

Abstract
NAD(P)H:quinone oxidoreductase1-null (NQO1-/-) mice exposed to 3 grays of γ-radiation demonstrated an increase in neutrophils, bone marrow hypercellularity, and enlarged lymph nodes and spleen. The spleen showed disrupted follicular structure, loss of red pulp, and granulocyte and megakarocyte invasion. Blood and histological analysis did not show any sign of infection in mice. These results suggested that exposure of NQO1-/- mice to γ-radiation led to myeloproliferative disease. Radiation-induced myeloproliferative disease was observed in 74% of NQO1-/- mice as compared to none in wild type mice. NQO1-/- mice exposed to γ-radiation also demonstrated tissues lymphoma (32%) and lung adenocarcinoma (84%). In contrast, only 11% wild type mice showed lymphoma and none showed lung adenocarcinoma. Exposure of NQO1-/- mice to γ-radiation resulted in reduced apoptosis in granulocytes and lack of induction of p53, p21, and Bax. NQO1-/- mice also demonstrated increased expression of myeloid differentiation factors C/EBPα and Pu.1. Intriguingly, exposure of NQO1-/- mice to γ-radiation failed to induce C/EBPα and Pu.1, as was observed in wild type mice. These results suggest that decreased p53/apoptosis and increased Pu.1 and C/EBPα led to myeloid hyperplasia in NQO1-/- mice. The lack of induction of apoptosis and differentiation contributed to radiation-induced myeloproliferative disease in NQO1-/- mice.
Keywords: NAD(P)H:quinone oxidoreductase1 (NQO1), NQO1 knockout mice, γ-radiation, apoptosis and differentiation, myeloproliferative diseases
Introduction
NAD(P)H:quinone oxidoreductase 1 (NQO1) is a cytosolic protein that catalyzes metabolism of quinones and their derivatives (1, 2). It has been shown that the two-electron reduction of quinones, catalyzed by NQO1, competes with the one-electron reduction catalyzed by cytochromes P450 and P450 reductase. This produces a comparatively stable hydroquinone that is removed by conjugation with glutathione, UDP-glucuronic acid etc (1-3). The two-electron reduction of quinones does not result in the formation of free radicals (semiquinones) and highly reactive oxygen species, hence protecting cells against the adverse effects of quinones and their derivatives (1-2). NQO1 activity is ubiquitously present in all tissues types (1, 2). NQO1 gene expression is induced in response to xenobiotics, antioxidants, oxidants, heavy metals, UV light, and ionizing radiation (1, 2, 4). Interestingly, NQO1 is part of an electrophilic and/or oxidative stress induced cellular defense mechanism that includes the induction of more than two dozen genes (1, 2, 4). The coordinated induction of defensive genes, including NQO1, provides necessary protection for cells against free radical damage, oxidative stress, and neoplasia.
Human NQO1 gene has been localized to chromosome 16q22 (5). A cytosine to thymidine (C-->T) polymorphism in exon 6 of human NQO1 gene produces a proline to serine (P187S) substitution that destabilizes and inactivates the enzyme (6, 7). The polymorphic NQO1 is rapidly degraded via ubiquitination and proteasome degradation (7). Individuals carrying both polymorphic genomic alleles are completely lacking in NQO1 activity, whereas individuals who are heterozygous with one polymorphic allele have low-to-intermediate NQO1 activity compared with wild type individuals (8). Approximately 2-4 % of human individuals are homozygous and 20-25% is heterozygous for this polymorphism (7-12). The frequency of NQO1 P187S is similar in Whites and African Americans, but higher in Hispanics and Asians (9-12). NQO1 P187S has been associated with greater risk of neutropenia in benzene-exposed adult Chinese workers (13) and is significantly overexpressed in radiation/chemotherapy-related (14) and de novo leukemias (15) in adults. Recently, Wiemels et al. (16) reported that NQO1 P187S conferred susceptibility to infant acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) with mixed lineage leukemia (MLL) translocations in a British population. More recently, Smith et al. reported similar findings in United States populations (17).
