

Abstract
Dynein is a minus-end directed microtubule motor that transports numerous cargoes throughout the cell. During mitosis, dynein motor activity is necessary for the positioning of spindle microtubules and has also been implicated in inactivating the spindle assembly checkpoint. Mutations in dynein motor and/or accessory proteins are associated with human disease, including cancer, and the delocalization of dynein from mitotic spindles has been correlated with an increased incidence of multipolar spindle formation in some cancer cells that contain supernumerary centrosomes. The high-risk human papillomavirus type 16 (HPV16) E7 oncoprotein induces centrosome overduplication and has been shown to cause multipolar mitotic spindle formation, a diagnostic hallmark of HPV-associated neoplasias. Here we show that HPV16 E7 expression leads to an increased population of mitotic cells with dynein delocalized from the mitotic spindle. This function maps to sequences of HPV16 E7 that are distinct from the region necessary for centrosome overduplication. However, contrary to previous reports, we provide evidence that dynein delocalization by HPV16 E7 is neither necessary nor sufficient to cause the formation of multipolar mitoses.
Keywords: Human papillomavirus, E7 oncoprotein, Centrosomes, Mitosis, Genomic Instability
Introduction
Dynein is a minus-end directed microtubule motor protein that is composed of several subunits. The approximately 520 kD heavy chain subunits are responsible for generating movement along the microtubule via ATPase activity and the approximately 74 kD intermediate chain subunits are thought to anchor dynein to its cargo (reviewed in 1). Dynein also contains smaller intermediate chain subunits and light chain subunits whose functions are not clearly understood, although it has been suggested that dynein light chains may also play an important role in diverse dynein-motor complex independent processes, such as apoptosis, enzyme regulation, transcriptional regulation and kidney development (2). The processivity of dynein is increased upon complex formation with the dynactin complex and dynactin is therefore commonly considered an obligatory cofactor (3). The dynein motor complex aids in the positioning of the Golgi complex and mitochondria, along with other organelles, and transports cargo from the endoplasmic reticulum, endosomes, and lysosomes (reviewed in 4, 5–7). Being a cytoplasmic motor complex, it likely plays an important role in transporting numerous cargoes and, additionally, dynein is also implicated in axonal transport in neurons (reviewed in 8). Furthermore, dynein transports proteins, such as pericentrin and γ-tubulin, which are necessary for centrosome assembly during interphase (9). In mitosis, the dynein motor complex positions mitotic spindle poles (reviewed in 10) and is responsible for targeting the nuclear mitotic apparatus protein 1 (NuMA) to spindle poles, where NuMA focuses and stabilizes microtubule ends and tethers them to the centrosomes (11, 12). Dynein is also recruited to kinetochores where it is reported to play a role in spindle assembly checkpoint inactivation by transporting checkpoint proteins away from properly attached kinetochores (13–15).
Likely due to the many integral functions of dynein, mutations in dynein motors and associated proteins have been linked to human diseases, including neurodegenerative disease (16) and cancer. Dynein light chain 1 (DLC1) expression is regulated by estrogen and associates with and acts as a transcriptional co-activator of the estrogen receptor (ER) (17). DLC1 is also a substrate of the p21-activated kinase 1 (PAK1) and PAK1-mediated DLC1 phosphorylation has been linked to proliferation of ER positive breast cancer cells (18). DLC1 appears to be frequently overexpressed in breast cancer and may contribute to accelerated cell cycle progression due to reduction of nuclear p21CIP1 and increased CDK2 activity (18, 19). Moreover, a genome-wide computer analysis of single nucleotide polymorphisms (SNP) in tumoral versus normal tissue identified a cancer specific DLC1 variant with a glycine to cysteine mutation at amino acid residue 79, which may affect ligand binding (20). Additionally, it was shown that in some cancer cell lines, there was in increased proportion of mitotic cells where dynein was delocalized from the mitotic spindle (21). In these cells, dynein delocalization was correlated with an increased incidence in multipolarity during mitosis and, therefore, dynein was purported to play a role in the coalescence of supernumerary centrosomes. Spindle multipolarity, together with centrosome amplification, has been observed in many tumor tissues including high-risk human papillomavirus (HPV)-associated lesions and cancers (22–24). Centrosome-associated mitotic abnormalities likely importantly contribute to the genomic instability that drives carcinogenesis (reviewed in 25, 26, 27). Thus, it is possible that the disruption of microtubule motors by high-risk HPV may contribute to HPV-associated tumorigenesis.
In concert with the high-risk HPV E6 oncoprotein, the high-risk HPV E7 oncoprotein is necessary for the establishment and maintenance of the transformed phenotype of HPV-associated neoplasias. High-risk HPV E6 and E7 disrupt the p53 and pRB tumor suppressor pathways, respectively. Additional mutations to the host genome are required for the malignant progression of HPV-associated premalignant lesions, however, and each of the viral oncoproteins actively contributes to the destabilization of the host genome through different mechanisms. The expression of high-risk HPV16 E7 results in the deregulation of the centrosome duplication cycle, allowing for excess rounds of centrosomal duplication to occur during one round of cell division (24, 28, 29). The accumulation of supernumerary centrosomes increases the frequency of aberrant mitoses and often causes the formation of multiple mitotic spindle poles. In fact, multipolar mitoses are a diagnostic hallmark used by pathologists to identify high-risk HPV-associated dysplasias (30). Due to the correlation between dynein delocalization and spindle multipolarity (21) and our observation that the recruitment of γ-tubulin to centrosomes, a dynein-dependent process (9), is altered in HPV16 E7-expressing cells (31), we were interested in determining whether HPV16 E7 affects dynein dynamics. Here we show that the expression of HPV16 E7 in cells leads to an increased population of mitotic cells where dynein is delocalized from the mitotic spindle. This function of HPV16 E7 is independent of its ability to cause centrosome overduplication and deregulate γ-tubulin dynamics, however, and maps to the carboxyl-terminus of HPV16 E7. Importantly, using primary human foreskin fibroblasts and keratinocytes as well as immortalized mouse embryo fibroblasts, we did not observe a correlation between dynein delocalization and the incidence of multipolar mitoses upon HPV16 E7 expression. Nevertheless, because dynein is involved in many cellular processes, it is possible HPV16 E7-mediated dynein delocalization may contribute to the many other functions of HPV16 E7, including cell cycle deregulation and abrogation of the spindle assembly checkpoint.
