

Abstract
Background
This study investigated the biodistribution and therapeutic efficacy of Lutetium-177 (177Lu) radiolabeled anti-Lewis Y monoclonal antibody hu3S193 radioimmunotherapy (RIT) in mice bearing prostate cancer xenografts. The ability of Epidermal Growth Factor Receptor (EGFR) tyrosine kinase inhibitor AG1478 and docetaxel chemotherapy to enhance the efficacy of RIT was also assessed in vivo.
Methods
The in vitro cytotoxicity of 177Lu labeled hu3S193 on Ley positive DU145 prostate cancer cells was assessed using proliferation assays, with induction of apoptosis measured by ELISA. The in vivo biodistribution and tumor localization of 177Lu-hu3S193 was assessed in mice bearing established DU145 tumor xenografts. The efficacy and maximum tolerated dose of 177Lu-hu3S193 RIT in vivo was determined by a dose escalation study. EGFR inhibitor AG1478 or docetaxel chemotherapy was administered at sub-therapeutic doses in conjunction with RIT in vivo.
Results
177Lu-hu3S193 mediated significant induction of cytotoxicity and apoptosis in vitro. In vivo analysis of 177Lu-hu3S193 biodistribution demonstrated specific targeting of DU145 prostate cancer xenografts, with maximal tumor uptake of 33.2 ± 3.9 %ID/g observed at 120 hr post injection. In RIT studies, 177Lu-hu3S193 caused specific and dose-dependent inhibition of prostate cancer tumor growth. A maximum tolerated dose of 350μCi was determined for 177Lu-hu3S193. Combination of 177Lu-hu3S193 RIT with EGFR inhibitor AG1478 or docetaxel chemotherapy both significantly improved efficacy.
Conclusions
177Lu-hu3S193 RIT is effective as a single agent in the treatment of Ley positive prostate cancer models. The enhancement of RIT by AG1478 or docetaxel indicates the promise of combined modality strategies.
Keywords: hu3S193, prostate cancer, radioimmunotherapy, combined modality
Introduction
The targeting of radioisotopes to tumor sites using monoclonal antibodies (mAbs) (Radioimmunotherapy, RIT) is a promising therapeutic approach, as the targeted radiation is able to kill cancer cells specifically and consistently (1–3). Prostate cancer is particularly suitable to RIT due to metastases to bone marrow and lymph nodes that are accessible to high levels of circulating mAbs, and also due to the typically small volume of disease that allows maximal tumor penetration of radiolabeled antibodies (4,5). Therapeutic efficacy has been observed in Phase I clinical trials of radiolabeled anti-prostate specific membrane antigen (PSMA) mAb J591, warranting the investigation of RIT in the treatment of prostate cancer (6,7).
As it has become apparent that RIT is most suited to treatment of low tumor burden, the selection of radioisotopes has become an important consideration, where physical properties such as path length, emission energy and physical half-life must correlate with a particular tumor size (8). The lanthanide radiometal 177Lu (Emax = 496keV) has been identified as having favorable characteristics for use in RIT including a 2mm path length suitable for treatment of micrometastases and small tumors <1cm while limiting irradiation of normal tissues (9).
Radioimmunotherapy of solid tumors has not had the same clinical success as achieved in hematological neoplasms, owing to the relative radioresistance of solid tumors, and an inability to deliver sufficient dose to tumor without substantial bone marrow toxicity (5,8,10). The challenges of solid tumor RIT in producing clinical responses has driven the investigation of various methods aimed at increasing the therapeutic efficacy of RIT including the combination of RIT with agents aiming to enhance radiosensitivity (8,11). Experience in external beam radiotherapy has identified a number of molecules and cellular pathways involved in mediating the cellular response to radiation including p53, EGFR, PI3K and Ras (12). The pharmacological modulation of these pathways has been identified as a method of increasing the efficacy of radiation therapies such as RIT (13).
The Epidermal Growth Factor Receptor (EGFR) and associated downstream signaling molecules have been identified as direct mediators of increased cellular resistance to radiation, with the mechanism suggested to include radiation induced ligand release and receptor signaling, resulting in the activation of transcription factors involved in enhanced tumor survival and DNA repair (14,15). Various strategies to abrogate EGFR mediated proliferation, survival and resistance in tumors have been investigated, including the blocking of ligand binding and receptor activation using anti-EGFR mAbs, and through the use of small molecular weight EGFR tyrosine kinase inhibitors (TKI) (16–18). Previous studies have explored the combination of EGFR inhibitors with radiotherapy and demonstrated enhancement of radiotherapy (19).
In addition to targeted molecular therapies, standard cytotoxic drugs have been investigated for their ability to enhance tumor response to radiation. These include taxanes such as docetaxel, for which the key mechanism of action involves microtubular stabilization resulting in mitotic block at the radiosensitive G2/M phase of the cell cycle (20,21). Synergistic enhancement of radiotherapy following combination with taxanes has been observed in prostate cancer using both external beam radiotherapy and RIT (22–24).
The unique specificity of the hu3S193 mAb for the Ley antigen makes it an attractive candidate for the delivery of radiation to Ley positive tumors. Significant expression of Ley has been observed on prostate cancer cells lines including the androgen independent lines DU145 and PC3 and the androgen dependent line LNCaP (25,26). Clinically, various studies have reported extensive expression of Ley in prostate tumor tissues, such as Martensson et. al. who observed extensive staining in 26 of 30 tumors (27). Further, highest expression of Ley in prostate cancer has been associated with poorly differentiated tumors and metastases (26). This widespread expression of Ley on prostate tumors and their metastases provides a solid rationale for the targeting of this tumor type with anti-Ley mAb hu3S193.
We have previously explored the use of 90Y-labeled hu3S193 in combination with paclitaxel and EGFR inhibition (28,29), however 90Y is not well suited to small volume disease due to the relatively long path-length of the emitted β-particles. Moreover, the utility of RIT with hu3S193 in prostate cancer where small volume disease is often clinically relevant has not previously been explored. This study is the first to assess the properties of hu3S193 radiolabeled with 177Lu, which may be better suited for RIT of small volume prostate cancer. Additionally, the mechanism of 177Lu-hu3S193 cytotoxicity was examined in this study through in-vitro analyses. The greatest potential for RIT lies in its combination with other therapeutic modalities (30). Subsequently, 177Lu-hu3S193 combined modality RIT (CMRIT) with either AG1478 (an EGFR TKI) or docetaxel was also explored.
Materials and Methods
Cell lines
The androgen independent DU145 prostate carcinoma cell line was obtained from American Type Culture Collection (ATCC, Manassas VA, USA). The colon carcinoma cell line SW1222 was obtained from the New York Branch of the Ludwig Institute for Cancer Research, New York NY, USA. Cells were grown in RPMI 1640 media supplemented with 10% v/v Fetal Calf Serum (CSL Ltd, Vic, Australia) 5% w/v Penicillin/Streptomycin (Penicillin G 5000 Units/mL/Streptomycin Sulphate 5000μg/mL, CSL, Parkville, Australia) and 5% L-Glutamine (200mM stock, JRH Biosciences, Lenexa KS, USA).
Antibody and radiolabelling
Humanized 3S193 (hu3S193), a CDR grafted IgG1 antibody specific for the Ley antigen (31), and isotype control huA33 (32) were produced by the Biological Production Facility, Ludwig Institute for Cancer Research (Melbourne, Australia). Lutetium-177 (177Lu) was obtained from Perkin-Elmer (Perkin Elmer Life and Analytical Sciences, Wellesley MA, USA). Radiolabeling of hu3S193 and huA33 mAbs with radioisotopes was achieved using the bifunctional metal ion chelate C-functionalized trans-cyclohexyl-diethylenetriaminepentaacetic acid (CHX-A″-DTPA) (33) using a modification of a procedure previously published (34).
