

Abstract
The impermeable nature of the cell membrane to peptides, proteins, DNA and oligonucleotides limits the therapeutic potential of these biological agents. However, the recent discovery of short cationic peptides that cross the plasma membrane efficiently is opening up new possibilities for the intracellular delivery of such agents. These peptides are commonly referred to as protein transduction domains (PTDs) and are successfully used to transport heterologous proteins, peptides and other types of cargo into cells. Several recent reports have used the membrane transducing technology in vivo to deliver biologically active cargo into various tissues. This review discusses the structure of the most commonly used PTDs and how their ability to transduce membranes is used to regulate biological functions. It also considers future directions and the potential of this technology to move from the laboratory into the clinic.
Defining the function of a protein or controlling its action requires cellular expression of mutant forms of the protein or silencing of the corresponding gene. Until recently, this was achieved by transgene expression, either using recombinant vectors or directly introducing proteins into cells by microinjection, electroporation or red-cell ghost fusion, all of which are cumbersome techniques requiring specialist equipment [1-3]. Now, a new, non-invasive technology for the direct delivery of biological material is emerging, following the discovery that certain proteins can enter cells in an unconventional way. The TAT protein, a transactivation factor from the human immunodeficiency virus 1 (HIV-1), was the first polypeptide shown to gain entry into cells when added exogenously to culture medium [4-6]. Other proteins reported to cross the plasma membrane belong to the homeoprotein family of transcription factors [7] such as the Drosophila Antennapedia (Antp) protein [7,8]. The herpes simplex virus VP22 has also been reported to cross the plasma membrane [9], although this result has recently been questioned [10]. The subsequent identification of short membrane-permeable cationic peptides derived from these proteins, and synthesis of other amphipathic peptides such as transportan [11,12], MAP [13] and KALA [14], has led to the development of a new technology for delivering biological agents into cells. These peptides are commonly known as protein transduction domains (PTDs) [15,16] or cell-penetrating peptides [17-19]. Hydrophobic domains corresponding to the leader sequence of proteins also cross membranes and have been used to introduce bioactive peptides into cells [20,21]. However, they might not be as potent as cationic PTDs and are only briefly mentioned here.
Peptides that transduce the plasma membrane
Peptides corresponding to amino acids 48–60 of the TAT protein [22] and a 16-omer derived from the third helix of the Antp homeodomain [8] were shown to translocate rapidly through the plasma membrane. Given that cationic residues in TAT and Antp PTDs are important for cellular uptake, poly-arginine (poly-R) and poly-lysine (poly-K) oligomers were tested for their ability to enter cells [23-26]. Internalization efficiency was found to depend on the length of the peptide backbone, and stretches of six (R6) to eight (R8) arginine residues showed the highest internalization potential [27,28]. Other groups selected cationic peptides from an M13 phage library displaying random, 12 amino-acid-long sequences [29], or synthesized amphipathic peptides such as transportan (a chimera of the N-terminal fragment of the neuropeptide galanin and of the wasp venom peptide mastoparan) [11], MAP [13], Pep-1 [30] or KALA [14]. Naturally derived and synthetic PTDs are all capable of cellular entry and a list of the most commonly used PTDs is given in Table 1.
Table 1.
Commonly used PTDsa
| Protein | PTD amino acid sequenceb |
|---|---|
| Cationic | |
| HIV-1 TAT (47–57) | YGRKKRRQRRR |
| Drosophila Antennapedia (43–58) | RQIKIWFQNRRMKWKK |
| Poly-arginine (R7) (synthetic) | RRRRRRR |
| PTD-5 (synthetic) | RRQRRTSKLMKR |
| Amphipathic | |
| Transportan (chimeric, galanin fragment plus mastoparan) | GWTLNSAGYLLGKINLKALAALAKKIL |
| KALA (synthetic) | WEAKLAKALAKALAKHLAKALAKALKACEA |
Abbreviations: HIV-1, human immunodeficiency virus 1; PTD, protein transduction domain.
Single-letter amino acid designation.