Materials and Methods
Cells
NIH 3T3 mouse embryo fibroblasts, HPV-negative cervical cancer cells C33a, and HPV-positive cervical cancer cells CaSki, SiHa, and HeLa were obtained from the ATCC and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% calf serum, penicillin (50 U/ml), and streptomycin (50 μg/ml). HEK293 human embryonic kidney cells were also obtained from the ATCC and were maintained in DMEM supplemented with 10% fetal bovine serum, penicillin (50 units/ml), and streptomycin (50 μg/ml). Normal oral keratinocytes (NOKs) immortalized by human telomerase (hTERT) +/− HPV16 E7 were previously described (32) and were maintained in keratinocyte-SFM supplemented with human recombinant epidermal growth factor 1–53 (EGF 1–53), bovine pituitary extract (BPE) (GIBCO Invitrogen), penicillin (50 U/ml), streptomycin (50 μg/ml), gentamicin (10 μg/ml), and amphotericin B (0.75 μg/ml).
NIH 3T3 cells with stable expression of empty vector (3T3-poz), C-terminally Flag- and HA-tagged HPV16 E7 (3T3-CE7), or C-terminally Flag- and HA-tagged HPV16 E7Δ21-24 (3T3-Δ21-24) were previously described (31). NIH 3T3 cells with stable expression of C-terminally Flag- and HA-tagged HPV16 E7H2G, E7H2P, E7Δ6-10, E7C24G, E7E26G, E7E46A, E7CVQ68-70AAA (E7CVQ), E7Δ79-83, and E7C91S [all in the poz-C vector (33, 34)] were made via cotransfections with pBudCE4.1 (Invitrogen) at a 5:1 ratio after selection with 1 mg/ml zeocin (Invitrogen) as previously described (31).
Primary human foreskin keratinocytes (HFKs) were isolated from anonymous newborn circumcisions and cultured as described previously (35). HFK cells stably expressing HPV16 E7 were created by transfecting primary HFK populations with p1435, a human β-actin HPV16 E7 expression plasmid, or p1318, the parental plasmid (36) as a control, together with pcDNA3.1 (Invitrogen) using the Amaxa Human Keratinocyte Nucleofector Kit (Amaxa Biosystems) per the manufacturer’s instructions. 0.2 mg/ml G418 was then used to select for transfected cells.
Primary human foreskin fibroblasts (HFFs) were also isolated from anonymous newborn circumcisions. After the epidermis was separated from the dermis, the dermis was minced and incubated in a 2 mg/ml collagenase solution in DMEM and trypsin (4:1) for two to four hours at 37°C. HFFs were maintained in DMEM supplemented with 10% calf serum, penicillin (50 U/ml), and streptomycin (50 μg/ml). HFF cells stably expressing wild type or mutant HPV16 E7 were generated from primary HFF populations infected with the following pBABE-puromycin based retroviral constructs: pBABE-puromycin (pBABE), pBABE-puromycin HPV16 E7 (pBABE-E7), pBABE-puromycin HPV16 E7Δ21-24 (pBABE-E7Δ21-24), and HPV16 E7Δ79-83 (pBABE-E7Δ79-83). Recombinant retroviruses were produced as previously described (32). Infections of 50% confluent HFFs were performed with a mixture of 2 ml viral supernatant, 8 μg/ml Polybrene, and 2 ml DMEM for 6 hours. The cells were maintained in DMEM supplemented with 10% calf serum, penicillin (50 U/ml), and streptomycin (50 μg/ml) following selection with 1 μg/ml puromycin.
Immunofluorescence
Cells were plated onto coverslips and were extracted, fixed, and stained as previously described (31). Primary antibodies used in these studies were anti-dynein (MAB1618, Chemicon International), anti-α-tubulin (ab18251, Abcam), and anti-γ-tubulin (H-183, Santa Cruz Biotechnology). Secondary antibodies used in these studies were Alexa Fluor 488 and Rhodamine Red-X (Invitrogen) and nuclei were counterstained with Hoechst 33258 dye. Immunofluorescence images were taken with a Nikon TE-300 inverted microscope equipped with phase and fluorescence optics.
Images were collected digitally using a progressive-scan, interline-cooled, charge-coupled device camera (Hamamatsu Corp.), analyzed with Metamorph software (Molecular Devices), and processed for presentation using Photoshop (Adobe Systems).
Western Blotting
Whole cell lysates were produced using ML buffer [20 mM Tris pH8, 1 mM EDTA, 300 mM NaCl, 0.5% NP-40, and protease inhibitors (Complete EDTA-free tablets; Roche Diagnostics)] and cleared by centrifugation (4 C, 20 min, 16,000 × g). 100μg aliquots of whole cell lysates were run on SDS 12% polyacrylamide gels. After electrophoresis, proteins were electrotransferred to a PVDF membrane (Millipore). The membrane was probed with anti-actin (MAB1501, Chemicon), anti-E7 (ED19), anti-HA (A190-108A, Bethyl Laboratories, Inc.) antibodies. Secondary antibodies used were horseradish peroxidase-linked anti-mouse or anti-rabbit IgG (GE Healthcare). Proteins were visualized using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Inc.) or SuperSignal ® West Dura Extended Duration Substrate (Thermo Scientific) and exposed on Kodak BioMax XAR film or electronically acquired with a Kodak Image Station 4000R equipped with Kodak Imaging Software, version 4.0.
RESULTS
HPV16 E7 expression leads to the delocalization of dynein from mitotic spindles
It was previously shown that the microtubule motor protein dynein is delocalized from mitotic spindles in some cancer cell lines and this was correlated to an increased incidence of multipolar metaphases in cells containing supernumerary centrosomes due to the abrogation of centrosome coalescence (21). Furthermore, dynein is responsible for the recruitment of γ-tubulin to centrosomes (9). Therefore, because HPV16 E7-expressing cells have increased levels of multipolar metaphases (37) and the recruitment of γ-tubulin to centrosomes is altered in the presence of HPV16 E7 (31), we wanted to determine if dynein is delocalized from mitotic spindles in HPV16 E7 expressing cells. Immunofluorescent staining for dynein, as well as the mitotic spindle marker γ-tubulin, was performed on mitotic NIH 3T3 cells that stably express control vector, 3T3-poz, or C-terminally HA/FLAG tagged HPV16 E7, 3T3-CE7. In mitotic cells, dynein localizes to mitotic spindles and, therefore, stains microtubules that emanate from mitotic spindle poles (Fig. 1A). Abnormal dynein localization, however, appeared punctate and did not largely localize to mitotic spindles (Fig. 1A). Mitotic cells that showed questionable, intermediate staining results were not included in this analysis. The incidence of dynein delocalization in the mitotic 3T3-CE7 cells was approximately 2.5-fold higher than in the 3T3-poz cells (21.6% as compared to 8.6%; p< 0.02, Student’s T-test) (Fig. 1B). Furthermore, normal oral keratinocytes (NOKs) immortalized by hTERT or primary human foreskin fibroblasts (HFFs) or keratinocytes (HFKs), each with the stable expression of HPV16 E7 at levels similar to HPV16 positive cervical carcinoma cells or control vector (data not shown), were examined to determine if this phenotype was also observed in human non-cancer derived cells. While the parental cells each had different base levels of abnormal dynein localization, HPV16 E7 expression in each of these cells resulted in an approximately 2-fold increase in the fraction of mitotic cells with delocalized dynein (2.2 fold in NOK cells, 23.4% as compared to 10.5%, (p<0.02); 2.1 fold in HFF cells, 15.2% as compared to 7.2%, (p<0.01); and 1.7 fold in HFK cells, 21.4% as compared to 12.3%, (p<0.02)) (Fig. 1B).
