modest reduction in β1 levels was observed, the adhesion of platelets to collagen under flow conditions was unaffected (Figure 4C). While integrin α2β1 function has not been the focus of this study, preliminary experiments indicate that ILK-deficient platelets are able to spread on collagen and form filopodia and lamellipodia; however, the timing of this compared with control platelets has not been assessed.
In other cell types ILK has been shown to bind directly to PINCH and parvin to form a complex known as the IPP that regulates integrin signaling17 and focal adhesion.19,22 Immunoblot analysis revealed that the expression of both proteins was reduced in the ILK-deficient platelets (Table 1), indicating that ILK is required to maintain full levels of these associated proteins, thus the phenotype may reflect the combined functions of these proteins.
ILK has also been shown to be involved in interactions between the plasma membrane and the cytoskeleton.47 Indeed this plasma membrane-cytoskeleton interaction with ILK has been reported to be essential for shape change, adhesion, and spreading in other cell types.48 We have found previously that stimulation of human platelets with CRP or thrombin results in the translocation of ILK from the cytoplasm to the plasma membrane where it colocalizes with integrin subunits and the membrane skeleton.2 This movement to the surface and possibly the movement of associated proteins is likely to be important for ILK function. In other cell types ILK has been shown to regulate such changes in cytoskeleton arrangement.1 We therefore examined the protein expression levels of cytoskeleton and integrin-associated proteins kindlin and talin that are implicated in integrin regulation.49,50 No difference in protein levels of kindlin or talin was observed in the ILK-deficient platelets, although talin showed a trend toward decreased expression. This suggests that these proteins are not involved in the functional defects observed in this study.
ILK-deficient platelets showed reduced aggregation in both washed platelets and in PRP over 5 minutes (Figure 1C-F), demonstrating the importance of ILK in platelet function. To begin to assess why ILK-deficient platelets showed reduced function, integrin αIIbβ3 surface exposure and affinity regulation were investigated. ILK depletion reduced slightly αIIbβ3 surface exposure (Figure 2A) despite normal levels of αIIbβ3 expression (Table 1). This is consistent with work on a skeletal muscle-restricted deletion of ILK that shows reduced localization of β1 integrin to myotendinous junctions.47 The activation of αIIbβ3 to a higher affinity binding state for fibrinogen was reduced markedly (data normalized for receptor numbers [Figure 2E]), although was not completely absent (Figure 2C-E), indicating that ILK has a significant role in αIIbβ3 activation. Clot retraction studies indicated no difference between the control and ILK-deficient platelets (data not shown), suggesting that the reduction in αIIbβ3 activation is unlikely to be explained by an outside-in signaling defect.
Defective platelet aggregation could also be caused by reduced secretion during activation. Platelet α- and dense-granule secretion by ILK-deficient platelets were therefore examined. Despite normal levels of P-selectin protein (Figure 3B) and normal platelet ultrastructure (Figure 3E), a dramatic reduction in agonist-induced α-granule secretion in the ILK KO platelets was revealed by examining P-selectin surface exposure (Figure 3A) and PF4 release (Figure 3C). This defect was seen even at high concentrations of agonists and suggests a central role for ILK in platelet activation. Dense granule secretion, however, was maintained at both high and low concentrations of agonist (Figure 3C,D). Taken together these results suggest that this is not a general disruption of the structural changes that occur upon platelet activation and that α-granule secretion is specifically effected by ILK depletion. Differential secretion of α- and dense-granules from platelets via the actin cytoskeleton has been recently reported.51 It is possible, therefore, that ILK plays a critical role in aspects unique to α-granule secretion.
The presence of fibrinogen (present in α-granules and plasma) did not rescue the reduction in aggregation observed in washed platelets (Figure 1E,F). These data suggest that the αIIbβ3 affinity up-regulation defect alone would cause a reduction in function. However, the increased lag time observed in the PRP and washed samples containing ILK-deficient platelets (Figure 1C-F) may be due to reduced α-granule secretion and further activation resulting from this.
The importance of ILK in thrombus generation in whole blood was investigated by studying thrombus formation on immobilized collagen under arterial flow conditions. The number of thrombi formed was not reduced (Figure 4C), suggesting that initial adhesion was not substantially affected by the absence of ILK. The depth of thrombi formed, however, was reduced in the blood of ILK-deficient mice (Figure 4B). This reduced thrombus growth is consistent with the reduced platelet αIIbβ3 activation observed (Figure 2B). The moderately increased bleeding in ILK-deficient mice (Figure 5) indicates the physiological importance of ILK for normal platelet function.