At present, how membrane-transducing proteins and peptide transporters are internalized is not clearly understood. Initially, internalization was reported to take place at 4°C and in the presence of endocytosis inhibitors, suggesting that these peptides do not use the conventional endocytic pathway to enter cells [8,27,28,31]. However, recent data challenge this view and show that internalization of TAT-, poly-R- and Antp-PTDs occurs, at least in part, via endocytosis [31-35]. An artefact created by cell fixation, during which cell-bound PTDs redistribute giving the impression of nuclear localization, might have contributed to the discrepancy in results seen between earlier and recent work [10,32,36,37]. Moreover, amphipathic PTDs, such as transportan, might use different mechanisms to enter cells [38,39]. It will be important in the future to determine conclusively the contribution of endocytic versus non-endocytic pathways during cell entry of individual PTDs (Figure 1).
Figure 1.
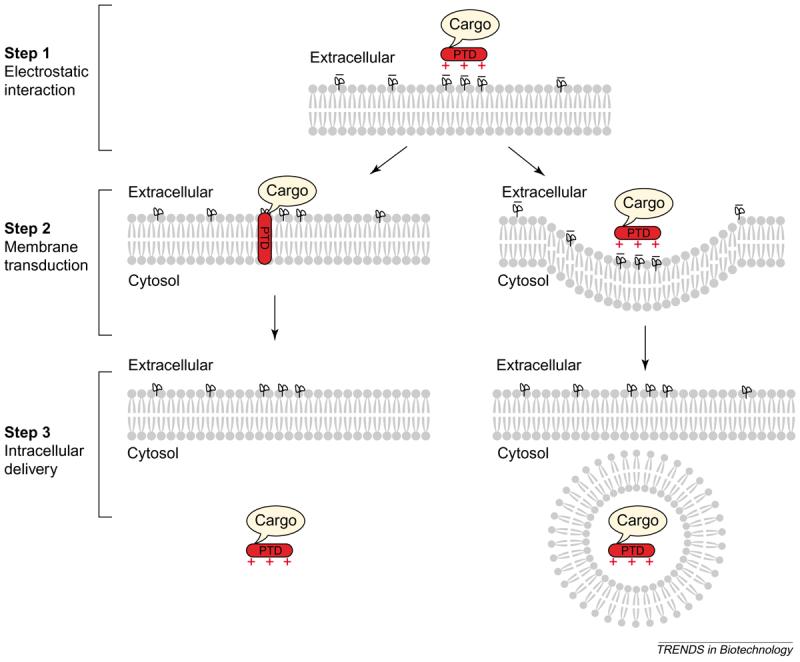
A three-step model of membrane transduction by cationic peptides. Step 1: electrostatic interactions between the positively charged protein transduction domain (PTD) (red +) and negative charges on the cell membrane (black −) promote binding of PTD to the cell surface. Step 2: the PTD penetrates the plasma membrane through an as-yet unknown mechanism, towing its attached cargo. Alternatively, the PTD–cargo complex is endocytosed by the cell. Step 3: intracellular delivery of the PTD–cargo complex.
In general, the process of membrane transduction can be divided into three steps (Figure 1). The first step probably involves ionic interactions between the basic groups of the PTD side-chains and negative charges on the plasma membrane. Evidence suggests a role for cell-surface heparan sulphate (HS) proteoglycans as receptors for TAT protein because soluble heparin or glycosaminoglycan lyases that specifically degrade HS chains hampered TAT uptake [28,40]. Similarly, TAT protein failed to transduce cells that were genetically defective in biosynthesis of fully sulphated HS [40], and transduction of poly-K-, Antp- and TAT-PTDs was reduced in mutant cell lines deficient in either HS or glycosaminoglycan synthesis [26,35]. This initial electrostatic interaction could lead to membrane penetration via novel, currently unknown mechanism(s), or the PTD and its associated cargo could enter cells via endocytosis (step 2). Studies on various permutations of the TAT- and poly-R-PTDs suggested that the guanidinium headgroups of the arginine residues play a role in facilitating cellular entry [24] and that only a subset of the headgroups in the peptide backbone is necessary for efficient uptake [25,41]. In the third step, the internalized PTD cargo exerts its biological function, although its potency probably depends on the route of internalization.
Interestingly, in certain cases, proteins genetically fused to the TAT PTD had higher biological activity when purified under denaturing conditions compared with native, correctly folded protein preparations [42,43]. This result suggests that unfolded TAT-fusion proteins cross the plasma membrane more effectively or, alternatively, that they acquire physiological function more readily once inside cells than proteins in their native conformation, which might be preferentially targeted for degradation [15]. However, this finding might not necessarily apply to all PTD-fusion proteins; poor membrane transduction by cationic PTDs has been reported where – although PTD proteins interacted with the cell membrane – internalization was observed only after prolonged treatment of the cells, presumably by endocytosis [37,44].