Immunoreactivity and affinity
Determination of the radiolabeled hu3S193 immunoreactivity was performed according to the ‘Lindmo’ cell binding assay, using Ley positive MCF-7 cells as previously described (35). The affinity constant (Ka) and the approximate number of antigen binding sites per cell were determined by Scatchard analysis (35)
Proliferation assay
Cells were plated at a density of 2 × 103 cells/well in TC Microwell 96 well plates (Nalge Nunclon, Denmark) by adding 0.1mL of a 2 × 104 cells/mL solution in to the appropriate wells, and then allowed to adhere overnight. The following day, cells were treated with 177Lu-hu3S193 or control 177Lu-huA33 at 5, 10, 20 and 40μCi/well. Corresponding control wells were treated with media alone, and unlabeled hu3S193 equal in concentration (~10μg/well) to radiolabeled mAb. After incubation of 120 hours, cell viability was determined using [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS, Cell Titre Aqueous One Solution Promega, Madison WI, USA) in the colorimetric proliferation assay according to the manufacturer’s protocol. 20μL of MTS reagent was added to control and treatment wells, incubated at 37°C for 2 hours and absorbance read at 490nm. The absolute absorbances for each group were determined by subtracting the background absorbance and the mean and standard deviation was determined for each treatment group.
Apoptosis cell death ELISA
Apoptosis resulting from treatment with 177Lu radiolabeled hu3S193 was assessed using the Cell Death Detection ELISAPLUS (Roche Diagnostics, Castle Hill, NSW, Australia), a quantitative sandwich ELISA, according to the manufacturer’s protocol. DU145 cells were prepared as for MTS cytotoxicity assay, and treated with radiolabeled hu3S193 for 120 hr.
Animal models and xenograft establishment
Tumor xenografts were established in male BALB/c nude (nu/nu) mice of 3 to 4 weeks of age, obtained from the Animal Resource Centre, Western Australia and maintained in autoclaved microisolator boxes housed in a positive pressure isolation unit (Techniplast Gazzada, Varese, Italy) for the duration of animal studies.
For the establishment of DU145 prostate xenografts, 10 × 106 cells were resuspended in 75μL media, and mixed with 75μL Matrigel Basement Membrane (BD Biosciences, Bedford MA, USA) (ratio 1:1) at 4°C before s.c. injection into the left inguinal line of the BALB/c nude mice. In the biodistribution studies, Ley-negative SW1222 colon carcinoma control tumors were established on the flank opposite the DU145 tumors by s.c. injection of 10 × 106 SW1222 cells in 150μL RPMI. Tumor growth was regularly measured using digital calipers and the formula; Tumor Volume (TV) = (Length × Width2)/2 of the tumor, where length was the longest axis and width the measurement at right angles to length (35). TV was expressed in mm3. All animal studies were approved by the Austin Health Animal Ethics Committee.
Biodistribution Study
The biodistribution of the 177Lu radiolabeled hu3S193 was assessed in mice bearing established DU145 xenografts (mean TV = 211.6 ± 56.6mm3). Mice were injected with 177Lu-hu3S193 (10μg protein, 14.8μCi activity) in 100μL saline by tail vein injection. At 10 min, 4, 24, 48, 72, 120, 168, 216 and 288 hr after post injection, groups of 5 mice were euthanized by Isofluorane anesthesia and then immediately bled via cardiac puncture. Tumors and normal tissues (liver, spleen, kidney, muscle, skin, bone, lung, heart, stomach, brain, small bowel and tail) were then resected and placed in individual γ-counter tubes. The activity of all samples were then counted on a dual gamma scintillation counter (Cobra II Auto Gamma; Packard Instruments), and the percent injected dose per gram (%ID/g) calculated. Results were expressed as Mean ± SD for each time point, and used in the calculation of tumor:blood (T:B) ratios.
Pharmacokinetic analysis
The pharmacokinetics of 177Lu-hu3S193 was calculated using the serum clearance data from the biodistribution study. A two compartment iv-bolus model with 1st order elimination was fitted to the data using the WinNonLin curve fitting program (WinNonLin Pro Node 5.0.1, Pharsight Co., Mountain View CA, USA).
Gamma Camera Imaging
Whole body imaging of mice was performed to identify in vivo localization of 177Lu-hu3S193. Mice were anesthetized with a mixture of 20mg/kg Xylazine/100mg/kg Ketamine, (10μL/g) by intraperitoneal injection, and placed under a Philips Axis gamma camera (Phillips Medical Systems, North Ryde NSW, Australia). Images of 20,000 counts were acquired at each time point, using a 128 × 128 matrix, and a zoom of 2. A standard equivalent to 10% injected dose was included in the field of view.
177Lu-hu3S193 dose titration studies
The therapeutic efficacy of 177Lu-CHX-A″-DTPA-hu3S193 alone was assessed in mice bearing established DU145 xenografts in order to determine the Maximal Tolerated Dose (MTD) of 177Lu-hu3S193. Mice (n = 6, TV = 123.2 ± 35.3mm3) received a single dose of 177Lu-hu3S193 (180μg protein) at doses of 100, 200, 350 and 500μCi. Separate groups received saline vehicle or 180μg unlabeled hu3S193 as controls in equivalent volumes to the radiolabeled antibodies.
177Lu-hu3S193 and AG1478 combined modality study
EGFR TKI AG1478 was combined with 177Lu-hu3S193 RIT to assess enhanced efficacy of RIT by EGFR inhibition. Mice (n = 6, TV = 144.8 ± 21.0mm3) were injected with a single dose 25, 50, 100 or 200μCi 177Lu-hu3S193 (80μg protein), or equivalent doses of huA33 isotype control mAb by tail vein injection, 10 days after establishment of DU145 xenografts. In combined modality groups, 400μg AG1478 was injected intraperitoneally for 5 consecutive days in a 7 day period, repeated over 2 weeks. The first dose was given 4 hr after injection of 177Lu-hu3S193. A dose of 400μg AG1478 has previously been determined to be sub-therapeutic in treatment of established xenografts, and therefore AG1478 was administered at this dose in combined modality groups (36). A group of mice received AG1478 alone to assess its therapeutic efficacy as a single agent.
Ki-67 Immuno-staining
Proliferation in xenografts was assessed by staining sections for the proliferation antigen Ki-67. Sections were incubated with rabbit anti-human Ki-67 monoclonal antibody (NeoMarkers, Fremont CA, USA) for 30 min at room temperature and processed further as described by Perera et. al (37). The percentage of Ki-67 positive nuclei in tumor sections was estimated by acquiring 10 high powered (400×) fields from each tumor section examined in each treatment group using Leica IM50 image acquisition software (Version 4.0, Leica Microsystems Imaging Solutions Ltd., Cambridge UK). Each image was then assessed for relative Ki-67 staining intensity from 3+ (very strong) to 1+ (weak) using the Leica QWin image analysis program (Version 3.1.0, Leica Microsystems Imaging Solutions Ltd., Cambridge UK). The total area of each section staining positive for Ki-67 was calculated, and a score with a maximum of 300 constructed. Mean scores for each treatment group were then collated for comparison.
Western Blotting
The levels of phosphorylated and total EGFR expressed in tumors were assessed by Western Blotting. Xenografts homogenates were prepared as detailed in Perera et. al (37). Xenograft samples were loaded into NuPage 4–12% Bis-Tris gels (Invitrogen™ Life Technologies, Mulgrave, Australia) and separated by SDS-PAGE at 120V for 90 min. Proteins were then transferred onto polyvinylidne difluoride (PVDF) Immobilon-P transfer membrane (Millipore Corporation, Bedford MA, USA), and subsequently blocked in 5% v/v HSA/PBS for 15 min at RT. Membranes were probed with goat anti-phosphotyrosine 1173 EGFR mAb (Santa Cruz biotechnology, Santa Cruz CA, USA) or murine anti-EGFR 528 mAb, and appropriate secondary anti-goat HRP conjugated antibody (Sigma Chemical Co., St Louis MO, USA) or anti-mouse HRP conjugated antibody (Chemicon International, Temecula CA, USA). The specific protein bands were visualized using enhanced chemiluminescence (ECL reagent, GE Healthcare Biosciences, Uppsala, Sweden), and exposure to Curix Ortho HT-G X-ray film (AGFA-Gevaert Ltd, Burwood, Australia).
177Lu-hu3S193 and docetaxel combined modality study
The enhancement of 177Lu-hu3S193 by combination with docetaxel chemotherapy was assessed in a CMRIT study, as docetaxel has previously been shown to enhance the anti-tumor efficacy of 90Y-DOTA-ChL6 RIT (23). This study was conducted in conjunction with the 177Lu-hu3S193 MTD dose study, with RIT alone groups serving as comparisons to groups receiving combined treatment. A single dose of 600μg docetaxel administered 24 hr after RIT was chosen as this dose and schedule had previously demonstrated significant efficacy (23). Groups of mice (n = 6, TV = 123.2 ± 35.3mm3) received single doses of 177Lu-hu3S193 RIT at 100, 200 or 350μCi followed by 600μg docetaxel (Taxotere®, Sanofi Aventis, Macquarie Park, Australia) injected i.p. 24 hr after injection of 177Lu-hu3S193. A further control group received 600μg docetaxel alone to allow comparison to CMRIT groups.