Figure 1.
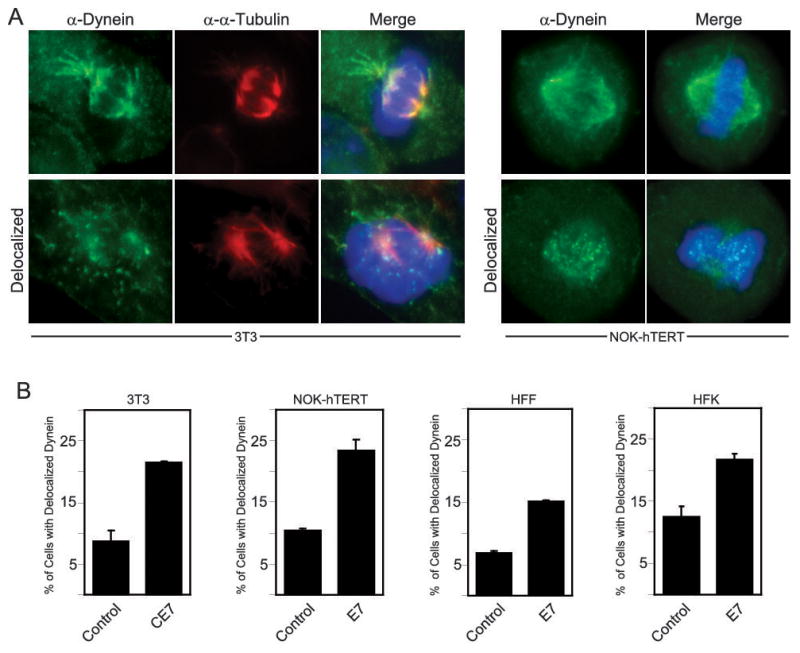
Dynein is delocalized from mitotic spindles in HPV16 E7-expressing cells. (A) Immunofluorescent staining of dynein (green) and α-tubulin (red). An example of normal dynein staining, on mitotic spindles, and abnormal dynein staining, punctate and away from spindles, is shown for both NIH 3T3 cells and NOK-hTERT cells. For each cell type, the example of normal dynein localization is an image of a control cell while the abnormal example is an image of an HPV16 E7-expressing cell. Normal and abnormal dynein localization was seen in both cell populations. (B) Bar graphs showing the percentage of mitotic cells with abnormal dynein localization in either NIH 3T3 cells stably expressing empty vector (control) or C-terminally HA/FLAG tagged HPV16 E7 (CE7) or NOK-hTERT cells, human foreskin fibroblasts (HFFs), or human foreskin keratinocytes (HFKs) stably expressing empty vector or HPV16 E7, as indicated. For the NIH 3T3 cells, the results represent averages from 2 independent experiments where >100 mitotic cells were counted per experiment. For the NOK-hTERT cells, the results represent averages from 3 independent experiments where 100–225 mitotic cells were counted per experiment. For HFF cells, the results represent averages from 2 independent experiments where >110 mitotic cells were counted per experiment. For HFK cells, the results represent averages from 3 independent experiments where >100 mitotic cells were counted per experiment. Error bars indicate the standard error between experiments.
Dynein delocalization occurs in HPV-positive cervical cancer cells
To determine if dynein delocalization was observed under conditions of biologically relevant HPV16 E7 expression, we compared the percentage of dynein delocalization in HPV16-positive cervical carcinoma cell lines CaSki and SiHa to HEK293 cells, which were previously shown to have predominantly normal dynein localization during mitosis (21), and to primary human foreskin keratinocytes (HFKs). We observed an increased incidence of delocalized dynein in the mitotic HPV-positive cervical cancer cells, 19.1% and 23.7% for CaSki and SiHa, respectively, as compared to the 8.5% and 6.1% seen in mitotic HEK293 and HFK cells, respectively (Fig. 2). Together, these results suggest that HPV16 E7-expression results in an increased incidence of dynein delocalized from the mitotic spindles and that a considerable fraction of mitotic HPV16-positive cancer cells contain delocalized dynein.
Figure 2.
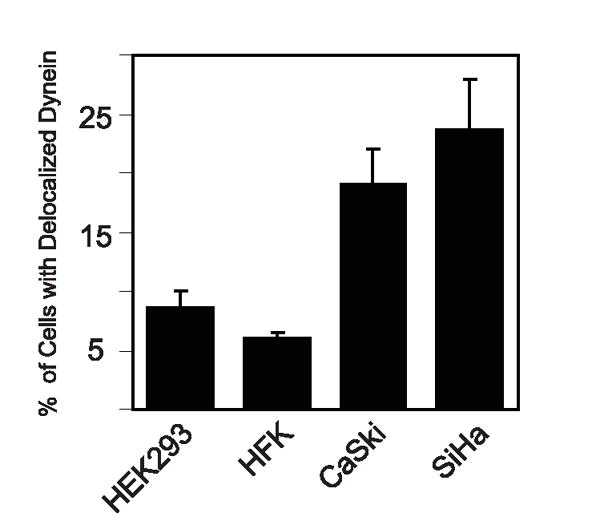
Dynein is delocalized from mitotic spindles in HPV-positive cervical carcinoma cells. Bar graph showing the percentage of mitotic cells with abnormal dynein localization in HPV-negative cells, HEK293 and primary human foreskin keratinocytes (HFKs), or HPV-positive cervical cancer cells, CaSki and SiHa. The results represent averages from 2 independent experiments where >100 mitotic cells were counted per experiment. Error bars indicate the standard error between experiments.