Introduction of heterologous proteins using PTDs
One crucial experiment linked the TAT PTD to heterologous proteins and demonstrated that these proteins gain entry into cells; this observation opened the way for the use of PTDs in biotechnology. Initially, it was demonstrated that truncated TAT protein (residues 37–72) chemically crosslinked to β-galactosidase (β-gal) or to horseradish peroxidase (HRP) was able to transduce various cell lines and primary cells with close to 100% of the cells becoming positive for β-gal or HRP activity [45]. When the minimal TAT sequence with PTD activity (amino acids 47–57) was identified and genetically fused to p27kip1 molecule, purified fusion-protein induced scattering of hepatocellular carcinoma cells, revealing a previously uncharacterized role of p27kip1 in cell migration [42].
Protein transduction technology has been used to regulate the nuclear factor-κB (NF-κB) signalling pathway. Fusion of TAT PTD to a non-degradable mutant of the inhibitor of κB α (IκBa) protein inhibited nuclear localization of NF-κB and prevented maturation of osteoclast precursors into osteoclasts [46]. Similar non-degradable TAT–IκBα mutants associated with endogenous NF-κB when added exogenously to cells in culture and inhibited its activity following stimulation of cells with pro-inflammatory cytokines [47]. Inhibition of this pathway was also achieved by a bioactive peptide delivered with the Antp PTD; this disrupted the integrity of the IκB kinase (IKK) complex at a step upstream of IκBα phosphorylation [48]. Similarly, the same peptide delivered with a different PTD (PTD-5) blocked the adverse effects of the pro-inflammatory cytokine IL-1β on islet cell function [49]. Finally, combinations of hydrophobic and nuclear localization sequence (NLS) peptides have been used to inhibit nuclear translocation of NF-κB [50-52].
Several other proteins in their wild-type or mutated forms have been transduced into cells following their fusion to PTDs. Transduction of eosinophils with dominant-negative Ras inhibited the high-affinity interaction of Mac-1 receptor to its ligand ICAM-1 [53], and Vocero-Akbani et al. [54] enhanced anti-viral responses to HIV by transducing cells with a modified caspase-3 polypeptide. The modified protein contained the cleavage site recognized by HIV protease, which remained inactive in healthy cells but was cleaved in HIV infected cells to release active caspase-3, resulting in apoptosis [54]. Other TAT- or Antp-transduced proteins include the Cdk4/6 inhibitor p16INK4a [55-58], p21(Waf1/Cip1) [59] and E2F [60] – all proteins involved in the cell cycle. One recent report describes TAT-assisted transduction of a single-chain Fv antibody that blocks the anti-apoptotic action of Bcl-2 protein [61]. Other PTDs used to introduce proteins or peptides into cells are summarized elsewhere [17]. Finally, PTDs have been also used to deliver drug carriers such as liposomes [62], DNA–DNA or liposome–DNA complexes [63,64], peptide nucleic acids [18,65] and phage particles [66].
PTD-mediated delivery in vivo
The therapeutic potential of protein transduction technology was highlighted by several recent reports that used the technology to deliver bioactive agents in vivo. The types of cargo delivered thus far are summarized in Table 2. The first reported delivery of heterologous proteins in vivo was achieved by injecting TAT(37–72)–β-gal conjugates intravenously into mice. Within 30 mins of administration, β-gal activity was detected in several tissues, with high activity seen in liver, spleen and heart, low activity in skeletal muscle and lung, and no activity in kidney and brain [45]. More dramatic results of β-gal delivery in vivo were reported by a β-gal fusion-protein to TAT(47–57) PTD that was expressed in bacteria and purified under denaturing conditions [67]. Analysis of tissue samples obtained at 4 h and 8 h post-intraperitoneal injection of TAT–β-gal into mice showed strong enzymatic activity in liver, spleen, lung, muscle fibres and heart. In contrast to the chemically conjugated β-gal, denatured TAT–β-gal protein was detected in the brain, suggesting that denatured TAT-fusion proteins show enhanced transduction of tissues compared with proteins prepared in their native form [67].