Statistical analysis and tumor response
Statistical analysis of therapy studies were carried out using Prism (Version 4.0 GraphPad Software Inc.). Unpaired two-tailed t tests were performed on mean (± SD) TVs at the termination of the PBS control group and at later time points where indicated. Animal survival was assessed by the Kaplan–Meier method and statistical analysis performed by Log-Rank testing using Prism. Significance was set at the 95% level, with results declared statistically significant if P < 0.05. In mice where a decrease of at least 50% in TV was observed for at least 7 days, a Partial Response (PR) was recorded. In mice where a complete regression was recorded for a 7 day period (i.e. TV reduction = 100%), a Complete Response (CR) was recorded (28).
Results
Immunoreactivity and affinity analysis of 177Lu-hu3S193
The immunoreactivity of 177Lu-hu3S193 for binding to DU145 was 50.4% as determined by the Lindmo assay. Binding affinity association constant (Ka) and the number of antigen binding sites per cell of 177Lu-hu3S193 was determined by Scatchard analysis. The Ka for DU145 was 0.6 × 107 M−1 and the number of binding sites per cell was 1.9 × 106.
177Lu-hu3S193 radioimmunotherapy induces cytotoxicity and apoptosis in vitro
The cytotoxicity of 177Lu-hu3S193 RIT was determined using the MTS proliferation assay following 120 hr incubation. A moderate and dose-dependent decrease in cell viability was observed following treatment of DU145 cells with 177Lu-hu3S193 (Fig. 1A). Significant inhibition of cell proliferation relative to untreated controls was apparent, and most evident following treatment of DU145 with 40μCi/well 177Lu-hu3S193 (P < 0.0001). Some cytotoxicity was evident following 40μCi/well 177Lu-huA33 although it was significantly less than achieved with the equivalent dose of 177Lu-hu3S193 (P = 0.007).
Figure 1.
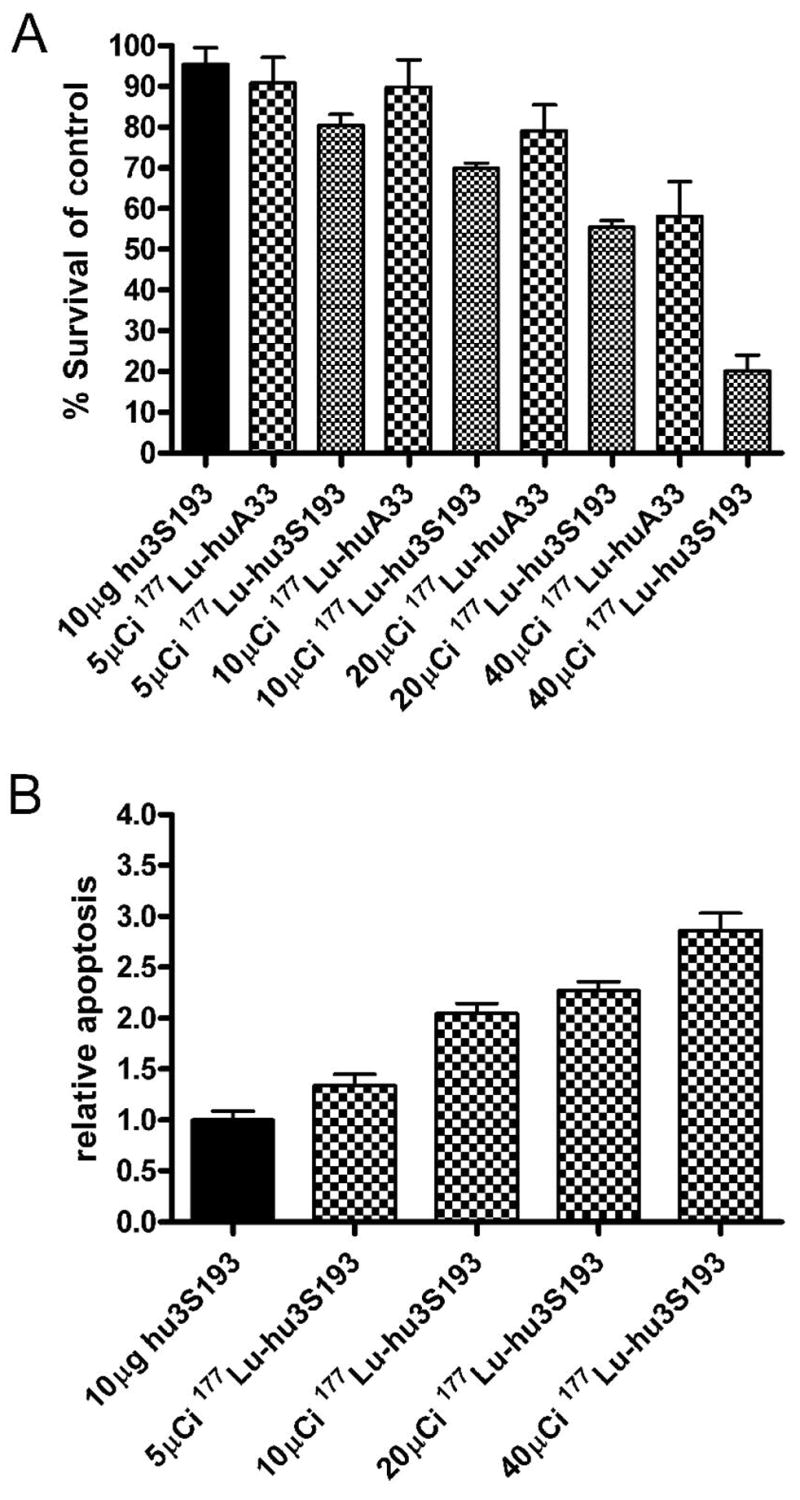
Cytotoxicity (A) and apoptosis (B) in DU145 cells following treatment with 177Lu-hu3S193 following 120 hrs incubation as indicated. Percentage survival was calculated using the formula [(A490treated/A490untreated control) × 100] (mean ± SD, n = 6). Apoptosis was determined by an ELISA assessing histone associated DNA fragmentation, and relative apoptosis determined using the formula (Apoptosistreated/Apoptosisuntreated control) (mean ± SD, n = 3).
The induction of apoptosis following 120 hr 177Lu-hu3S193 was also assessed in vitro. As observed in for the proliferation assay, 177Lu-hu3S193 caused a moderate and dose dependent increase in apoptosis, which was most significant at the 40μCi/well dose (P < 0.0001) relative to unlabeled hu3S193 treated cells (Fig 1B).
Biodistribution of 177Lu-hu3S193 in vivo
The biodistribution of 177Lu-hu3S193 was assessed in DU145 xenografted BALB/c nude mice over 288 hr. Maximal tumor uptake of 177Lu-hu3S193 in DU145 xenografts was 33.2 ± 3.9 %ID/g at 120 hr pi (Fig. 2A). Maximal tumor uptake in control Ley negative SW1222 tumors at this time was 4.6 ± 0.4 %ID/g, indicating specificity of DU145 uptake. Blood clearance of 177Lu-hu3S193 occurred at moderate rate, with pharmacokinetic analysis determining T½α of 3.4 ± 0.6 hr, a terminal T½β of 211.9 ± 24.5 hr and a clearance rate of 16.9 ± 1.43 mL/hr. Normal tissue accumulation of 177Lu-hu3S193 was negligible, with values lower than 10%ID/g in all tissues except blood at the time corresponding to maximal tumor uptake. Tumor targeting by 177Lu-hu3S193 was confirmed by gamma camera imaging in mice. As seen in Fig. 2B, clear localization of 177Lu-hu3S193 to DU145 tumors was observed.
Figure 2.

(A) Biodistribution of 177Lu-hu3S193 in blood (■), DU145 tumors (▲) and control SW122 tumors (▼) in mice. (means ± SD %ID/g, n = 5). (B) Gamma camera imaging of 177Lu-hu3S193 tumor localization obtained 120 hours post injection. (C) Surface rendered CT scan of a mouse bearing a DU145 tumor xenograft. (D) Gamma camera image and CT overlay demonstrating localization of 177Lu-hu3S193 to DU145 tumor xenograft (white arrow).