HPV16 E7-induced dynein delocalization is not dependent on HPV16 E7-mediated centrosome overduplication
The expression of HPV16 E7 causes centrosome overduplication in primary human cells (24, 38) and HPV16 E7 associates with and deregulates γ-tubulin (31); both of these activities rely upon amino acid residues 21–24 of HPV16 E7 (31, 39). To determine whether abnormal dynein localization as a result of HPV16 E7 expression was related to the ability of HPV16 E7 to induce aberrant centrosome duplication and/or to subvert γ-tubulin dynamics, we generated a panel of stable NIH 3T3 based cell lines to map the domain(s) of E7 necessary for dynein delocalization. The NIH 3T3 cell line was chosen because it is an immortalized non-cancer derived cell line with a low level of abnormal dynein localization during mitosis (Fig. 1B). Furthermore, as reported previously (31), HPV16 E7 expression in NIH 3T3 cells induces a 3.3-fold increase in cells with supernumerary centrosomes, whereas expression of the HPV16 E7Δ21-24 mutant does not. Therefore, NIH 3T3 cells with the stable expression of various previously characterized HPV16 E7 mutants, each containing C-terminal HA/FLAG tags, were established and analyzed for the incidence of abnormal dynein localization as well as for the incidence of supernumerary centrosomes. As was previously shown (31, 39), HPV16 E7Δ21-24 was unable to cause centrosome overduplication (Fig. 3A). Interestingly, however, expression of HPV16 E7Δ21-24 induced dynein delocalization with a similar efficiency as wild type E7 (2.2 fold versus 2.5 fold, respectively, over control cells). In contrast, the C-terminal mutants, E7CVQ68-70AAA (hereby called E7CVQ), E7Δ79-83, and E7C91S each displayed a statistically significant (1.6 fold, 2.0 fold, 1.8 fold, respectively; p<0.05, Student’s T-test) decrease in the ability to cause dynein delocalization as compared to wild type HPV16 E7. Interestingly, although the E7CVQ, E7Δ79-83, and E7C91S mutants also display a decreased ability to cause centrosome overduplication as compared to wild type HPV16 E7 (1.9 fold, p=0.02; 1.5 fold, p=0.04; and 1.8 fold, p=0.02, respectively), their capacity to induce supernumerary centrosomes was still significantly higher compared to the E7Δ 21-24 mutant (1.8 fold, p=0.03; 2.4 fold, p=0.0001; and 2.0 fold, p=0.02, respectively) (Fig. 3A). Hence, the ability of HPV16 E7 to disrupt dynein localization in mitotic cells maps to the C-terminus of HPV16 E7.
Figure 3.
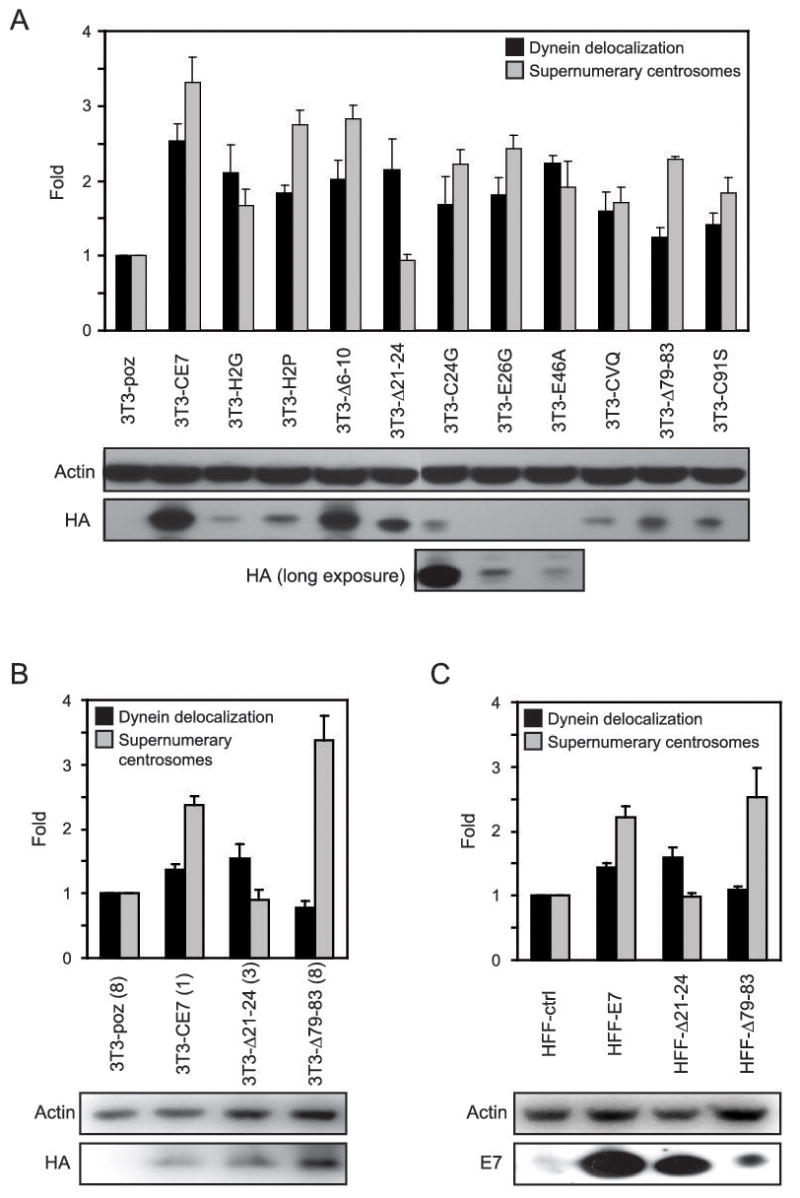
HPV16 E7-mediated dynein delocalization and centrosome overduplication are separate events. (A) Bar graph showing the fold increase in either dynein delocalization (black bars) or supernumerary centrosomes (grey bars) in NIH 3T3 cells with stable expression of the indicated HPV16 E7 plasmids as compared to the empty vector control. For dynein delocalization counts, the results represent averages from 2 independent experiments where >125 cells were counted per experiment. For supernumerary centrosome counts, the results represent averages from 3 independent experiments where >150 cells were counted per experiment. Error bars indicate the standard error between experiments. Western blots provided beneath the graph show the expression levels of the various cell lines, where HA antibody detects the HA/FLAG tagged E7 proteins; actin is shown as a loading control. (B and C) As described in (A). Cell lines analyzed are clonal populations of the 3T3-poz, 3T3-CE7, 3T3-Δ 21-24, and 3T3-Δ 79-83 cell lines or primary human foreskin fibroblasts (HFFs) stably expressing empty vector (ctrl), wild type E7, or the E7 Δ21-24 or E7 Δ79-83 mutants, as indicated. For dynein delocalization counts, the results represent averages from 2 independent experiments where >100 cells were counted per experiment. For supernumerary centrosome counts, the results represent averages from 2 independent experiments where >200 cells were counted per experiment. Error bars indicate the standard error between experiments. Western blots are provided to show expression levels of the various HPV16 E7 constructs; actin is shown as a loading control.