NQO1-/- mice were generated (18). Mice deficient in NQO1 gene expression were born and developed normal, indicating that NQO1 does not play a role in mouse development. Further studies on NQO1-/- mice have revealed altered intracellular redox status and altered metabolism of carbohydrates, fatty acids, and nucleotides, and reduced accumulation of abdominal fat with age (19). In addition, studies demonstrated that loss of NQO1 gene expression in NQO1-/- mice led to myelogenous hyperplasia of bone marrow and increased sensitivity of NQO1-/- mice to menadione-induced hepatic damage (20). NQO1-/- mice also demonstrated benzene toxicity (21) and significantly increased sensitivity to skin carcinogenesis in response to benzo(a)pyrene (22) and dimethylbenzanthracene (23). NQO1-/- mice showed lower levels of tumor suppressor protein p53 and decreased apoptosis in bone marrow and skin (20, 24, 25).
The high frequency of P187S alleles present in spontaneous and radiation/chemotherapeutic drug-induced leukemia, combined with myeloid hyperplasia in NQO1-/- mice, raised interesting questions regarding the role of NQO1 in protection against de novo and radiation/chemotherapy-related leukemia. We used NQO1-/- mice to investigate in vivo role of NQO1 in radiation-induced leukemia. A majority of NQO1-/- mice upon exposure to γ-radiation developed myeloproliferative disease. This was evident from increased neutrophils, bone marrow hypercellularity, enlarged lymph nodes and spleen, disrupted follicular structure, and loss of red pulp in spleen, and granulocyte and megakarocyte invasion of spleen. NQO1-null mice exposed to γ-radiation also demonstrated tissues lymphoma and lung adenocarcinoma. In contrast, only a few wild type mice showed lymphoma and none showed lung adenocarcinoma. Further investigation revealed that exposure of NQO1-/- mice to γ-radiation resulted in reduced apoptosis in granulocytes and lack of induction of p53, p21, and Bax. In addition, the NQO1-/- mice demonstrated increased expression of myeloid differentiation factors Pu.1 and C/EBPα as compared to wild type mice. Interestingly, exposure of NQO1-/- mice to γ-radiation failed to induce C/EBPα and Pu.1, which was observed in wild type mice. These results suggest that increased C/EBPα and Pu.1 led to myeloid hyperplasia, and lack of induction of apoptosis and differentiation factors contributed to radiation-induced myeloproliferative disease in NQO1-/- mice.
Material and Methods
Flow analysis of bone marrow and blood from wild type and NQO1-/- mice
Six to nine week old wild type and age-matched NQO1-/- mice were anesthetized using Ketamine (80 mg/kg)/Xylazine (16 mg/kg) mix. 0.5 ml blood was collected by cardiac stick in EDTA-coated tubes to avoid clotting. Mice were sacrificed by decapitation and femurs were surgically removed and cut at the ends. The bone marrow was flushed gently with cold sterile PBS. After two PBS washes, cells were resuspended in the Annexin binding buffer to a concentration of 1×106 cells/ml. Cells were then incubated with Annexin V-FITC (apoptotic marker), and myeloid lineage specific differentiation marker Gr-1 antibody. Cells were fixed in 4% paraformaldehyde. Myeloid cells and apoptosis were measured using Coulter EPICS XL-MCL Flow Cytometer (Beckman-Coulter Co, Miami, FL). In related experiment, 100 μl of the blood was added to 1 μl Annexin V-FITC, and 2.5 μl Gr-1 antibody (0.2 mg/ml); gently vortexed and incubated on ice in the dark for 30 min. Red Blood Corpuscles were hemolyzed and fixed using Coulter Q-prep, and analyzed using Coulter EPICS XL-MCL Flow Cytometer.
Flow analysis of bone marrow and blood from wild type and NQO1-/- mice after γ-irradiation. Flow cytometry analysis of blood and bone marrow after γ-irradiation
Six to nine week old wild type and NQO1-/- mice were irradiated with 3 grays of γ-radiation (Gammacell 1000: Cesium-137, Nordion International, Ontario, Canada). Forty eight hours later mice were anesthetized and blood was collected by cardiac stick in EDTA-coated tubes to avoid clotting. Mice after collection of blood were sacrificed by decapitation. Femur bones were removed and bone marrow cells were flushed out with cold PBS. After two PBS washes, the cells were resuspended in the Annexin binding buffer to a concentration of 1×106 cells/ml.