177Lu-hu3S193 radioimmunotherapy inhibits the growth of DU145 xenografts
The MTD of 177Lu-hu3S193 was determined in male BALB/c nude mice, with a single tail vein injection of radioconjugate given 12 days after establishment of tumor where mean DU145 TV was 123.2 ± 35.3mm3. Doses of 100, 200, 350 and 500μCi of 177Lu-hu3S193 were administered. The growth curves and Kaplan-Meier survival plots are shown in Fig. 3A and 3B respectively. Significant radiotoxicity was apparent in mice which received doses of 177Lu-hu3S193 above 350μCi, with severe weight loss (>15%) and hemorrhagic rash observed in 4/6 mice receiving 500μCi 177Lu-hu3S193. At doses at or below 350μCi 177Lu-hu3S193, only mild (~10%) transient weight loss was observed, and no other deaths occurred. The MTD of 177Lu-hu3S193 in this study was deemed to be 350μCi.
Figure 3.
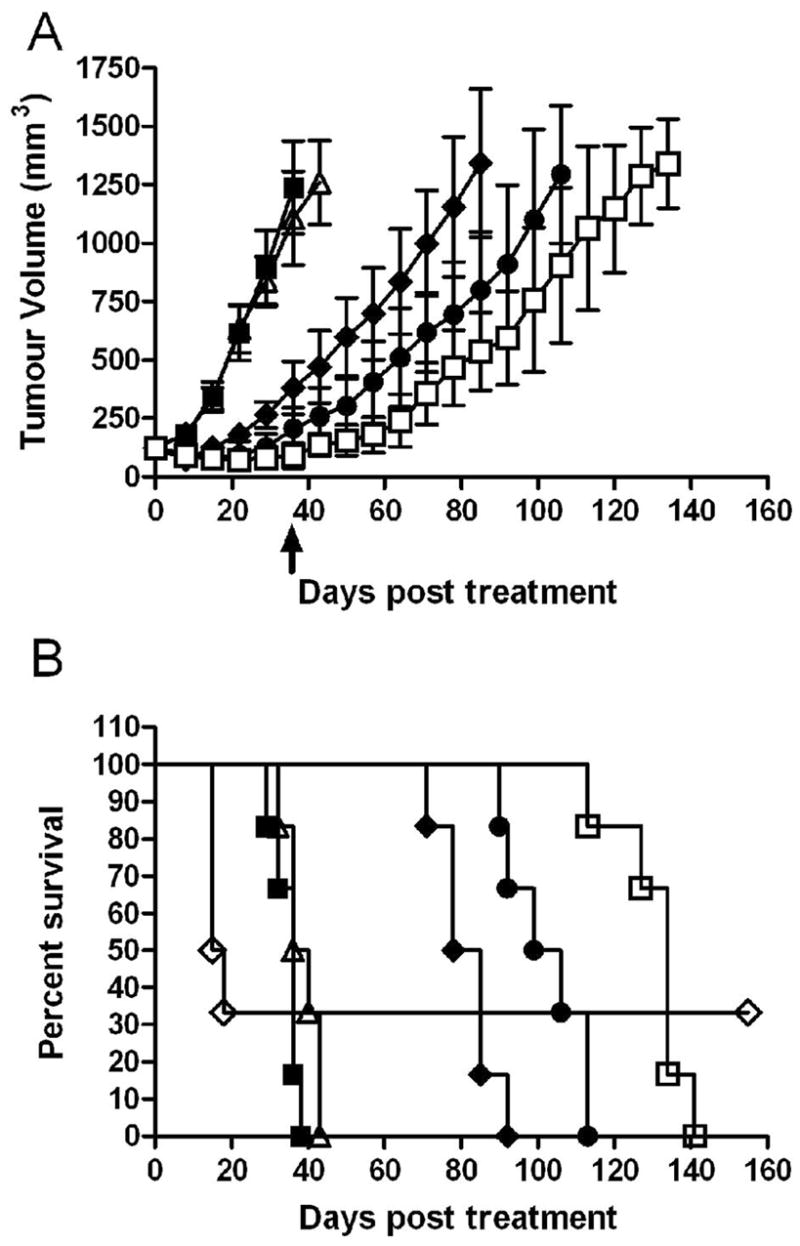
DU145 xenograft growth curves (mm3) (A) and Kaplan-Meier survival curves (B) in Maximum Tolerated Dose study in response to single dose 177Lu-hu3S193 RIT. Mice received saline vehicle (■), 180μg hu3S193 (△), 100μCi 177Lu-hu3S193 (◆), 200μCi 177Lu-hu3S193 (●), 350μCi 177Lu-hu3S193 (□) and 500μCi 177Lu-hu3S193 (◇), (Mean ± SD, n = 6). Statistical significance was assessed at termination of placebo control mice on day 36 (arrow).
Rapid tumor growth was observed in mice treated with saline placebo or 180μg of hu3S193, and all mice had been euthanized by Day 36 and 43 respectively due to excessive TV. In mice receiving 177Lu-hu3S193 alone, clear dose-dependent delay of tumor growth was apparent, with tumors treated with 350μCi significantly (P = 0.0002) smaller than those treated with 100μCi as determined at Day 36 of the study. Moderate retardation of DU145 tumor growth was observed following 100μCi 177Lu-hu3S193, although all tumors continued to grow, and mice were euthanized between Day 72 and 92 of the study. A 49 day survival advantage following 100μCi 177Lu-hu3S193 relative to unlabeled hu3S193 was apparent (P = 0.0007). Tumors in mice treated with higher doses of 200 and 350μCi 177Lu-hu3S193 demonstrated further slower growth, and were significantly smaller than those in unlabeled hu3S193 treated mice (P < 0.0001 for both 200 and 350μCi). Mice treated with 200μCi or 350μCi 177Lu-hu3S193 survived until Day 133 and Day 150 of the study respectively, representing significant increases in survival compared to control mice (P = 0.0007 for both 200 and 350μCi). A PR was observed in 2/6 mice treated with 350μCi 177Lu-hu3S193, although no responses were observed at the lower doses.
177Lu-hu3S193 and AG1478 have additive effects on established DU145 xenografts
Results of the 177Lu-hu3S193 and AG1478 CMRIT study are shown in Fig. 4. Unlabeled hu3S193 (80μg) did not demonstrate inhibition of tumor growth. Treatment with 25 and 50μCi 177Lu-hu3S193 did not significantly inhibit tumor growth, although marked suppression of tumor growth was observed in tumors treated with 100 (P = 0.002) and 200μCi (P < 0.0001) 177Lu-hu3S193 relative to unlabeled hu3S193 treated tumors. No inhibition of tumor growth was observed at any dose of isotype control 177Lu-huA33. The 400μg dose of AG1478 did not cause any significant delay of DU145 tumor growth alone. CMRIT with AG1478 enhanced the efficacy of 177Lu-hu3S193 at each of the doses investigated. Tumors in CMRIT groups at doses of 25, 50, 100 and 200μCi were all significantly smaller than tumors in the equivalent 177Lu-hu3S193 groups (P = 0.0056, P = 0.0001, P = 0.0002, P = 0.0067 respectively at Day 32) (Fig. 4A–D). A PR was observed in two animals that received 200μCi 177Lu-hu3S193 and AG1478. Enhancement of efficacy was maintained, with tumors in CMRIT groups at doses of 100 and 200μCi significantly smaller than tumors treated with 177Lu-hu3S193 alone at day 50 of the study (P = 0.0026 and P = 0.019 respectively).
Figure 4.

DU145 xenograft growth curves (mm3) in 177Lu-hu3S193 and AG1478 Combined Modality Radioimmunotherapy study in mice receiving saline vehicle (■), 80μg hu3S193 (△), 400μg AG1478 (▼), 177Lu-hu3S193 (◇), 177Lu-huA33 (□) and 177Lu-hu3S193 + AG1478 (●) at doses of (A) 25μCi, (B) 50μCi, (C) 100μCi and (D) 200μCi radiolabeled mAb, (Mean ± SD, n = 6). Statistical significance was assessed at termination of placebo control mice on day 32 (arrow).
Kaplan-Meier survival curves in the 177Lu-hu3S193 and AG1478 Combined Modality Radioimmunotherapy study. Mice in (E) received saline vehicle (■), 80μg hu3S193 (▲) or 400μg AG1478 (▼). The survival curves for mice that received 177Lu-hu3S193 alone at doses of 25μCi (■) or 50μCi (▼) or 177Lu-hu3S193 + AG1478 at doses of 25μCi (□) or 50μCi (▽) are shown in (F). Note that higher dose groups were excluded as AG1478 did not alter survival in these groups.