Table 2.
Types of cargo delivered using protein transduction technologya
| PTD used | Cargo | Tissue investigated | Refs |
|---|---|---|---|
| TAT, Antp | Peptides | Endothelium, inflammatory cells | [48,68] |
| TAT, Antp, poly-arginine, PTD-5 | Proteins | Multiple (heart, liver, spleen, lung, kidney, muscle, etc.) | [45,67,70-72] |
| TAT | Liposomes | Tumour cells (in vitro) | [62] |
| TAT | Nanoparticles | Stem cells | [78] |
| TAT, poly-arginine, transportan | Nucleic acids | Epithelium, lung carcinoma (in vivo) and tumour cells (in vitro) | [14,63-65] |
| TAT, Antp | Recombinant virus | Endothelium, muscle | [76] |
| TAT | Recombinant phage | Cell lines (in vitro) | [66] |
| Poly-arginine | Small chemical compound | Skin | [77] |
Abbreviation: Antp, Drosophila Antennapedia protein; PTD, protein transduction domain.
The Antp internalization region was used to introduce the scaffolding domain (residues 82–101) of caveolin-1 into cells to regulate endothelial nitric oxide (NO) synthase action [68]. This chimeric peptide was taken up efficiently by endothelial cells resulting in acetyl-choline-induced vasodilation and NO production, and its administration in mice suppressed acute inflammation and vascular leak [68]. Intravenous administration of a membrane-permeable IκBα protein inhibited NF-κB activity and diminished infiltration of inflammatory cells in a rat pleurisy model (Blackwell, N.M. et al., unpublished). Another group used purified TAT–Bcl-xL protein to transduce primary neurons in culture and to diminish staurosporin-induced apoptosis. Intraperitoneal injection of TAT–Bcl-xL resulted in transduction of neurons in the brain as early as 1–2 h post-injection, and in a murine model of focal ischaemia, TAT–Bcl-xL decreased cerebral infarction by up to 40% [69]. Therefore, PTD-mediated protein transduction can be used in vivo to regulate the activity of crucial signalling pathways known to operate in pathological conditions.
PTD-assisted induction of DNA recombination in vivo was assessed by a chimera of Cre recombinase fused to the membrane translocation sequence (MTS) from Kaposis fibroblast growth factor (FGF-4) and the NLS from SV40 large T-antigen [70]. The NLS–Cre–MTS fusion protein induced DNA recombination in up to 70–80% of the cells in vitro, and intraperitoneal injection of the protein into loxP transgenic mice induced DNA recombination in all tissues examined, including heart, kidney, lung, spleen, liver and brain [70]. A second study compared FGF-4 MTS (hydrophobic), SV40 NLS (basic) and TAT PTD (basic) for their ability to promote cellular uptake of Cre and found that TAT PTD was the most potent, resulting in >95% recombination efficiency in embryonic stem cells and primary spleenocytes ex vivo [71]. However, in this report, intraperitoneal injection of TAT–Cre protein failed to induce significant recombination in vivo with only some activity observed at the site of injection, suggesting limited diffusion of the protein to surrounding tissues. The reason for the opposing in vivo results reported by the two papers is unclear.
Poor in vivo transduction has also been reported for muscle tissue. Although efficient in transducing cultured myoblasts, a TAT–GFP protein led only to few positive fibres when injected intravenously or subcutaneously into mice [72]. Instead, there was strong fluorescence of the extracellular matrix (ECM) suggesting that the poor transduction of muscle fibres could be caused by TAT–GFP binding to ECM components [72].
In the work discussed so far, transduction of tissues in vivo was achieved by injection of purified PTD-protein. In an alternative strategy, TAT-β-glucuronidase – a lysosomal enzyme that is deficient in the disease mucopolysaccharidosis VII – was expressed as a transgene from recombinant viral vectors, and its bio-distribution examined in enzyme-deficient mice [73]. Liver sections analysed 10 days after intravenous injection of recombinant virus showed enzyme activity throughout the parenchyma, suggesting diffusion of the expressed TAT-fusion protein to surrounding tissue. Enzyme activity was also detected in tissues such as spleen, kidney, heart, lung and skeletal muscle [73]. By contrast, two other groups reported lack of intercellular transport of virally expressed TAT–GFP fusion protein either in vivo or in vitro [74,75], and TAT PTD did not enhance the immunogenicity of another protein when it was expressed de novo as TAT-fusion protein [74]. These contradictory results could be attributed to the production of variable levels of protein or that the protein partners fused to TAT PTD (β-glucuronidase versus GFP) have a role in determining how well the protein is secreted from producing cells or taken up by neighbouring cells. Nevertheless, these differences highlight that generalizations should be avoided.