100 μl of the blood was added to a 5 ml glass tube containing 1 ml Annexin V-FITC, and 2.5 μl Gr-1 antibody (0.2 mg/ml), gently vortexed and incubated on ice in the dark for 30 min. Red blood corpuscles were hemolyzed and white blood cells were fixed using Coulter Q-prep, and analyzed using Coulter EPICS XL-MCL Flow Cytometer. In related experiments, the bone marrow cells were incubated with Annexin V-FITC, and PE-labeled anti-Gr-1 antibody by procedure as described above for blood cells. Assays for the determination of myeloid and apoptotic cells were essentially performed using Coulter EPICS XL-MCL Flow Cytometer.
PI staining of bone marrow cells after γ-irradiation
Six to nine week old wild type and NQO1-/- mice were anaesthetized using Ketamine (80 mg/kg)/Xylazine (16 mg/kg) mix. Mice were then irradiated with sublethal dose of 3 grays of γ-radiation. Forty-eight hours later, mice were sacrificed. Bone marrow cells were flushed out with cold PBS and resuspended in 2 ml of cold saline. Cells were fixed by adding 5 ml of 90% cold ethanol drop wise and left for 30 minutes at room temperature. Each sample was stained with 1 ml propidium iodide 50 μg/ml (Sigma Chemical Company, St. Louis, MO). 100 μl of 1 mg/ml RNase was added to each sample and incubated for 30 minutes at 37°C. Samples were analyzed using Coulter EPICS XL-MCL Flow Cytometer (Beckman-Coulter Co, Miami, FL).
PI staining of bone marrow cells after ex vivo γ-irradiation
Six to nine week old wild type and NQO1-/- mice were sacrificed and their femurs obtained. The bones were cut and marrow flushed out gently with RPMI 10% FBS with antibiotics. Cells were either unirradiated or irradiated with 3 grays of γ-radiation, and cultured in 24 well plates. Forty-eight hours later, cells were collected, washed with cold PBS, fixed in alcohol, and stained with propidium iodide as described above.
PI staining of isolated myeloid cells after γ-irradiation
Wild type and NQO1-/- mice were sacrificed. Bone marrow cells were flushed out with RPMI medium with 10% FBS and antibiotics. Bone marrow cells were stained with Gr-1 antibody. Cells were sorted using Coulter Epics ALTRA flow cytometer (Beckman-Coulter Co, Miami, FL). Isolated myeloid cells were irradiated with 3 grays of γ-radiation; and then cultured in 6 well plates for 48 hours. Cells were washed in cold PBS, fixed in alcohol and stained with propidium iodide. Both irradiated and non-irradiated samples were then analyzed using Coulter EPICS XL-MCL Flow Cytometer.
Western analysis of the bone marrow
Six to nine week old wild type and NQO1-/- mice were irradiated with 3 grays of γ-radiation. Twelve hours later, mice were sacrificed. Femurs were obtained. The bones were cut on both ends. Marrow was flushed with cold buffer containing 50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycolate, 0.1% SDS, 0.5% Triton X-100, and protease inhibitor cocktail (Roche, Basel, Switzerland). One hundred micrograms of each bone marrow lysate was loaded and separated on 12 % polyacrylamide gels, blotted on ECL membrane, and probed with antibodies against p21, Bax (BD Pharmingen, San Diego, CA), p53 (CM5 antibodies, Novacastra, UK), NQO1 (generated in our lab), C/EBPα, Pu.1 (Santa Cruz, Santa Cruz, CA) and actin (Sigma Chemical Company, St. Louis, MO).