Combination of AG1478 with 177Lu-hu3S193 was also observed to prolong the relative survival of these mice significantly compared to 177Lu-hu3S193 alone at the 25 and 50μCi dose (25μCi; P = 0.001, 50μCi; P = 0.0012, Fig. 4F). No signs of acute toxicity were evident, although mice treated with AG1478 alone did demonstrate ~10% weight loss. However this weight loss was transient and combination of AG1478 with 177Lu-hu3S193 did not lead to heightened toxicities, as mice treated with combined agents survived until the completion of the study.
AG1478 inhibits EGFR phosphorylation and decreases proliferation in vivo
The effect of treatments on DU145 tumor proliferation was assessed by determining the expression of the Ki-67 proliferation marker in tumors following completion of treatment on Day 14 (Fig. 5A). 200μCi 177Lu-hu3S193 and 400μg AG1478 alone were both observed to cause a significant (P < 0.0001) decrease in expression of Ki-67 relative to that in tumors treated with 80μg hu3S193. However, the greatest decrease in Ki-67 was observed in tumors following CMRIT treatment, with a mean composite score of 40.55 ± 22.55 relative to the mean score of 214.83 ± 45.27 for unlabeled hu3S193. Ki-67 expression of CMRIT treated tumors was also significantly (P < 0.0001) reduced compared to the mean scores of either respective single treatment
Figure 5.
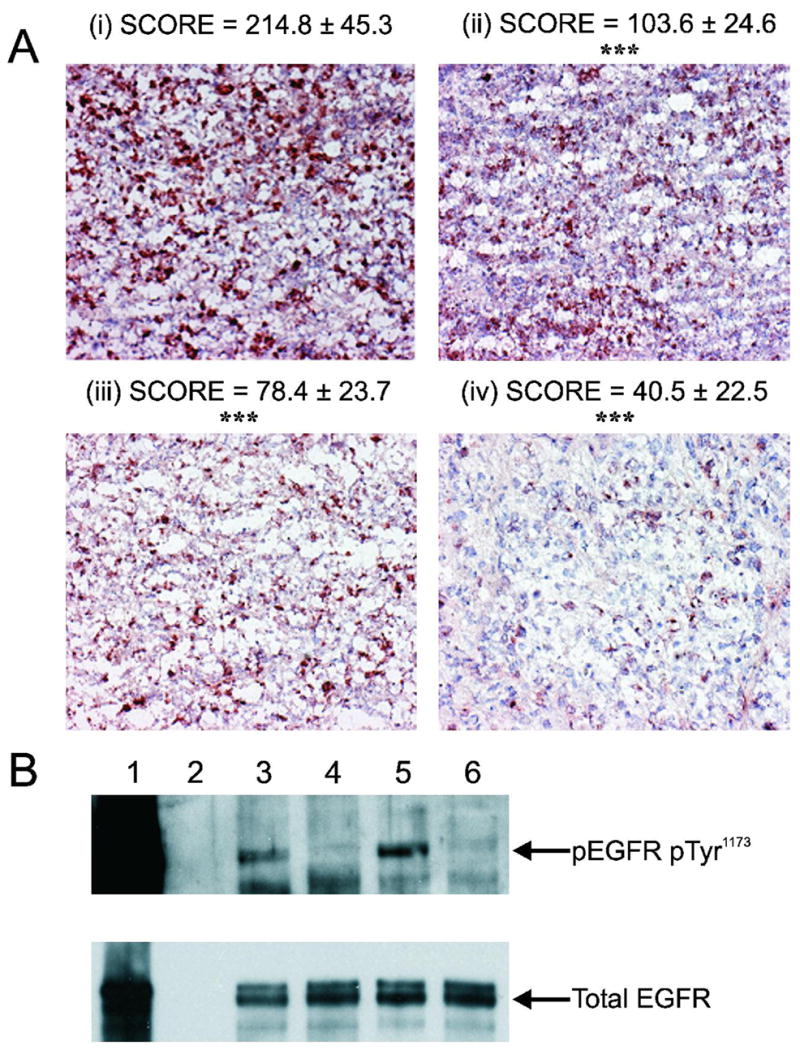
Expression of Ki-67 proliferation antigen and EGFR in DU145 tumors from the 177Lu-hu3S193 and AG1478 Combined Modality Radioimmunotherapy study. (A) Representative images of Ki-67 proliferation antigen expression in tumors following treatments with 80μg hu3S193 (i), 400μg AG1478 (ii), 200μCi 177Lu-hu3S193 (iii) and 200μCi 177Lu-hu3S193 + AG1478 (iv) (400X magnification). Relative expression of each treatment group was determined by analyzing 11 distinct fields from 2 different tumors and calculating a composite score as detailed in Materials and Methods. Significant reduction (P < 0.0001) in Ki-67 tumor expression relative to unlabeled hu3S193 control is indicated by ***. (B) Expression of phospho(Tyr1173) and total EGFR in vivo following treatments. Lanes represent a 100ng/mL EGF stimulated A431 positive control sample (1), blank (2) and tumor samples treated with 80μg hu3S193 (3), 400μg AG1478 (4), 200μCi 177Lu-hu3S193 (5) and 200μCi 177Lu-hu3S193 + AG1478 (6). Note that lane 1 is over-exposed in order to detect EGFR in the in vivo samples.
The ability of AG1478 to inhibit EGFR phosphorylation in vivo was assessed by western blotting DU145 tumor homogenates using the same tumors used in analysis of Ki-67 expression (Fig. 5B). Whereas significant expression of phosphorylated EGFR was evident in tumors treated with 200μCi 177Lu-hu3S193, it is completely abolished in tumors treated with 200μCi 177Lu-hu3S193 in combination with AG1478.
Docetaxel enhances the 177Lu-hu3S193 induced regression of established DU145 xenografts
The ability of docetaxel to enhance the efficacy of 177Lu RIT was assessed at the 177Lu-hu3S193 dose levels of 100, 200 and 350μCi (Fig. 6). When administered as a single agent, docetaxel caused moderate retardation of DU145 tumor growth, with significant difference in tumor volume (P = 0.001) compared to saline control at Day 36. Additionally, docetaxel treatment increased survival of mice to 57 Days relative to the 36 Day survival of saline treated mice. Mice treated with 100μCi 177Lu-hu3S193 and docetaxel survived until the end of the study, with significantly improved survival over mice treated with RIT alone (P = 0.0006) or docetaxel alone (P = 0.0008).
Figure 6.

DU145 xenograft curves (mm3) in 177Lu-hu3S193 and docetaxel Combined Modality Radioimmunotherapy study in mice receiving saline vehicle (■), 180μg hu3S193 (△), 600μg docetaxel (▼), 177Lu-hu3S193 (◇) and 177Lu-hu3S193 + docetaxel (●) at doses of (A) 100μCi, (B) 200μCi and (C) 350μCi of radiolabelled mAb, (Mean ± SD, n = 6). Statistical significance was assessed at termination of placebo control mice on day 36 (arrow).
Kaplan-Meier survival curves in the 177Lu-hu3S193 and docetaxel Combined Modality Radioimmunotherapy study. Mice received saline vehicle (■), 180μg hu3S193 (▲), 600μg docetaxel (▼), and 177Lu-hu3S193 without (D) and with docetaxel (E) at doses of 100μCi 177Lu-hu3S193 (◆), 200μCi 177Lu-hu3S193 (●) and 350μCi 177Lu-hu3S193 (□)
At the higher doses of 177Lu-hu3S193 RIT, more profound efficacy was apparent following CMRIT, with 83.3% (5/6) mice treated with 200μCi 177Lu-hu3S193 and docetaxel having a PR at Day 36 of the study. Additionally, survival of these mice was superior to docetaxel alone (P = 0.0008), although not to RIT alone as these mice also survived to Day 113. The greatest efficacy of CMRIT was observed at the 350μCi dose, indicating a dose-dependent response. At this dose 50% of mice had a PR to combined treatment, and 50% achieved a CR, with two of these maintained for the duration of the study to day 150. Significant reduction in tumor volume compared to RIT alone (P = 0.0039), and increase in survival relative to docetaxel alone (P = 0.0008) was observed.