Since ILK appeared to possess a central role in the platelet activation process, it was important to establish that cell signaling was triggered following exposure to agonists. The phosphorylation of PLCγ2 and the mobilization of calcium from internal stores following stimulation of the collagen receptor GPVI are events up-stream of granule secretion and integrin activation. Upon stimulation with collagen (or CRP) both events appear unaffected by ILK depletion (Figure 6), indicating that early collagen signaling events are not dependent on the presence of ILK and that the apparent defect in platelet signaling lies further downstream.
The changes to platelet signaling and function described here, as a result of ILK deficiency, may be as a direct result of loss of ILK kinase activity or ILK protein-protein interactions (or a combination of both of these). In other cells ILK has been shown to form associations with a complex of molecules, components of which include PINCH and parvin, that together with ILK regulate integrin signaling17,19,22 and have been shown to be necessary for the formation of focal adhesions.22,25 Through these and other interactions ILK is implicated in many processes including cytoskeletal arrangement1,15 cell adhesion, migration, survival, and proliferation.52 Further work will be required to establish the basis of the defects observed in ILK-deficient mice, and whether these result from disruption of protein complex formation and/or kinase activity.
Our data provide compelling evidence that ILK has a role in the regulation of platelet function and suggests a dual role for ILK in the regulation of platelet function through the control of α-granule secretion and the affinity modulation of αIIbβ3.
Acknowledgments
The authors dedicate this work to the memory of Ms Susan Berresford for her valued contribution to this study.
The authors thank Molecular Devices in Wokingham and Dr Amanda Sadler for support and advice regarding calcium assays using mouse platelets, and Dr Chris Stain (Center for Advanced Microscopy, University of Reading) for advice on TEM imaging.
This work was supported by grants from the Wellcome Trust (072498/z/03/z and 082338) and the British Heart Foundation (FS/03/076/15916, RG/05/007, and PG/2001/143). Support for engineering of the ILK-floxed mice was provided by a grant from the Shriners of North America to R.S.-A.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.L.T., T.S., J.M.S., P.A.J., S.J., N.E.B., and J.M.G. participated in designing and performing the research; R.S.-A., J.F., and S.D. contributed transgenic mice used to create the conditional KO system; and K.L.T. and J.M.G. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Katherine L. Tucker, School of Biological Sciences, University of Reading, Reading, Berkshire, RG6 6AJ, United Kingdom; e-mail: k.l.tucker@rdg.ac.uk.
References
- 1.Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, et al. Regulation of cell adhesion and anchorage-dependent growth by a new b1-integrin-linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- 2.Stevens JM, Jordan PA, Sage T, Gibbins JM. The regulation of integrin-linked kinase in human platelets: evidence for involvement in the regulation of integrin alpha 2 beta 1. J Thromb Haemost. 2004;2:1443–1452. doi: 10.1111/j.1538-7836.2004.00870.x. [DOI] [PubMed] [Google Scholar]
- 3.White DE, Coutu P, Shi YF, et al. Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev. 2006;20:2355–2360. doi: 10.1101/gad.1458906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bendig G, Grimmler M, Huttner IG, et al. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 2006;20:2361–2372. doi: 10.1101/gad.1448306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, Huang XN, Yan W, et al. Role of the integrin-linked kinase/PINCH1/alpha-parvin complex in cardiac myocyte hypertrophy. Lab Invest. 2005;85:1342–1356. doi: 10.1038/labinvest.3700345. [DOI] [PubMed] [Google Scholar]
- 6.Lu H, Fedak PW, Dai X, et al. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation. 2006;114:2271–2279. doi: 10.1161/CIRCULATIONAHA.106.642330. [DOI] [PubMed] [Google Scholar]