Flow cytometry/histology/cytogenetic analysis of mice one year after exposure to γ-irradiation
Seven week old wild type and NQO1-/- mice were anaesthetized using ketamine (80 mg/kg)/ Xylazine (16 mg/kg) mix and exposed to 0, 1, or 3 grays of γ-radiation. Each group contained 20 mice. Mice were then fed autoclaved food and water to avoid infectious complications. One year after exposure mice were analyzed for signs of myeloproliferation. Mice were euthanized and blood samples were collected by cardiac stick for complete blood counts (CBCs) analysis, Wright stained blood smear preparation, and flow cytometry analysis using PE-labeled anti-Gr-1 antibody. Both femurs were obtained from each mouse. One femur was decalcified for histology analysis and one was cut on both ends, bone marrow cells were flushed in ice cold PBS for flow cytometry analysis (as mentioned above). Spleens were split into halves, one half for histology and the other for flow cytometry analysis.
The bones were placed in 10% neutral buffered formalin (Fischer Scientific, Houston, TX) for 24 hours. After this time period, the bones were decalcified in TBD-2 Decalcifier (23% Formic Acid + 9% Sodium Citrate) (Shandon Lipshaw, Pittsburgh, PA) for 24 hours. The tissues were then embedded in paraffin and cut into 4 μm sections. The sections were placed onto slides and stained with hematoxylin and eosin (H&E) (Richard-Allan Scientific, Kalamazoo, MI). In addition to bone marrow, the lung, liver, kidneys, thymus, and lymph nodes were also obtained for histological analysis. All tissue samples were fixed in 10% neutral-buffered formalin solution. Tissue samples were embedded in paraffin. Sections were cut and mounted on glass slides, stained with H&E, and analyzed under light microscopy.
A few of the wild type and NQO1-/- male and female mice were injected intraperitoneally with 0.2 ml of Colcemid (2.0 mg/ml stock solution) one hour prior to killing. Bone marrow was aspirated in a hypotonic solution (0.075M KCl) with the help of a syringe fitted with 25 gauge needle. Cell clumps were broken into single cell suspension by mild vortexing. Cells were suspended in KCl solution for 15-20 minutes at room temperature, fixed in acetic acid:methanol (1:3 by volume) and finally dropped onto glass slides for air-dried preparations. G-banding was performed following standard laboratory procedures (26). Banded chromosomes were classified following the standard nomenclature of the Committee on Standardized Genetic Nomenclature for Mice (27). An average of 15-20 G-banded metaphases were photographed and complete karyotypes prepared from each animal using a Genetiscan (Perceptive System, Inc., Houston, TX). Additional 10 to 15 conventionally Giemsa-stained metaphase spreads from each animal were evaluated for any chromatid or chromosome-type aberrations and for the determination of model chromosome number.
Results
Myeloid hyperplasia in NQO1-/- mice
Flow cytometry analysis of bone marrow cells showed a significant increase in myeloid cell marker Gr-1-positive cells in NQO1-/- mice compared to wild type mice (Fig. 1A, left panel). Analysis of fifteen mice in each group revealed that Gr-1 positive myeloid cells increased from 18% in wild type to 30% in NQO1-/- mice (Fig. 1A, middle panel). The analysis also showed increase in Gr-1 positive cells in peripheral blood (Fig. 1B, left and middle panels). We combined myeloid cell marker Gr-1 with apoptotic marker annexin V to determine if the loss of NQO1 had an effect on apoptosis of myeloid cells. Indeed, less apoptosis of myeloid cells was observed in NQO1-/- mice bone marrow, as compared to wild type mice (Fig. 1A, right panel). Further analysis also showed lower apoptosis in peripheral blood granulocytes (Fig. 1B, right panel). These results indicate that the loss of NQO1 in mice caused myeloid hyperplasia of the bone marrow and significant increase in blood granulocytes. The decrease in apoptosis in myeloid cells and granulocytes might have contributed to the myeloid hyperplasia in NQO1-/- mice.
Figure 1. Flow cytometry analysis of wild type and NQO1-/- mice bone marrow and blood.