Although the expression levels of the wild type and mutant HPV16 E7-expressing cells differed somewhat, expression of HPV16 E7 in the various cell lines was detectable via Western blotting (Fig. 3A) and, more importantly, variations in expression levels did not appear to directly influence the phenotypes examined. Nevertheless, to confirm that the observed phenotypes are not a consequence of expression levels, we generated single cell clones from the 3T3-poz, 3T3-CE7, 3T3-Δ21-24, and 3T3-Δ79-83 cell lines and examined clones that expressed the E7 constructs at similarly low levels (3T3-CE7(1), 3T3-Δ21-24(3), and 3T3-Δ79-83(8)); the 3T3-poz clone, 3T3-poz(8), was chosen at random (Fig. 3B). The expression of HPV16 E7 in the 3T3-CE7(1) cell line was quantified and is 4 fold lower than that of the pooled 3T3-CE7 cell line (data not shown). Again, the same pattern of activity is observed in the cells expressing wild type HPV16 E7 or the E7Δ21-24 and E7Δ79-83 mutants. The expression of wild type HPV16 E7 in the 3T3-CE7(1) clonal cell line results in a 2.4 fold increase in cells with supernumerary centrosomes (p=0.01) and a 1.4 fold increase in mitotic cells with delocalized dynein (p=0.05) (Fig. 3B). We suspect that the more modest fold increase in dynein delocalization observed with the clonal cell line expressing HPV16 E7 as compared to the pooled population (Fig. 3A) is due to a slightly higher basal level of dynein delocalization detected in the 3T3-poz(8) cell line, which may be inherent to the specific clone; this supports the notion that pooled populations of cell lines are likely more reliable because variations among single cells are averaged out over the entire population. Regardless, in comparison to the control 3T3-poz(8) cells, the 3T3-E7Δ21-24(3) cell line displayed a 1.5 fold increase in the amount of mitotic cells with delocalized dynein (p=0.1) while an increase in cells with supernumerary centrosomes was not observed (0.9 fold, p=0.6) (Fig. 3B). Conversely, in comparison to the control 3T3-poz(8) cells, the 3T3-E7Δ79-83(8) cell line did not display an increase in the amount of mitotic cells with delocalized dynein (0.8 fold, p=0.2), yet contained a 3.4 fold increased amount of cells with supernumerary centrosomes (p=0.02) (Fig. 3B). The fact that the same mapping pattern was observed with pooled populations that contained varied expression of the HPV16 E7 constructs as compared to clonal populations expressing similar levels of HPV16 E7 provides further support for the notion that the observed phenotypes are not consequences of the levels of HPV16 E7 expression.
Additionally, in order to ensure that wild type HPV16 E7 as well as the E7Δ21-24 and E7Δ79-83 mutants displayed these observed activities in human cells, we examined primary human foreskin fibroblasts (HFFs) with the stable expression of control vector, wild type HPV16 E7, or the E7Δ21-24 or E7Δ79-83 mutants. Once again, the same pattern of activity is observed whereby the HFF cells expressing wild type HPV16 E7 display a 2.2 fold (p=0.02) increase in cells with supernumerary centrosomes and a 1.4 fold (p=0.02) increase in mitotic cells where dynein is delocalized from the mitotic spindle (Fig. 3C). In cells expressing the HPV16 E7Δ21-24 mutant, a 1.6 fold (p=0.05) increase in mitotic cells with delocalized dynein is observed, while there is no detectable increase in cells with supernumerary centrosomes (1 fold, p=0.9) (Fig. 3C). In cells expressing the HPV16 E7Δ79-83 mutant, a 2.5 fold (p=0.07) increase in cells with supernumerary centrosomes is observed, yet there is no appreciable increase in the amount of mitotic cells with delocalized dynein (1.1 fold, p=0.2) (Fig. 3C). These data confirm our finding that the ability of HPV16 E7 to disrupt dynein localization in mitotic cells maps to a separate region of HPV16 E7 as that responsible for the deregulation of centrosome duplication.
Dynein delocalization in HPV16 E7-expressing cells does not clearly correlate with induction of multipolar metaphases
As mentioned above, the delocalization of dynein from mitotic spindles in some cancer cells has been associated with an increased incidence of multipolar mitoses (21). To investigate whether HPV16 E7-mediated dynein delocalization contributes to the formation of multipolar mitoses in HPV16 E7-expressing cells, we utilized NIH3T3 cells and primary human foreskin fibroblasts and keratinocytes. HPV16 E7 has previously been shown to induce centrosome overduplication in NIH 3T3 cells and in primary cells (31, 38), as confirmed here (Fig. 3C); in the primary HFKs utilized in the following experiment, the stable expression of HPV16 E7 resulted in a 2.2 fold increase in cells with supernumerary centrosomes (p=0.0004). Furthermore, HPV16 E7 has been implicated in the induction of multipolar mitoses in U2OS osteosarcoma cells (24). To determine whether there was a correlation between multipolar mitoses and dynein delocalization, it was first necessary to investigate whether there was an increased incidence of multipolar metaphases in HPV16 E7-expressing NIH3T3 cells, HFFs, or HFKs. We examined at least 200 metaphases each for control and HPV16 E7 expressing cells. In the stable NIH 3T3 cells, we did not detect a noticeable difference between control 3T3-poz (4/200; 2%) and 3T3-CE7 cells (5/200; 2.5%) (Fig. 4A). With HFFs, we again did not observe a single case of multipolar mitosis in either control or HPV16 E7-expressing cells. In the HFK cells, the expression of HPV16 E7 resulted in a slight increase in the incidence of multipolar mitoses (4/225; 1.78%) as compared to the empty vector control (2/225; 0.89%), however this difference was not statistically significant (p=0.7, Fisher’s exact test). Moreover, at least some of the previously observed NOK cells as well as the primary HFK cells undergoing multipolar mitoses exhibited normal, mitotic spindle-associated dynein staining (Fig. 4B and data not shown), suggesting that dynein delocalization induced as a consequence of HPV16 E7 expression is not sufficient to generate multipolar mitoses and, conversely, that multipolar mitoses can arise in cells with normal dynein localization. Taken together, these results suggest that multipolar mitoses do not automatically ensue in cells with supernumerary centrosomes that exhibit increased dynein delocalization during mitosis.
Figure 4.