Discussion
Radioimmunotherapy offers a therapeutic strategy for the selective delivery of radiation to tumors, while limiting the systemic effects of radiation on normal tissues (30). The radioisotope 177Lu has been identified as a promising alternative to established β-emitters such as 131I and 90Y, with physical properties amenable to RIT of small lesions where clinical efficacy of RIT is most likely (5,38,39). The limited efficacy of RIT in the treatment of solid tumors has lead to the investigation of various methods for improving the therapeutic efficacy of RIT (8). Combined modality RIT, where the mAb targeted radiation is combined with a disparate therapeutic that aims to enhance the radiation induced tumor killing, is currently under investigation using a number of agents (21,28,29).
The effects of 177Lu-hu3S193 upon in vitro cytotoxicity and induction of apoptosis in DU145 was examined in order to assess the efficacy of RIT prior to in vivo studies. Dose-dependent cytotoxicity was apparent. Cytotoxicity was dependent upon 177Lu as unlabeled hu3S193 failed to elicit any effect. However, the antibody dose levels used in these studies was low, and the lack of efficacy here does not reflect the cytotoxic properties of unlabeled hu3S193 that have been observed due to potent complement-dependent and antibody-dependent cellular cytotoxicity activity (31). Non-specific “cross-fire” irradiation, where cells may be irradiated by an adjacent unbound radioconjugate, most likely explains the limited cytotoxicity observed following higher doses of 177Lu-huA33. The specificity of 177Lu-hu3S193 is apparent by the consistently more potent cytotoxicity observed relative to the equivalent radiation dose of 177Lu-huA33.
177Lu-hu3S193 was observed to mediate significant (P < 0.0001) apoptosis of DU145 cells, indicating apoptosis is at least partly responsible for the observed cytotoxicity. These results support the view that the induction of apoptosis following accumulation of irreversible indirect DNA damage is the principal mechanism of cell death following β-particle emitter irradiation (40).
The localization of 177Lu-hu3S193 to tumors was assessed in vivo by biodistribution studies. Maximal tumor uptake was generally comparable to hu3S193 uptake previously observed following radiolabeling with either 131I, 111In or 90Y in MCF-7 breast or A431 squamous cancer xenograft models (28,29,35). Uptake of 177Lu-hu3S193 was relatively homogenous in Ley positive areas of tumors as assessed by immunohistochemistry and autoradiography (data not shown), and combined with gamma camera imaging, indicate that 177Lu-hu3S193 is able to successfully target and irradiate Ley positive tumors. Hu3S193 has been assessed in phase I/II trials in patients with Ley expressing tumors, and has demonstrated excellent targeting of tumors without any normal tissue binding (41,42).
Single agent 177Lu-hu3S193 demonstrated significant anti-tumor toxicity and effected a significant improvement in survival relative to mice treated with unlabeled hu3S193. Evident toxicity was observed in 4/6 mice which received doses of 500μCi 177Lu-hu3S193 and subsequently, 350μCi is the apparent MTD of 177Lu-hu3S193 in this model. Similar results were observed in the first therapeutic examination of 177Lu in vivo in a study assessing the anti-TAG-72 mAb 177Lu-CC49 in LS174T colon cancer xenografted mice, where 4/9 mice survived following 500μCi, while all mice treated with 350μCi 177Lu-CC49 survived (43).
We have previously demonstrated the enhancement of 90Y-hu3S193 RIT in combination with the EGFR TKI AG1478 in A431 squamous cell carcinoma xenografts known to over express EGFR (29). The current study explored the efficacy of 177Lu-hu3S193 RIT in combination with AG1478 in established DU145 prostate cancer xenografts expressing moderate amounts of EGFR. Super-additive enhancement of 177Lu-hu3S193 anti-tumor effects by AG1478 were observed at all 177Lu-hu3S193 doses investigated, with the greatest significance observed at the 50μCi dose (P = 0.0001). Other studies have observed enhancement of RIT following combination with anti-EGFR mAbs 425 and cetuximab, providing further rationale for combination of RIT with anti-EGFR therapeutics (21,44).
Analysis of tumor homogenates demonstrated that AG1478 inhibited activation of EGFR in combined treatment tumors whereas robust expression of phosphorylated EGFR was observed in tumors treated with 177Lu-hu3S193 alone. The inhibition of EGFR activation following tumor irradiation may be a mechanism for the enhanced anti-tumor efficacy of CMRIT. Further analysis of tumor expression of Ki-67 proliferation marker by immunohistochemistry demonstrated that the most significant decrease in tumor proliferation was observed following combination treatment. Additionally, AG1478 was observed to enhance 177Lu-hu3S193 cytotoxicity and apoptosis in vitro (data not shown).
Enhancement of 177Lu-hu3S193 anti-tumor activity with the taxane docetaxel was also explored, as docetaxel is widely used in the treatment of solid tumors such as prostate cancer, in addition to having recognized radiosensitizing properties (20,45). Upon combination of 177Lu-hu3S193 with docetaxel in vivo, marked enhancement of DU145 tumor radiosensitivity was observed, resulting in significantly reduced tumor volume in all mice receiving combined therapies. Complete responses were observed in 3/6 mice receiving 350μCi 177Lu-hu3S193 and docetaxel. Although Stromberg et. al reported that the related taxane paclitaxel failed to radiosensitize DU145 cells in vitro, our studies clearly demonstrate radiosensitization of DU145 xenografts by docetaxel chemotherapy (46). Docetaxel may be a more potent radiosensitizer than paclitaxel, as suggested in a study by O’Donnell et. al, where superior anti-tumor efficacy of 90Y-ChL6 RIT combined with docetaxel was observed relative to a parallel study assessing 90Y-ChL6 RIT and paclitaxel at equivalent doses (23).
The principal mechanism of action of docetaxel radiosensitization is thought to involve microtubule stabilization and cell cycle arrest at the radiosensitive G2/M phase mediating an increased induction of apoptosis (45,47). Docetaxel induced radiosensitization and apoptosis is also likely to result from its phosphorylation and inactivation of the anti-apoptotic protein Bcl-2 (48). Significant enhancement of hu3S193 RIT by paclitaxel in previous studies of CMRIT, and clinical assessment of RIT combined with paclitaxel provide further rationale for the current exploration of 177Lu-hu3S193 with taxane chemotherapy (28,49,50).
Conclusions
The current study is the first to investigate the efficacy of 177Lu-hu3S193 RIT, with in vitro analysis demonstrating that 177Lu-hu3S193 mediates apoptosis in prostate cancer cells. In vivo studies demonstrated specific targeting and therapeutic efficacy against prostate cancer xenografts. CMRIT studies demonstrate the enhancement of 177Lu-hu3S193 using either the EGFR TKI AG1478 or docetaxel chemotherapy. Collectively, these results indicate the promise of 177Lu-hu3S193 RIT alone and in CMRIT for the treatment of Ley positive prostate cancer and other Ley positive malignancies.
Acknowledgments
We thank the staff from the Department of Nuclear Medicine, Austin Hospital, for their assistance in the CT scanning and gamma camera imaging of animals and staff of the Tumour Targeting Program for assistance in biodistribution studies. This work was supported in part by an NH&MRC Program Grant No. 280912 and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Financial Support: MPK was supported by a Melbourne Research Scholarship, University of Melbourne, Melbourne, Australia. IDD is supported in part by a Victorian Cancer Agency Clinician Researcher Fellowship. MWB is supported by the Intramural Research Program of the NIH, National Cancer Institute, Centre for Cancer Research.