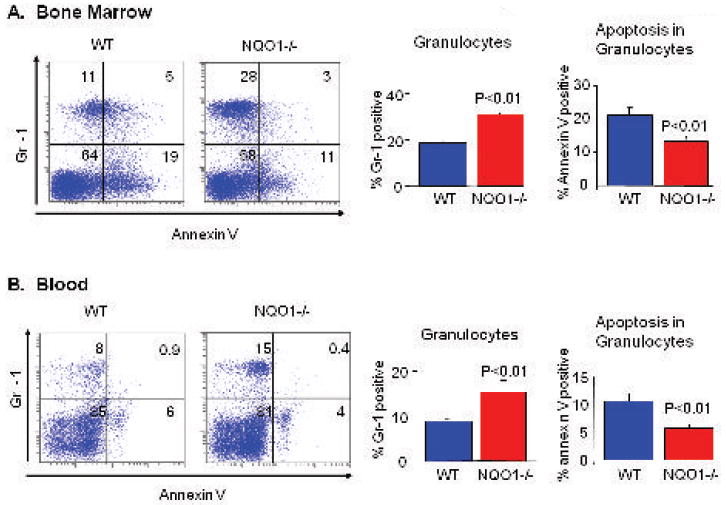
Six to nine week old wild type and NQO1-/- mice were euthanized and blood collected by cardiac stick. Red Blood Corpuscles were hemolyzed and white blood cells were fixed using Coulter Q-prep, and analyzed using Coulter EPICS XL-MCL Flow Cytometer. The mice were sacrificed and femurs surgically removed. Bone marrow cells were flushed with sterile cold PBS. Cells were stained with PE-labeled anti-Gr-1 antibody (myeloid cell marker) and Annexin V-FITC (apoptosis). Assay for the determination of myeloid cells and apoptosis in myeloid cells were essentially performed as described by the manufacturer and measured using Coulter EPICS XL-MCL Flow Cytometer. A). Bone Marrow and B). Blood. Left Panels: Representative samples. Middle Panels: Percentage of myeloid cells in bone marrow and granulocytes in peripheral blood, average of 15 mice. Roght Panels. Percentage of apoptosis in myeloid cells, average of 15 mice.
Myeloid cells resistance to γ-radiation induced apoptosis in NQO1-/- mice
We exposed wild type and NQO1-/- mice to a sublethal dose of 3 grays γ-radiation to determine the short-term effect of radiation on myeloid cell survival and apoptosis. Forty eight hours later, we analyzed the peripheral blood and bone marrow for γ-radiation-induced apoptosis. PE-labeled anti-Gr-1 (myeloid cell marker) and FITC-labeled annexin V (apoptosis marker) analysis revealed that exposure to γ-radiation increased apoptosis in peripheral blood granulocytes and bone marrow myeloid cells in both wild type and NQO1-/- mice (Fig. 2A and B). However, the magnitude of increase in apoptosis of blood granulocytes and bone marrow myeloid cells were significantly lower in NQO1-/- mice, as compared to wild type mice. γ-irradiation more than doubled the percentage of apoptosis in myeloid cells from 20% to 43% in wild type bone marrow, but only increase apoptosis in NQO1-/- myeloid cells from 14% to 18% (Fig. 2B). In similar experiment, the propidium iodide (PI) staining also showed significantly lower apoptosis in NQO1-/- bone marrow cells as compared to wild type mice (Fig. 2C, left panel). Apoptotic cells with condensed chromatin appear in the sub-G region after PI staining. In an active tissue like the bone marrow, apoptotic cells with condensed chromatin will be rapidly phagocytosed. Therefore, we replaced the in vivo irradiation experiment with an ex vivo irradiation. Bone marrow cells were collected and irradiated with 3 grays of γ-radiation, cultured for 48 hours, and then stained with propidium iodide. Ex vivo experiment showed similar results as observed in the in vivo experiment (Fig. 2C, right panel). Lower apoptosis was observed in NQO1-/- mice total bone marrow cells, as compared to wild type mice bone marrow. To further confirm the relative resistance of NQO1-/- myeloid cells to γ-radiation-induced apoptosis, we isolated myeloid cells from wild type and NQO1-/- mice using FACS (Fluorescence Activated Cell Sorting) after labeling with PE-labeled anti-Gr-1 antibodies. Isolated myeloid cells were irradiated with 3 grays of γ-radiation, cultured for 48 hours, and then analyzed by propidium iodide staining. Again NQO1-/- myeloid cells showed high resistance to γ-radiation-induced apoptosis as compared to wild type myeloid cells (Fig. 2D). These results revealed that bone marrow myeloid cells deficient in NQO1 are relatively resistant to γ-radiation-induced apoptosis.