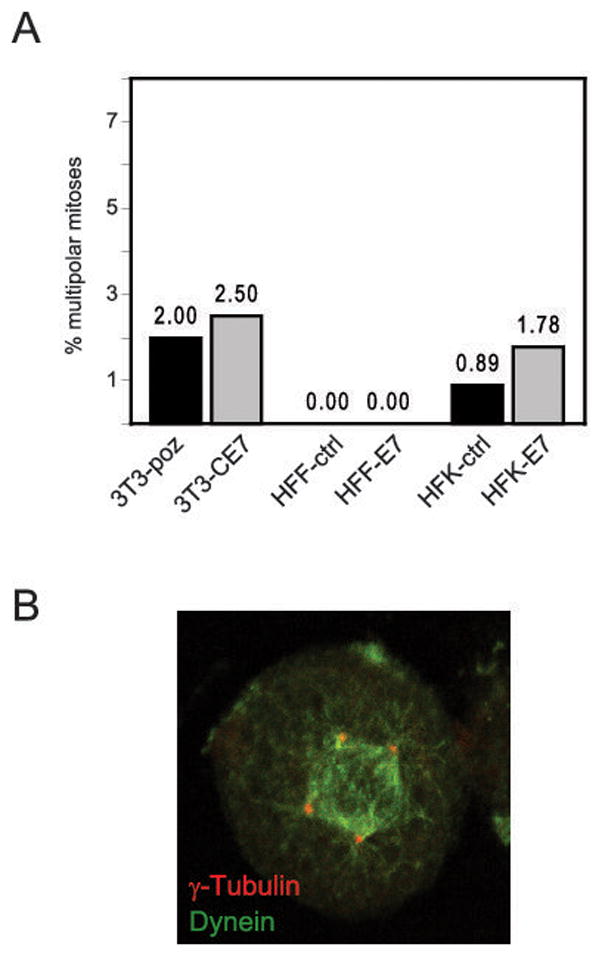
HPV16 E7-mediated dynein delocalization does not correlate with an increased incidence of multipolarity. (A) Bar graph depicting the percentage of multipolar mitoses in either control or HPV16 E7-expressing NIH 3T3, human foreskin fibroblast (HFF) or human foreskin keratinocyte (HFK) cells, as indicated. For 3T3 and HFF cells, 200 mitotic cells were analyzed each, and for HFK cells, 225 mitotic cells were analyzed. (B) Immunofluorescent staining of dynein (green) and γ-tubulin (red) in a multipolar mitotic NOK-hTERT+16E7 cell.
DISCUSSION
Recently, the deregulation of dynein localization was implicated in the formation of multipolar mitoses (21). Additionally, the recruitment of γ-tubulin onto centrosomes is mediated by dynein (9). In light of these observations and given that we have previously shown that the expression of HPV16 E7 leads to centrosome overduplication and mitotic abnormalities associated with multipolar metaphases (24, 37, 38), and that HPV16 E7 expression alters the recruitment of γ-tubulin to centrosomes (31), we investigated whether dynein was delocalized from mitotic spindles in HPV16 E7-expressing cells. After examining various cell types, including primary and immortalized cells, we conclude that the expression of HPV16 E7 leads to a higher frequency of mitotic cells with dynein delocalized from mitotic spindles as compared to the empty vector control. Furthermore, the capacity to delocalize dynein from mitotic spindles maps to the C-terminus of HPV16 E7, which is distinct from the amino terminal domain that has previously been identified as a major determinant for the induction of supernumerary centrosomes (24, 38) and subversion of γ-tubulin dynamics (31). Interestingly, however, HPV16 E7-induced dynein delocalization does not correlate with multipolar spindle formation, even in cell lines that display supernumerary centrosomes; this unexpected result will be discussed further below.
Dynein is a microtubule motor that is clearly important for numerous cellular processes, therefore making it difficult to determine what, if any, is the primary purpose behind the deregulation of dynein localization by HPV16 E7. One of the key roles of HPV16 E7 in the viral life cycle is the establishment of a DNA-replication competent cellular milieu, which is achieved via the deregulation of the cell cycle at various stages. HPV16 E7-mediated pRB degradation promotes S-phase entry and is also predicted to disrupt mitotic checkpoints through aberrant Mad2 expression (40). However, because dynein is implicated in the inactivation of the spindle assembly checkpoint at kinetochores (13–15), it would seem that dynein delocalization may lead to a prolonged mitotic checkpoint. Nonetheless, one could argue that allowing cells to aberrantly proceed into anaphase could be detrimental to the host cells and, thus, to the virus. Many viruses must maintain a sensitive balance within a cell whereby they utilize numerous cellular components but ensure that the cell has sufficient resources to survive for the duration of infection. Therefore, the delocalization of dynein by HPV16 E7 may help dampen the abrogation of the mitotic spindle assembly checkpoint by HPV16 E7 (41).
Because dynein is a minus-end directed motor, it is responsible for transporting many proteins and/or complexes to the centrosomes, where the minus-ends of microtubules are anchored. Centrosomes have previously been suggested to act as a scaffold for proteasomal machinery (42, 43) and the dynein complex transports aggregates of misfolded proteins to the centrosome to facilitate degradation (44). The centrosome-associated proteasome is not limited to the degradation of misfolded proteins (43), however, and it is therefore likely that other cargoes of the dynein motor complex are also degraded at the centrosome. Interestingly, it was shown that preventing the degradation of mps1 at the centrosome causes centrosome reduplication (45), suggesting that degradation of proteins specifically at the centrosome may be an important step in various tightly coordinated cellular processes. It is interesting to note that the carboxyl terminal mutants have a slightly decreased ability to cause centrosome overduplication as compared to wild type HPV16 E7, and it will be interesting to determine whether dynein delocalization also contributes at least partially to HPV16 E7-mediated centrosome overduplication. Furthermore, it remains possible that the delocalization of dynein interferes with the degradation of other proteins that are important to cell cycle regulation and, thus, contributes to HPV16 E7-mediated disruption of the cell cycle.