Pre-incubation of recombinant virus with Antp PTD facilitated gene delivery in vitro and in vivo. In cultured cells, viral transduction was increased by 5–10-fold, as determined by GFP expression [76]. In mice with hind limb ischaemia, the angiogenic potential of the vascular endothelial growth factor delivered by a viral vector was increased approximately threefold in the presence of Antp peptide [76]. Therefore, cell-permeable peptides can enhance viral cell entry, thus improving the efficacy of therapeutically relevant genes in vivo.
Membrane-transducing peptides could improve the absorption of drugs with poor tissue penetration such as cyclosporin A (CsA), which is ineffective as a local anti-inflammatory agent owing to its poor absorption by the skin. CsA–arginine heptamers (r7–CsA) penetrated the epidermis and dermis, reached dermal T-lymphocytes, and inhibited cutaneous inflammation [77]. Such conjugates also penetrated human skin, which is significantly thicker than mouse, suggesting the potential use of r7–CsA as a topical anti-inflammatory drug in the clinic [77]. In another report, magnetic nanoparticles coated with TAT PTD transduced haematopoietic CD34+ cells and neural progenitor cells [78]. The particles were retained intracellularly for several days to enable monitoring of the labelled cells in vivo [78]. Localization and retrieval of cell populations could facilitate analysis of stem-cell–organ interactions, thereby advancing the therapeutic use of stem cells.
Future prospects
Considerable in vitro and in vivo experimental data are now available to establish protein transduction technology as a useful tool in biological research. With the completion of the human genome project, protein transduction technology has the potential of becoming a useful tool in determining the function of newly identified proteins by providing a non-invasive and efficient alternative to DNA transfection. However, for this to become reality, it will be important to better understand the process of cell entry, which might differ among the various PTD subgroups.
Given that certain considerations are addressed, this technology could be useful in the clinic as well as in the regulation of signal transduction pathways that are involved in pathological disorders. Today, viral vectors are used to introduce genes into cells, but with the drawbacks of simultaneously introducing unwanted and/or harmful genetic material and having inadequate control over expression levels of the transgene. By contrast, PTD-mediated delivery offers temporal and reversible regulation of a biological process by directly controlling intracellular levels of inhibitors. Administration of inhibitors can be stopped and, if necessary, resumed later to avoid detrimental side-effects.
There are several unanswered questions and possible limitations to the use of PTD technology in in vivo applications. A major disadvantage is lack of targeting specificity. Therefore, for each case, it will be important to establish not only that the PTD-chimera has a beneficial effect on diseased cells but also that it has no adverse effects on healthy tissue. Alternatively, cells in some organs or tissues might be less amenable to transduction as has been reported for brain and muscle [45,72]. As mentioned earlier, another limitation is our incomplete knowledge of the mechanisms used by PTDs to gain entry into cells. Further research is needed in this area because understanding the molecular aspects of this process could open the way for the synthesis of simpler and possibly more potent membrane transducers. Immunogenicity of the PTD itself or its cargo could be another obstacle to the routine use of this technology in the clinic and thorough studies are needed to assess the extent of this problem. Finally, it is important to understand the pharmacokinetics of in vivo protein transduction.
Despite these drawbacks, membrane-transduction technology could offer an easy alternative for manipulating intracellular biological processes in vitro and in vivo. But can this technology deliver in the clinic? The answer to this question might not be far off.
Acknowledgements
I would like to thank Nathan Blackwell, Derek Gilroy, Phupinder Sembi, Pat Ozegbe and anonymous reviewers for helpful suggestions. P.S.K. is supported by a Career Development Award from the Wellcome Trust (Ref. No. 58408).