- 7.Gkretsi V, Bowen WC, Yang Y, Wu C, Michalopoulos GK. Integrin-linked kinase is involved in matrix-induced hepatocyte differentiation. Biochem Biophys Res Commun. 2007;353:638–643. doi: 10.1016/j.bbrc.2006.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naska S, Park KJ, Hannigan GE, Dedhar S, Miller FD, Kaplan DR. An essential role for the integrin-linked kinase-glycogen synthase kinase-3 beta pathway during dendrite initiation and growth. J Neurosci. 2006;26:13344–13356. doi: 10.1523/JNEUROSCI.4462-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terpstra L, Prud'homme J, Arabian A, et al. Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol. 2003;162:139–148. doi: 10.1083/jcb.200302066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attwell S, Roskelley C, Dedhar S. The integrin-linked kinase (ILK) suppresses anoikis. Oncogene. 2000;19:3811–3815. doi: 10.1038/sj.onc.1203711. [DOI] [PubMed] [Google Scholar]
- 11.Persad S, Dedhar S. The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Rev. 2003;22:375–384. doi: 10.1023/a:1023777013659. [DOI] [PubMed] [Google Scholar]
- 12.Marotta A, Parhar K, Owen D, Dedhar S, Salh B. Characterisation of integrin-linked kinase signalling in sporadic human colon cancer. Br J Cancer. 2003;88:1755–1762. doi: 10.1038/sj.bjc.6600939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Chen K, Tu Y, Wu C. Distinct roles of two structurally closely related focal adhesion proteins, alpha-parvins and beta-parvins, in regulation of cell morphology and survival. J Biol Chem. 2004;279:41695–41705. doi: 10.1074/jbc.M401563200. [DOI] [PubMed] [Google Scholar]
- 14.Pasquet JM, Noury M, Nurden AT. Evidence that the platelet integrin alphaIIb beta3 is regulated by the integrin-linked kinase, ILK, in a PI3-kinase dependent pathway. Thromb Haemost. 2002;88:115–122. [PubMed] [Google Scholar]
- 15.Yamaji S, Suzuki A, Kanamori H, et al. Possible role of ILK-affixin complex in integrin-cytoskeleton linkage during platelet aggregation. Biochem Biophys Res Commun. 2002;297:1324–1331. doi: 10.1016/s0006-291x(02)02381-1. [DOI] [PubMed] [Google Scholar]
- 16.Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- 18.Lynch DK, Ellis CA, Edwards PA, Hiles ID. Integrin-linked kinase regulates phosphorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene. 1999;18:8024–8032. doi: 10.1038/sj.onc.1203258. [DOI] [PubMed] [Google Scholar]
- 19.Li F, Zhang Y, Wu C. Integrin-linked kinase is localized to cell-matrix focal adhesions but not cell-cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats. J Cell Sci. 1999;112(Pt 24):4589–4599. doi: 10.1242/jcs.112.24.4589. [DOI] [PubMed] [Google Scholar]
- 20.Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung-Hagesteijn C, Mahendra A, Naruszewicz I, Hannigan GE. Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin-linked kinase, ILK1. EMBO J. 2001;20:2160–2170. doi: 10.1093/emboj/20.9.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tu Y, Li F, Goicoechea S, Wu C. The LIM-only protein PINCH directly interacts with integrin-linked kinase and is recruited to integrin-rich sites in spreading cells. Mol Cell Biol. 1999;19:2425–2434. doi: 10.1128/mcb.19.3.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Chen K, Guo L, Wu C. Characterization of PINCH-2, a new focal adhesion protein that regulates the PINCH-1-ILK interaction, cell spreading, and migration. J Biol Chem. 2002;277:38328–38338. doi: 10.1074/jbc.M205576200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Guo L, Chen K, Wu C. A critical role of the PINCH-integrin-linked kinase interaction in the regulation of cell shape change and migration. J Biol Chem. 2002;277:318–326. doi: 10.1074/jbc.M108257200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Chen K, Tu Y, et al. Assembly of the PINCH-ILK-CH-ILKBP complex precedes and is essential for localization of each component to cell-matrix adhesion sites. J Cell Sci. 2002;115:4777–4786. doi: 10.1242/jcs.00166. [DOI] [PubMed] [Google Scholar]
- 26.Zervas CG, Gregory SL, Brown NH. Drosophila integrin-linked kinase is required at sites of integrin adhesion to link the cytoskeleton to the plasma membrane. J Cell Biol. 2001;152:1007–1018. doi: 10.1083/jcb.152.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Troussard AA, Mawji NM, Ong C, Mui A, St-Arnaud R, Dedhar S. Conditional knock-out of integrin-linked kinase demonstrates an essential role in protein kinase B/Akt activation. J Biol Chem. 2003;278:22374–22378. doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- 28.Friedrich EB, Liu E, Sinha S, et al. Integrin-linked kinase regulates endothelial cell survival and vascular development. Mol Cell Biol. 2004;24:8134–8144. doi: 10.1128/MCB.24.18.8134-8144.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persad S, Attwell S, Gray V, et al. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase - Critical roles for kinase activity and amino acids arginine 211 and serine 343. J Biol Chem. 2001;276:27462–27469. doi: 10.1074/jbc.M102940200. [DOI] [PubMed] [Google Scholar]