References
- 1.Scott AM, Welt S. Antibody-based immunological therapies. Curr Opin Immunol. 1997;9(5):717–722. doi: 10.1016/s0952-7915(97)80054-4. [DOI] [PubMed] [Google Scholar]
- 2.Chong G, Lee FT, Hopkins W, Tebbutt N, Cebon JS, Mountain AJ, Chappell B, Papenfuss A, Schleyer P, U P, Murphy R, Wirth V, Smyth FE, Potasz N, Poon A, Davis ID, Saunder T, O’Keefe GJ, Burgess AW, Hoffman EW, Old LJ, Scott AM. Phase I Trial of 131I-huA33 in Patients with Advanced Colorectal Carcinoma. Clin Cancer Res. 2005;11(13):4818–4826. doi: 10.1158/1078-0432.CCR-04-2330. [DOI] [PubMed] [Google Scholar]
- 3.Bethge WA, Sandmaier BM. Targeted cancer therapy and immunosuppression using radiolabeled monoclonal antibodies. Semin Oncol. 2004;31(1):68–82. doi: 10.1053/j.seminoncol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Smith-Jones PM. Radioimmunotherapy of prostate cancer. Q J Nucl Med Mol Imaging. 2004;48(4):297–304. [PubMed] [Google Scholar]
- 5.Scott AM. Radioimmunotherapy of Prostate Cancer: Does Tumor Size Matter? J Clin Oncol. 2005;23(21):4567–4569. doi: 10.1200/JCO.2005.01.903. [DOI] [PubMed] [Google Scholar]
- 6.Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ, Bander NH. Phase I trial of yttrium-90-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for androgen-independent prostate cancer. J Clin Oncol. 2004;22(13):2522–2531. doi: 10.1200/JCO.2004.09.154. [DOI] [PubMed] [Google Scholar]
- 7.Bander NH, Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ. Phase I trial of 177Lu-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J Clin Oncol. 2005;23(21):4591–4601. doi: 10.1200/JCO.2005.05.160. [DOI] [PubMed] [Google Scholar]
- 8.Jhanwar YS, Divgi C. Current status of therapy of solid tumors. J Nucl Med. 2005;46(Suppl 1):141S–150S. [PubMed] [Google Scholar]
- 9.Stein R, Govindan SV, Chen S, Reed L, Richel H, Griffiths GL, Hansen HJ, Goldenberg DM. Radioimmunotherapy of a human lung cancer xenograft with monoclonal antibody RS7: evaluation of (177)Lu and comparison of its efficacy with that of (90)Y and residualizing (131)I. J Nucl Med. 2001;42(6):967–974. [PubMed] [Google Scholar]
- 10.Divgi C. Editorial: What Ails Solid Tumor Radioimmunotherapy? Cancer Biotherapy & Radiopharmaceuticals. 2006;21(2):81. doi: 10.1089/cbr.2006.21.81. [DOI] [PubMed] [Google Scholar]
- 11.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 12.Chinnaiyan P, Allen GW, Harari PM. Radiation and new molecular agents, part II: targeting HDAC, HSP90, IGF-1R, PI3K, and Ras. Semin Radiat Oncol. 2006;16(1):59–64. doi: 10.1016/j.semradonc.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Ma BB, Bristow RG, Kim J, Siu LL. Combined-modality treatment of solid tumors using radiotherapy and molecular targeted agents. J Clin Oncol. 2003;21(14):2760–2776. doi: 10.1200/JCO.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 14.Yacoub A, McKinstry R, Hinman D, Chung T, Dent P, Hagan MP. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat Res. 2003;159(4):439–452. doi: 10.1667/0033-7587(2003)159[0439:egfair]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 15.Hagan M, Yacoub A, Dent P. Ionizing radiation causes a dose-dependent release of transforming growth factor alpha in vitro from irradiated xenografts and during palliative treatment of hormone-refractory prostate carcinoma. Clin Cancer Res. 2004;10(17):5724–5731. doi: 10.1158/1078-0432.CCR-04-0420. [DOI] [PubMed] [Google Scholar]
- 16.Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z, Skrinos E, Murone C, Saunder TH, Chappell B, Papenfuss AT, Poon AM, Hopkins W, Smyth FE, MacGregor D, Cher LM, Jungbluth AA, Ritter G, Brechbiel MW, Murphy R, Burgess AW, Hoffman EW, Johns TG, Old LJ. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A. 2007;104(10):4071–4076. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33(4):369–385. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Baselga J, Arribas J. Treating cancer’s kinase ‘addiction’. 2004;10(8):786–787. doi: 10.1038/nm0804-786. [DOI] [PubMed] [Google Scholar]
- 19.She Y, Lee F, Chen J, Haimovitz-Friedman A, Miller VA, Rusch VR, Kris MG, Sirotnak FM. The epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 selectively potentiates radiation response of human tumors in nude mice, with a marked improvement in therapeutic index. Clin Cancer Res. 2003;9(10 Pt 1):3773–3778. [PubMed] [Google Scholar]
- 20.Choy H. Combining taxanes with radiation for solid tumors. Int J Cancer. 2000;90(3):113–127. [PubMed] [Google Scholar]
- 21.Burke PA, DeNardo SJ, Miers LA, Kukis DL, DeNardo GL. Combined modality radioimmunotherapy. Promise and peril Cancer. 2002;94(4 Suppl):1320–1331. doi: 10.1002/cncr.10303. [DOI] [PubMed] [Google Scholar]
- 22.Kumar P. A new paradigm for the treatment of high-risk prostate cancer: radiosensitization with docetaxel. Rev Urol. 2003;5(Suppl 3):S71–77. [PMC free article] [PubMed] [Google Scholar]
- 23.O’Donnell RT, DeNardo SJ, Miers LA, Lamborn KR, Kukis DL, DeNardo GL, Meyers FJ. Combined modality radioimmunotherapy for human prostate cancer xenografts with taxanes and 90Y-DOTA-peptide-ChL6. Prostate. 2002;50(1):27–37. doi: 10.1002/pros.10029. [DOI] [PubMed] [Google Scholar]
- 24.Zhang AL, Russell PJ, Knittel T, Milross C. Paclitaxel enhanced radiation sensitization for the suppression of human prostate cancer tumor growth via a p53 independent pathway. The Prostate. 2007;67(15):1630–1640. doi: 10.1002/pros.20638. [DOI] [PubMed] [Google Scholar]
- 25.Boghaert ER, Sridharan L, Armellino DC, Khandke KM, DiJoseph JF, Kunz A, Dougher MM, Jiang F, Kalyandrug LB, Hamann PR, Frost P, Damle NK. Antibody-targeted chemotherapy with the calicheamicin conjugate hu3S193-N-acetyl gamma calicheamicin dimethyl hydrazide targets Lewisy and eliminates Lewisy-positive human carcinoma cells and xenografts. Clin Cancer Res. 2004;10(13):4538–4549. doi: 10.1158/1078-0432.CCR-04-0037. [DOI] [PubMed] [Google Scholar]
- 26.Myers RB, Srivastava S, Grizzle WE. Lewis Y antigen as detected by the monoclonal antibody BR96 is expressed strongly in prostatic adenocarcinoma. J Urol. 1995;153(5):1572–1574. [PubMed] [Google Scholar]
- 27.Martensson S, Bigler SA, Brown M, Lange PH, Brawer MK, Hakomori S. Sialyl-Lewis(x) and related carbohydrate antigens in the prostate. Hum Pathol. 1995;26(7):735–739. doi: 10.1016/0046-8177(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 28.Kelly MP, Lee FT, Smyth FE, Brechbiel MW, Scott AM. Enhanced efficacy of 90Y-radiolabeled anti-Lewis Y humanized monoclonal antibody hu3S193 and paclitaxel combined-modality radioimmunotherapy in a breast cancer model. J Nucl Med. 2006;47(4):716–725. [PubMed] [Google Scholar]
- 29.Lee FT, Mountain AJ, Kelly MP, Hall C, Rigopoulos A, Johns TG, Smyth FE, Brechbiel MW, Nice EC, Burgess AW, Scott AM. Enhanced efficacy of radioimmunotherapy with 90Y-CHX-A″-DTPA-hu3S193 by inhibition of epidermal growth factor receptor (EGFR) signaling with EGFR tyrosine kinase inhibitor AG1478. Clin Cancer Res. 2005;11(19 Pt 2):7080s–7086s. doi: 10.1158/1078-0432.CCR-1004-0019. [DOI] [PubMed] [Google Scholar]