References
- 1.Gomperts BD, Fernandez JM. Techniques for membrane permeabilization. Trends Biochem. Sci. 1985;10:414–417. [Google Scholar]
- 2.Chang DC, et al. Guide to Electroporation and Electrofusion. Academic Press; 1991. [Google Scholar]
- 3.Celis JE, et al. Microinjection and Organelle Transplantation Techniques. Academic Press; 1986. [Google Scholar]
- 4.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 5.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 6.Mann DA, Frankel AD. Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J. 1991;10:1733–1739. doi: 10.1002/j.1460-2075.1991.tb07697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prochiantz A. Messenger proteins: homeoproteins, TAT and others. Curr. Opin. Cell Biol. 2000;12:400–406. doi: 10.1016/s0955-0674(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 8.Derossi D, et al. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 9.Elliott G, O'Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88:223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 10.Lundberg M, Johansson M. Is VP22 nuclear homing an artifact? Nat. Biotechnol. 2001;19:713–714. doi: 10.1038/90741. [DOI] [PubMed] [Google Scholar]
- 11.Pooga M, et al. Cell penetration by transportan. FASEB J. 1998;12:67–77. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 12.Pooga M, et al. Cellular translocation of proteins by transportan. FASEB J. 2001;15:1451–1453. doi: 10.1096/fj.00-0780fje. [DOI] [PubMed] [Google Scholar]
- 13.Oehlke J, et al. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta. 1998;1414:127–139. doi: 10.1016/s0005-2736(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 14.Wyman TB, et al. Design, synthesis, and characterization of a cationic peptide that binds to nucleic acids and permeabilizes bilayers. Biochemistry. 1997;36:3008–3017. doi: 10.1021/bi9618474. [DOI] [PubMed] [Google Scholar]
- 15.Schwarze SR, et al. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol. 2000;10:290–295. doi: 10.1016/s0962-8924(00)01771-2. [DOI] [PubMed] [Google Scholar]
- 16.Wadia JS, Dowdy SF. Protein transduction technology. Curr. Opin. Biotechnol. 2002;13:52–56. doi: 10.1016/s0958-1669(02)00284-7. [DOI] [PubMed] [Google Scholar]
- 17.Lindgren M, et al. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 18.Gariepy J, Kawamura K. Vectorial delivery of macromolecules into cells using peptide-based vehicles. Trends Biotechnol. 2001;19:21–28. doi: 10.1016/s0167-7799(00)01520-1. [DOI] [PubMed] [Google Scholar]
- 19.Langel U. Cell-Penetrating Peptides, Processes and Applications. CRC Press; 2002. [Google Scholar]
- 20.Hawiger J. Cellular import of functional peptides to block intracellular signaling. Curr. Opin. Immunol. 1997;9:189–194. doi: 10.1016/s0952-7915(97)80134-3. [DOI] [PubMed] [Google Scholar]
- 21.Hawiger J. Noninvasive intracellular delivery of functional peptides and proteins. Curr. Opin. Chem. Biol. 1999;3:89–94. doi: 10.1016/s1367-5931(99)80016-7. [DOI] [PubMed] [Google Scholar]
- 22.Vives E, et al. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 23.Buschle M, et al. Transloading of tumor antigen-derived peptides into antigen-presenting cells. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3256–3261. doi: 10.1073/pnas.94.7.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wender PA, et al. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothbard JB, et al. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J. Med. Chem. 2002;45:3612–3618. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- 26.Mai JC, et al. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J. Biol. Chem. 2002;277:30208–30218. doi: 10.1074/jbc.M204202200. [DOI] [PubMed] [Google Scholar]
- 27.Futaki S, et al. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki T, et al. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 29.Mi Z, et al. Characterization of a class of cationic peptides able to facilitate efficient protein transduction in vitro and in vivo. Mol. Ther. 2000;2:339–347. doi: 10.1006/mthe.2000.0137. [DOI] [PubMed] [Google Scholar]
- 30.Morris MC, et al. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 31.Vives E, et al. TAT peptide internalization: seeking the mechanism of entry. Curr. Protein Pept. Sci. 2003;4:125–132. doi: 10.2174/1389203033487306. [DOI] [PubMed] [Google Scholar]