- 30.Hanks SKaH T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 31.Sakai T, Li S, Docheva D, et al. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003;17:926–940. doi: 10.1101/gad.255603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibbins JM. Platelet adhesion signalling and the regulation of thrombus formation. J Cell Sci. 2004;117:3415–3425. doi: 10.1242/jcs.01325. [DOI] [PubMed] [Google Scholar]
- 33.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–887. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung SM, Moroi M. Platelets interact with soluble and insoluble collagens through characteristically different reactions. J Biol Chem. 1998;273:14827–14837. doi: 10.1074/jbc.273.24.14827. [DOI] [PubMed] [Google Scholar]
- 35.Nurden AT. Glanzmann thrombasthenia. Orphanet J Rare Dis. 2006;1:10. doi: 10.1186/1750-1172-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 37.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 38.Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 39.Morton LF, Hargreaves PG, Farndale RW, Young RD, Barnes MJ. Integrin alpha 2 beta 1-independent activation of platelets by simple collagen-like peptides: collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for alpha 2 beta 1-independent platelet reactivity. Biochem J. 1995;306:337–344. doi: 10.1042/bj3060337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbins JM, Briddon S, Shutes A, et al. The p85 subunit of phosphatidylinositol 3-kinase associates with the Fc receptor gamma-chain and linker for activitor of T cells (LAT) in platelets stimulated by collagen and convulxin. J Biol Chem. 1998;273:34437–34443. doi: 10.1074/jbc.273.51.34437. [DOI] [PubMed] [Google Scholar]
- 41.Cicmil M, Thomas JM, Leduc M, Bon C, Gibbins JM. Platelet endothelial cell adhesion molecule-1 signalling inhibits the activation of human platelets. Blood. 2002;99:137–144. doi: 10.1182/blood.v99.1.137. [DOI] [PubMed] [Google Scholar]
- 42.Kulkarni S, Nesbitt WS, Dopheide SM, Hughan SC, Harper IS, Jackson SP. Techniques to examine platelet adhesive interactions under flow. In: Gibbins JM, Mahaut-Smith MP, editors. Platelets and Megakaryocytes: Functional Assays. Vol 1. Totowa, New Jersey: Humana Press; 2004. pp. 165–186. [DOI] [PubMed] [Google Scholar]
- 43.White JG. Electron microscopy methods for studying platelet structure and function. In: Gibbins JM, Mahaut-Smith MP, editors. Methods in Molecular Biology: Platelets and Megakaryocytes: Functional Assays. Vol 1. Totowa, NJ: Humana Press; 2004. pp. 47–63. [DOI] [PubMed] [Google Scholar]
- 44.Hamilton DL, Abremski K. Site-specific recombination by the bacteriophage P1 lox-Cre system: Cre-mediated synapsis of two lox sites. J Mol Biol. 1984;178:481–486. doi: 10.1016/0022-2836(84)90154-2. [DOI] [PubMed] [Google Scholar]
- 45.Hall MA, Curtis DJ, Metcalf D, et al. The critical regulator of embryonic hematopoiesis SCL, is vital in the adult for megakaryopoiesis, erythropoiesis, and lineage choice in CFU-S12. Proc Natl Acad Sci U S A. 2003;100:992–997. doi: 10.1073/pnas.0237324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furie B, Furie BC, Flaumenhaft R. A journey with platelet P-selectin: the molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost. 2001;86:214–221. [PubMed] [Google Scholar]
- 47.Wang HV, Chang LW, Brixius K, et al. Integrin-linked kinase stabilizes myotendinous junctions and protects muscle from stress-induced damage. J Cell Biol. 2008;180:1037–1049. doi: 10.1083/jcb.200707175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boulter E, Van Obberghen-Schilling E. Integrin-linked kinase and its partners: a modular platform regulating cell-matrix adhesion dynamics and cytoskeletal organization. Eur J Cell Biol. 2006;85:255–263. doi: 10.1016/j.ejcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Petrich BG, Marchese P, Ruggeri ZM, et al. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J Exp Med. 2007;204:3103–3111. doi: 10.1084/jem.20071800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 51.Flaumenhaft R, Dilks JR, Rozenvayn N, Monahan-Earley RA, Feng D, Dvorak AM. The actin cytoskeleton differentially regulates platelet alpha-granule and dense-granule secretion. Blood. 2005;105:3879–3887. doi: 10.1182/blood-2004-04-1392. [DOI] [PubMed] [Google Scholar]
- 52.Grashoff C, Aszodi A, Sakai T, Hunziker EB, Fassler R. Integrin-linked kinase regulates chondrocyte shape and proliferation. EMBO Rep. 2003;4:432–438. doi: 10.1038/sj.embor.embor801. [DOI] [PMC free article] [PubMed] [Google Scholar]