The delocalization of dynein was shown to correlate with the increased incidence of multipolar mitoses in certain cancer cell lines (21). It was purported that dynein was somehow involved in the process of centrosome coalescence (21), whereby supernumerary centrosomes are clustered together to ensure for the formation of a bipolar spindle during mitosis (46, 47). Therefore, we hypothesized that the delocalization of dynein in HPV16 E7-expressing cells may contribute to disruption of centrosomal coalescence and to the emergence of abnormal, multipolar mitoses in HPV16 E7-expressing cells. An increased incidence of multipolar metaphases was previously detected in U2OS osteosarcoma cells expressing HPV16 E7 and it was shown that this was an immediate effect as evidenced by the fact that multipolarity was increased in cells 48 hours after expression of HPV16 E7 (24). In both primary human cells and immortalized mouse cells expressing HPV16 E7, however, we did not notice a significantly increased frequency of multipolar mitotic cells (Fig 4A). The reason for the differences in our observations may be due to the fact that we analyzed cells that have not been in culture for extended periods of time (post-HPV16 E7-expression) and, therefore, have not acquired necessary downstream alterations that may play a role in the formation of multipolar spindles. Although it was shown that HPV16 E7 could increase the frequency of multipolar metaphases 48 hours post transfection, this experiment was performed in U2OS osteosarcoma cells that may already have a compromised cellular environment that is permissive to multipolar spindle formation. In fact, preliminary experiments suggested that U2OS cells have a higher population of mitotic cells with delocalized dynein than HEK293 cells (data not shown) and we therefore decided not to use these cells for studies regarding the effect of HPV16 E7-expression on dynein localization. Accordingly, our results agree with the observation that HPV16 E7 expression in primary human foreskin keratinocytes does not result in a dramatic increase of multipolar mitotic cells (24). Importantly, our data show that, in cells containing supernumerary centrosomes, the emergence of multipolar metaphases is not an obligatory result of dynein delocalization and that multipolarity can occur even in the presence of properly localized dynein. As such, this finding is consistent with results reported by Rebacz and colleagues who showed that griseofulvin, a compound isolated from Penicillium, inhibited centrosome coalescence without affecting the localization of dynein to mitotic spindles (48).
Overall, our results do not rule out the possibility that the increased delocalization of dynein from mitotic spindles by HPV16 E7 plays a role in HPV16 E7-induced spindle multipolarity, but suggest that it may need to work in concert with other cellular alterations that occur after extended cell divisions. Additionally, the fact that there appeared to be a synergistic effect on spindle multipolarity when both HPV16 E6 and E7 were expressed in primary human foreskin keratinocytes (24) suggests that the effects of HPV16 E7 on dynein delocalization may need to compile with the effects of HPV16 E6 expression in order to induce abnormal multipolar metaphases. For example, if HPV16 E7-mediated dynein delocalization indeed contributes to multipolar spindle formation, the expression of HPV16 E6 may be necessary to disrupt cellular checkpoints that would normally render a cell intolerant to forming and/or maintaining multiple mitotic spindle poles. Furthermore, HPV16 E6-mediated cytokinesis defects may cause a cell to have extra cellular factors or chromosomal DNA that would somehow alter cellular signals and, together with HPV16 E7-mediated dynein delocalization, would now allow for multipolar spindle formation. For instance, in a cell that contained 4N DNA content due to failed cytokinesis, presumably as a consequence of the loss of p53 tumor suppressor activity (49), extra unattached kinetochores may emit signals that promote the formation of superfluous spindle poles to accommodate the increased number of chromosomes. Ultimately, we have shown that, contrary to previous reports (21), the delocalization of dynein from mitotic spindles is neither necessary nor sufficient to cause the formation of multipolar mitoses. It will be interesting to determine if the mutants of HPV16 E7 that have a diminished ability to cause dynein delocalization are still able to enhance the frequency of multipolarity in HPV16 E6-expressing cells.
The potential lack of multipolar mitoses at early stages of persistent viral infection does not rule out the possibility that HPV16 E7-mediated centrosome overduplication contributes to genomic instability, however, because even coalesced supernumerary centrosomes may still lead to chromosome missegregation during mitosis. Indeed, in HPV-associated anal neoplasias, it was shown that there was a significant population of “pseudo-bipolar” mitoses where extra centrosomes were clustered together (50). Because pseudo-bipolar anaphases were seen more frequently than multipolar anaphases, it was proposed that pseudo-bipolar mitoses more likely contributed to chromosomal instability than their multipolar counterparts (50). Overall, it would appear that, while multipolar mitoses may be a good marker for premalignant neoplasias, it may not be a required step and thereby should not be considered a primary mechanistic determinant for the establishment of genomic instability in HPV-associated neoplasias. Furthermore, these findings suggest that the mechanism of centrosomal coalescence may not be adequate to ensure proper chromosome segregation in cells that contain supernumerary centrosomes.
Acknowledgments
PHS grants T32CA009031 (C.L.N), R01 CA066980, CA081135 (K.M.) and ACS PF-07-072-01-MBC (M.E.M-D.)
We are grateful to Hiroyuki Hayakawa for assistance with preliminary experiments and for helpful discussions.
References
- 1.Asai DJ, Koonce MP. The dynein heavy chain: structure, mechanics and evolution. Trends Cell Biol. 2001;11:196–202. doi: 10.1016/s0962-8924(01)01970-5. [DOI] [PubMed] [Google Scholar]
- 2.Barbar E. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry. 2008;47:503–8. doi: 10.1021/bi701995m. [DOI] [PubMed] [Google Scholar]
- 3.King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2:20–4. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- 4.Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–75. doi: 10.1111/j.1600-0854.2007.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barr FA, Egerer J. Golgi positioning: are we looking at the right MAP? J Cell Biol. 2005;168:993–8. doi: 10.1083/jcb.200501088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murshid A, Presley JF. ER-to-Golgi transport and cytoskeletal interactions in animal cells. Cell Mol Life Sci. 2004;61:133–45. doi: 10.1007/s00018-003-3352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirokawa N, Noda Y, Okada Y. Kinesin and dynein superfamily proteins in organelle transport and cell division. Curr Opin Cell Biol. 1998;10:60–73. doi: 10.1016/s0955-0674(98)80087-2. [DOI] [PubMed] [Google Scholar]
- 8.Susalka SJ, Pfister KK. Cytoplasmic dynein subunit heterogeneity: implications for axonal transport. J Neurocytol. 2000;29:819–29. doi: 10.1023/a:1010995408343. [DOI] [PubMed] [Google Scholar]
- 9.Young A, Dictenberg JB, Purohit A, Tuft R, Doxsey SJ. Cytoplasmic dynein-mediated assembly of pericentrin and gamma tubulin onto centrosomes. Mol Biol Cell. 2000;11:2047–56. doi: 10.1091/mbc.11.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp DJ, Rogers GC, Scholey JM. Roles of motor proteins in building microtubule-based structures: a basic principle of cellular design. Biochim Biophys Acta. 2000;1496:128–41. doi: 10.1016/s0167-4889(00)00014-8. [DOI] [PubMed] [Google Scholar]