- 32.Richard JP, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 33.Drin G, et al. Studies on the internalisation mechanism of cationic cell-penetrating peptides. J. Biol. Chem. doi: 10.1074/jbc.M303938200. in press. [DOI] [PubMed] [Google Scholar]
- 34.Fittipaldi A, et al. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 tat fusion proteins. J. Biol. Chem. doi: 10.1074/jbc.M303045200. in press. [DOI] [PubMed] [Google Scholar]
- 35.Console S, et al. Antennapedia and HIV TAT ‘protein transduction domains’ promote endocytosis of high Mr cargo upon binding to cell surface glycosaminoglycans. J. Biol. Chem. doi: 10.1074/jbc.M301726200. in press. [DOI] [PubMed] [Google Scholar]
- 36.Lundberg M, Johansson M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem. Biophys. Res. Commun. 2002;291:367–371. doi: 10.1006/bbrc.2002.6450. [DOI] [PubMed] [Google Scholar]
- 37.Lundberg M, et al. Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther. 2003;8:143–150. doi: 10.1016/s1525-0016(03)00135-7. [DOI] [PubMed] [Google Scholar]
- 38.Hallbrink M, et al. Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta. 2001;1515:101–109. doi: 10.1016/s0005-2736(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 39.Lindgren M, et al. Translocation properties of novel cell penetrating transportan and penetratin analogues. Bioconjug. Chem. 2000;11:619–626. doi: 10.1021/bc990156s. [DOI] [PubMed] [Google Scholar]
- 40.Tyagi M, et al. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001;276:3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 41.Ho A, et al. Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo. Cancer Res. 2001;61:474–477. [PubMed] [Google Scholar]
- 42.Nagahara H, et al. Transduction of full-length TAT fusion proteins into mammalian cells: TAT–p27Kip1 induces cell migration. Nat. Med. 1998;4:1449–1452. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 43.Becker-Hapak M, et al. TAT-mediated protein transduction into mammalian cells. Methods. 2001;24:247–256. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- 44.Falnes PO, et al. Ability of the Tat basic domain and VP22 to mediate cell binding, but not membrane translocation of the diphtheria toxin A-fragment. Biochemistry. 2001;40:4349–4358. doi: 10.1021/bi002443l. [DOI] [PubMed] [Google Scholar]
- 45.Fawell S, et al. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. U. S. A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abu-Am Y, et al. TAT fusion proteins containing tyrosine 42-deleted IκBα arrest osteoclastogenesis. J. Biol. Chem. 2001;276:30499–30503. doi: 10.1074/jbc.M104725200. [DOI] [PubMed] [Google Scholar]
- 47.Kabouridis PS, et al. Inhibition of NF-κB activity by a membrane-transducing mutant of IκBα. J. Immunol. 2002;169:2587–2593. doi: 10.4049/jimmunol.169.5.2587. [DOI] [PubMed] [Google Scholar]
- 48.May MJ, et al. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 49.Rehman KK, et al. Protection of islets by in situ peptide-mediated transduction of the IκB kinase inhibitor NEMO-binding domain peptide. J. Biol. Chem. 2003;278:9862–9868. doi: 10.1074/jbc.M207700200. [DOI] [PubMed] [Google Scholar]
- 50.Fujihara SM, Nadler SG. Intranuclear targeted delivery of functional NF-κB by 70 kDa heat shock protein. EMBO J. 1999;18:411–419. doi: 10.1093/emboj/18.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujihara SM, et al. A D-amino acid peptide inhibitor of NF-κB nuclear localization is efficacious in models of inflammatory disease. J. Immunol. 2000;165:1004–1012. doi: 10.4049/jimmunol.165.2.1004. [DOI] [PubMed] [Google Scholar]
- 52.Yan Liu X, et al. Peptide-directed suppression of a pro-inflammatory cytokine response. J. Biol. Chem. 2000;275:16774–16778. doi: 10.1074/jbc.C000083200. [DOI] [PubMed] [Google Scholar]
- 53.Myou S, et al. Blockade of focal clustering and active conformation in β2-integrin-mediated adhesion of eosinophils to intercellular adhesion molecule-1 caused by transduction of HIV TAT-dominant negative Ras. J. Immunol. 2002;169:2670–2676. doi: 10.4049/jimmunol.169.5.2670. [DOI] [PubMed] [Google Scholar]
- 54.Vocero-Akbani AM, et al. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat. Med. 1999;5:29–33. doi: 10.1038/4710. [DOI] [PubMed] [Google Scholar]