- 30.Wong JYC. Systemic targeted radionuclide therapy: Potential new areas. International Journal of Radiation Oncology*Biology*Physics. 2006;66(2 Supplement 1):S74–S82. doi: 10.1016/j.ijrobp.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 31.Scott AM, Geleick D, Rubira M, Clarke K, Nice EC, Smyth FE, Stockert E, Richards EC, Carr FJ, Harris WJ, Armour KL, Rood J, Kypridis A, Kronina V, Murphy R, Lee FT, Liu Z, Kitamura K, Ritter G, Laughton K, Hoffman E, Burgess AW, Old LJ. Construction, production, and characterization of humanized anti-Lewis Y monoclonal antibody 3S193 for targeted immunotherapy of solid tumors. Cancer Res. 2000;60(12):3254–3261. [PubMed] [Google Scholar]
- 32.Scott AM, Lee F-T, Jones R, Hopkins W, MacGregor D, Cebon JS, Hannah A, Chong G, U P, Papenfuss A, Rigopoulos A, Sturrock S, Murphy R, Wirth V, Murone C, Smyth FE, Knight S, Welt S, Ritter G, Richards E, Nice EC, Burgess AW, Old LJ. A Phase I Trial of Humanized Monoclonal Antibody A33 in Patients with Colorectal Carcinoma: Biodistribution, Pharmacokinetics, and Quantitative Tumor Uptake. Clin Cancer Res. 2005;11(13):4810–4817. doi: 10.1158/1078-0432.CCR-04-2329. [DOI] [PubMed] [Google Scholar]
- 33.Wu C, Kobayashi H, Sun B, Yoo TM, Paik CH, Gansow OA, Carrasquillo JA, Pastan I, Brechbiel MW. Stereochemical influence on the stability of radiometal complexes in vivo. Synthesis and evaluation of the four stereoisomers of 2-(p-nitrobenzyl)-trans-CyDTPA. Bioorg Med Chem. 1997;5(10):1925–1934. doi: 10.1016/s0968-0896(97)00130-2. [DOI] [PubMed] [Google Scholar]
- 34.Nikula TK, Curcio MJ, Brechbiel MW, Gansow OA, Finn RD, Scheinberg DA. A rapid, single vessel method for preparation of clinical grade ligand conjugated monoclonal antibodies. Nucl Med Biol. 1995;22(3):387–390. doi: 10.1016/0969-8051(94)00126-5. [DOI] [PubMed] [Google Scholar]
- 35.Clarke K, Lee FT, Brechbiel MW, Smyth FE, Old LJ, Scott AM. In vivo biodistribution of a humanized anti-Lewis Y monoclonal antibody (hu3S193) in MCF-7 xenografted BALB/c nude mice. Cancer Res. 2000;60(17):4804–4811. [PubMed] [Google Scholar]
- 36.Johns TG, Luwor RB, Murone C, Walker F, Weinstock J, Vitali AA, Perera RM, Jungbluth AA, Stockert E, Old LJ, Nice EC, Burgess AW, Scott AM. Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc Natl Acad Sci U S A. 2003;100(26):15871–15876. doi: 10.1073/pnas.2036503100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perera RM, Narita Y, Furnari FB, Gan HK, Murone C, Ahlkvist M, Luwor RB, Burgess AW, Stockert E, Jungbluth AA, Old LJ, Cavenee WK, Scott AM, Johns TG. Treatment of human tumor xenografts with monoclonal antibody 806 in combination with a prototypical epidermal growth factor receptor-specific antibody generates enhanced antitumor activity. Clin Cancer Res. 2005;11(17):6390–6399. doi: 10.1158/1078-0432.CCR-04-2653. [DOI] [PubMed] [Google Scholar]
- 38.Behr TM, Liersch T, Greiner-Bechert L, Griesinger F, Behe M, Markus PM, Gratz S, Angerstein C, Brittinger G, Becker H, Goldenberg DM, Becker W. Radioimmunotherapy of small-volume disease of metastatic colorectal cancer. Cancer. 2002;94(4 Suppl):1373–1381. doi: 10.1002/cncr.10308. [DOI] [PubMed] [Google Scholar]
- 39.Schott ME, Schlom J, Siler K, Milenic DE, Eggensperger D, Colcher D, Cheng R, Kruper WJ, Jr, Fordyce W, Goeckeler W. Biodistribution and preclinical radioimmunotherapy studies using radiolanthanide-labeled immunoconjugates. Cancer. 1994;73(3 Suppl):993–998. doi: 10.1002/1097-0142(19940201)73:3+<993::aid-cncr2820731337>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 40.Friesen C, Lubatschofski A, Kotzerke J, Buchmann I, Reske SN, Debatin KM. Beta-irradiation used for systemic radioimmunotherapy induces apoptosis and activates apoptosis pathways in leukaemia cells. Eur J Nucl Med Mol Imaging. 2003;30(9):1251–1261. doi: 10.1007/s00259-003-1216-z. [DOI] [PubMed] [Google Scholar]
- 41.Krug LM, Milton DT, Jungbluth AA, Chen LC, Quaia E, Pandit-Taskar N, Nagel A, Jones J, Kris MG, Finn R, Smith-Jones P, Scott AM, Old L, Divgi C. Targeting Lewis Y (Ley) in small cell lung cancer with a humanized monoclonal antibody, hu3S193: a pilot trial testing two dose levels. J Thorac Oncol. 2007;2(10):947–952. doi: 10.1097/JTO.0b013e3181560dcc. [DOI] [PubMed] [Google Scholar]
- 42.Scott AM, Tebbutt N, Lee F-T, Cavicchiolo T, Liu Z, Gill S, Poon AMT, Hopkins W, Smyth FE, Murone C, MacGregor D, Papenfuss AT, Chappell B, Saunder TH, Brechbiel MW, Davis ID, Murphy R, Chong G, Hoffman EW, Old LJ. A Phase I Biodistribution and Pharmacokinetic Trial of Humanized Monoclonal Antibody Hu3s193 in Patients with Advanced Epithelial Cancers that Express the Lewis-Y Antigen. Clin Cancer Res. 2007;13(11):3286–3292. doi: 10.1158/1078-0432.CCR-07-0284. [DOI] [PubMed] [Google Scholar]
- 43.Schlom J, Siler K, Milenic DE, Eggensperger D, Colcher D, Miller LS, Houchens D, Cheng R, Kaplan D, Goeckeler W. Monoclonal antibody-based therapy of a human tumor xenograft with a 177Lu-labeled immunoconjugate. Cancer Res. 1991;51(11):2889–2896. [PubMed] [Google Scholar]
- 44.van Gog FB, Brakenhoff RH, Stigter-van Walsum M, Snow GB, van Dongen GA. Perspectives of combined radioimmunotherapy and anti-EGFR antibody therapy for the treatment of residual head and neck cancer. Int J Cancer. 1998;77(1):13–18. doi: 10.1002/(sici)1097-0215(19980703)77:1<13::aid-ijc3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 45.Gligorov J, Lotz JP. Preclinical pharmacology of the taxanes: implications of the differences. Oncologist. 2004;9(Suppl 2):3–8. doi: 10.1634/theoncologist.9-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- 46.Stromberg JS, Lee YJ, Armour EP, Martinez AA, Corry PM. Lack of radiosensitization after paclitaxel treatment of three human carcinoma cell lines. Cancer. 1995;75(9):2262–2268. doi: 10.1002/1097-0142(19950501)75:9<2262::aid-cncr2820750912>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 47.Dunne AL, Mothersill C, Robson T, Wilson GD, Hirst DG. Radiosensitization of colon cancer cell lines by docetaxel: mechanisms of action. Oncol Res. 2004;14(9):447–454. doi: 10.3727/0965040041791455. [DOI] [PubMed] [Google Scholar]
- 48.Ganansia-Leymarie V, Bischoff P, Bergerat JP, Holl V. Signal transduction pathways of taxanes-induced apoptosis. Curr Med Chem Anticancer Agents. 2003;3(4):291–306. doi: 10.2174/1568011033482422. [DOI] [PubMed] [Google Scholar]
- 49.Richman CM, DeNardo SJ, O’Donnell RT, Yuan A, Shen S, Goldstein DS, Tuscano JM, Wun T, Chew HK, Lara PN, Kukis DL, Natarajan A, Meares CF, Lamborn KR, DeNardo GL. High-Dose Radioimmunotherapy Combined with Fixed, Low-Dose Paclitaxel in Metastatic Prostate and Breast Cancer by Using a MUC-1 Monoclonal Antibody, m170, Linked to Indium-111/Yttrium-90 via a Cathepsin Cleavable Linker with Cyclosporine to Prevent Human Anti-mouse Antibody. Clin Cancer Res. 2005;11(16):5920–5927. doi: 10.1158/1078-0432.CCR-05-0211. [DOI] [PubMed] [Google Scholar]
- 50.Meredith RF, Alvarez RD, Partridge EE, Khazaeli MB, Lin C-Y, Macey DJ, Austin JM, Kilgore LC, Grizzle WE, Schlom J, LoBuglio AF. Intraperitoneal Radioimmunochemotherapy of Ovarian Cancer: A Phase I Study. Cancer Biotherapy & Radiopharmaceuticals. 2001;16(4):305–315. doi: 10.1089/108497801753131381. [DOI] [PubMed] [Google Scholar]