- 11.Merdes A, Ramyar K, Vechio JD, Cleveland DW. A complex of NuMA and cytoplasmic dynein is essential for mitotic spindle assembly. Cell. 1996;87:447–58. doi: 10.1016/s0092-8674(00)81365-3. [DOI] [PubMed] [Google Scholar]
- 12.Merdes A, Heald R, Samejima K, Earnshaw WC, Cleveland DW. Formation of spindle poles by dynein/dynactin-dependent transport of NuMA. J Cell Biol. 2000;149:851–62. doi: 10.1083/jcb.149.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffis ER, Stuurman N, Vale RD. Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J Cell Biol. 2007;177:1005–15. doi: 10.1083/jcb.200702062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howell BJ, McEwen BF, Canman JC, et al. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol. 2001;155:1159–72. doi: 10.1083/jcb.200105093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojcik E, Basto R, Serr M, Scaerou F, Karess R, Hays T. Kinetochore dynein: its dynamics and role in the transport of the Rough deal checkpoint protein. Nat Cell Biol. 2001;3:1001–7. doi: 10.1038/ncb1101-1001. [DOI] [PubMed] [Google Scholar]
- 16.Gerdes JM, Katsanis N. Microtubule transport defects in neurological and ciliary disease. Cell Mol Life Sci. 2005;62:1556–70. doi: 10.1007/s00018-005-5007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rayala SK, den Hollander P, Balasenthil S, Yang Z, Broaddus RR, Kumar R. Functional regulation of oestrogen receptor pathway by the dynein light chain 1. EMBO Rep. 2005;6:538–44. doi: 10.1038/sj.embor.7400417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vadlamudi RK, Bagheri-Yarmand R, Yang Z, et al. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004;5:575–85. doi: 10.1016/j.ccr.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 19.den Hollander P, Kumar R. Dynein light chain 1 contributes to cell cycle progression by increasing cyclin-dependent kinase 2 activity in estrogen-stimulated cells. Cancer Res. 2006;66:5941–9. doi: 10.1158/0008-5472.CAN-05-3480. [DOI] [PubMed] [Google Scholar]
- 20.Aouacheria A, Navratil V, Wen W, et al. In silico whole-genome scanning of cancer-associated nonsynonymous SNPs and molecular characterization of a dynein light chain tumour variant. Oncogene. 2005;24:6133–42. doi: 10.1038/sj.onc.1208745. [DOI] [PubMed] [Google Scholar]
- 21.Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127–9. doi: 10.1126/science.1104905. [DOI] [PubMed] [Google Scholar]
- 22.Pihan GA, Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003;63:1398–404. [PubMed] [Google Scholar]
- 23.Gisselsson D, Palsson E, Yu C, Mertens F, Mandahl N. Mitotic instability associated with late genomic changes in bone and soft tissue tumours. Cancer Lett. 2004;206:69–76. doi: 10.1016/j.canlet.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 24.Duensing S, Lee LY, Duensing A, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97:10002–07. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkley BR, Goepfert TM. Supernumerary centrosomes and cancer: Boveri’s hypothesis resurrected. Cell Motil Cytoskeleton. 1998;41:281–8. doi: 10.1002/(SICI)1097-0169(1998)41:4<281::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 26.Saunders W. Centrosomal amplification and spindle multipolarity in cancer cells. Semin Cancer Biol. 2005;15:25–32. doi: 10.1016/j.semcancer.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen CL, Hayakawa H, Munger K. Activities of human papillomavirus oncoproteins that contribute to genomic instability and carcinogenic progression. In: Norrild B, editor. Human Papillomavirus Gene Regulation and Transformation. Kerala, India: Transworld Research Network; 2007. pp. 139–64. [Google Scholar]
- 28.Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. The forkhead-associated domain protein Cep170 interacts with Polo-like kinase 1 and serves as a marker for mature centrioles. Mol Biol Cell. 2005;16:1095–107. doi: 10.1091/mbc.E04-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spardy N, Duensing A, Charles D, et al. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J Virol. 2007;81:13265–70. doi: 10.1128/JVI.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winkler B, Crum CP, Fujii T, et al. Koilocytotic lesions of the cervix. The relationship of mitotic abnormalities to the presence of papillomavirus antigens and nuclear DNA content. Cancer. 1984;53:1081–7. doi: 10.1002/1097-0142(19840301)53:5<1081::aid-cncr2820530511>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen CL, Eichwald C, Nibert ML, Munger K. Human papillomavirus type 16 E7 oncoprotein associates with the centrosomal component gamma-tubulin. J Virol. 2007;81:13533–43. doi: 10.1128/JVI.01669-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piboonniyom S, Duensing S, Swilling NW, Hinds PW, Münger K. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res. 2003;63:476–83. [PubMed] [Google Scholar]
- 33.Huh K, Zhou X, Hayakawa H, et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81:9737–47. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huh KW, DeMasi J, Ogawa H, Nakatani Y, Howley PM, Munger K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc Natl Acad Sci U S A. 2005;102:11492–7. doi: 10.1073/pnas.0505337102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones DL, Alani RM, Münger K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes & Development. 1997;11:2101–11. doi: 10.1101/gad.11.16.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417–21. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duensing S, Münger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Canc Res. 2002;62:7075–82. [PubMed] [Google Scholar]
- 38.Duensing S, Duensing A, Crum CP, Munger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61:2356–60. [PubMed] [Google Scholar]
- 39.Duensing S, Munger K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol. 2003;77:12331–5. doi: 10.1128/JVI.77.22.12331-12335.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernando E, Nahle Z, Juan G, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- 41.Thomas JT, Laimins LA. Human papillomavirus oncoproteins E6 and E7 independently abrogate the mitotic spindle checkpoint. J Virol. 1998;72:1131–37. doi: 10.1128/jvi.72.2.1131-1137.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wigley WC, Fabunmi RP, Lee MG, et al. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145:481–90. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. Activity and regulation of the centrosome-associated proteasome. J Biol Chem. 2000;275:409–13. doi: 10.1074/jbc.275.1.409. [DOI] [PubMed] [Google Scholar]
- 44.Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- 45.Kasbek C, Yang CH, Yusof AM, Chapman HM, Winey M, Fisk HA. Preventing the degradation of mps1 at centrosomes is sufficient to cause centrosome reduplication in human cells. Mol Biol Cell. 2007;18:4457–69. doi: 10.1091/mbc.E07-03-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ring D, Hubble R, Kirschner M. Mitosis in a cell with multiple centrioles. J Cell Biol. 1982;94:549–56. doi: 10.1083/jcb.94.3.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- 48.Rebacz B, Larsen TO, Clausen MH, et al. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res. 2007;67:6342–50. doi: 10.1158/0008-5472.CAN-07-0663. [DOI] [PubMed] [Google Scholar]
- 49.Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 rrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 50.Duensing A, Chin A, Wang L, Kuan SF, Duensing S. Analysis of centrosome overduplication in correlation to cell division errors in high-risk human papillomavirus (HPV)-associated anal neoplasms. Virology. 2008;372:157–64. doi: 10.1016/j.virol.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]