- 55.Ezhevsky SA, et al. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10699–10704. doi: 10.1073/pnas.94.20.10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lissy NA, et al. TCR antigen-induced cell death occurs from a late G1 phase cell cycle check point. Immunity. 1998;8:57–65. doi: 10.1016/s1074-7613(00)80458-6. [DOI] [PubMed] [Google Scholar]
- 57.Kato D, et al. Features of replicative senescence induced by direct addition of Antennapedia–p16INK4A fusion protein to human diploid fibroblasts. FEBS Lett. 1998;427:203–208. doi: 10.1016/s0014-5793(98)00426-8. [DOI] [PubMed] [Google Scholar]
- 58.Fahraeus R, et al. Characterization of the cyclin-dependent kinase inhibitory domain of the INK4 family as a model for a synthetic tumour suppressor molecule. Oncogene. 1998;16:587–596. doi: 10.1038/sj.onc.1201580. [DOI] [PubMed] [Google Scholar]
- 59.Mutoh M, et al. A p21(Waf1/Cip1)carboxyl-terminal peptide exhibited cyclin-dependent kinase-inhibitory activity and cytotoxicity when introduced into human cells. Cancer Res. 1999;59:3480–3488. [PubMed] [Google Scholar]
- 60.Lissy NA, et al. A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature. 2000;407:642–645. doi: 10.1038/35036608. [DOI] [PubMed] [Google Scholar]
- 61.Cohen-Saidon C, et al. A novel strategy using single chain antibody to show the importance of Bcl-2 in mast cell survival. Blood. doi: 10.1182/blood-2002-12-3921. in press. [DOI] [PubMed] [Google Scholar]
- 62.Torchilin VP, et al. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2001;98:8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rudolph C, et al. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J. Biol. Chem. 2003;278:11411–11418. doi: 10.1074/jbc.M211891200. [DOI] [PubMed] [Google Scholar]
- 64.Torchilin VP, et al. Cell transfection in vitro and in vivo with nontoxic TAT peptide–liposome–DNA complexes. Proc. Natl. Acad. Sci. U. S. A. 2003;100:1972–1977. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pooga M, et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 1998;16:857–861. doi: 10.1038/nbt0998-857. [DOI] [PubMed] [Google Scholar]
- 66.Eguchi A, et al. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J. Biol. Chem. 2001;276:26204–26210. doi: 10.1074/jbc.M010625200. [DOI] [PubMed] [Google Scholar]
- 67.Schwarze SR, et al. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 68.Bucci M, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 69.Cao G, et al. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jo D, et al. Epigenetic regulation of gene structure and function with a cell-permeable Cre recombinase. Nat. Biotechnol. 2001;19:929–933. doi: 10.1038/nbt1001-929. [DOI] [PubMed] [Google Scholar]
- 71.Peitz M, et al. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc. Natl. Acad. Sci. U. S. A. 2002;99:4489–4494. doi: 10.1073/pnas.032068699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caron NJ, et al. Intracellular delivery of a Tat–eGFP fusion protein into muscle cells. Mol. Ther. 2001;3:310–318. doi: 10.1006/mthe.2001.0279. [DOI] [PubMed] [Google Scholar]
- 73.Xia H, et al. The HIV Tat protein transduction domain improves the biodistribution of β-glucuronidase expressed from recombinant viral vectors. Nat. Biotechnol. 2001;19:640–644. doi: 10.1038/90242. [DOI] [PubMed] [Google Scholar]
- 74.Leifert JA, et al. Full-length proteins attached to the HIV tat protein transduction domain are neither transduced between cells, nor exhibit enhanced immunogenicity. Gene Ther. 2002;9:1422–1428. doi: 10.1038/sj.gt.3301819. [DOI] [PubMed] [Google Scholar]
- 75.Cashman SM, et al. Evidence of protein transduction but not intercellular transport by proteins fused to HIV tat in retinal cell culture and in vivo. Mol. Ther. 2003;8:130–142. doi: 10.1016/s1525-0016(03)00131-x. [DOI] [PubMed] [Google Scholar]
- 76.Gratton JP, et al. Cell-permeable peptides improve cellular uptake and therapeutic gene delivery of replication-deficient viruses in cells and in vivo. Nat. Med. 2003;9:357–363. doi: 10.1038/nm835. [DOI] [PubMed] [Google Scholar]
- 77.Rothbard JB, et al. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000;6:1253–1257. doi: 10.1038/81359. [DOI] [PubMed] [Google Scholar]
- 78.Lewin M, et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]