Figure 2. In vivo and ex vivo analysis of peripheral blood and bone marrow cells after γ-irradiation.
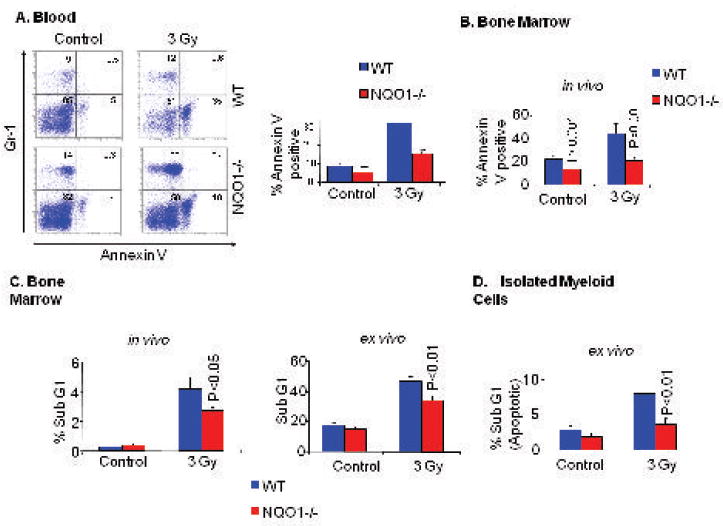
A/B). In vivo analysis of wild type and NQO1-/- mice peripheral blood and bone marrow. Mice were irradiated with 3 grays of γ-radiation. Forty-eight hours later, mice were euthanized and blood collected by cardiac stick. Mice were sacrificed and femur bones removed. Blood and bone marrow were analyzed by incubation with PE-labeled anti-Gr-1 antibody (myeloid cell marker) and Annexin V-FITC (apoptosis) and Flow cytometry. A). Left Panel: Blood representative samples are shown. A). Right Panel: A bar diagram for blood analysis from 15 mice in each group is shown. B). The bar diagram for bone marrow analysis from 15 mice is shown. C). In vivo and ex vivo analysis of wild type and NQO1-/- mice bone marrow by propidium iodide (PI) staining. Mice were irradiated (in vivo) or mice were sacrificed and their bone marrow collected and then irradiated with 3 grays of γ-radiation (in vitro). The cells were cultured in 24 well plates for forty-eight hours. PI staining: Cells were fixed by adding 5 ml of 90% cold ethanol. Each sample was stained with 1 ml propidium iodide 50 g/ml. 100 μl of 1 mg/ml Rnase were added and incubated for 30 minutes at 37°C. Samples were analyzed using Coulter EPICS XL-MCL Flow Cytometer. Apoptotic cells appear in the sub-G1 region. Percentage of cells in the sub-G1 region for ten wild type and ten NQO1-/- mice were analyzed with standard t-test for statistical significance. D). Bone marrow cells were stained for Gr-1 in RPMI medium. Myeloid cells were sorted with FACS, irradiated, cultured for 48 hours in RPMI/10%FBS medium. Myeloid cells were then washed twice and fixed with 90% cold ethanol. Each sample was stained with propidium iodide 50 μg/ml. Samples were then analyzed using Coulter EPICS XL-MCL Flow Cytometer. Apoptotic cells appear in the sub-G1 region. Average percentages of cells in sub-G1 region of five mice are presented.
Myeloproliferative diseases in NQO1-/- mice after exposure to γ-radiation
Resistance to γ-radiation-induced apoptosis may help NQO1-/- myeloid cells to escape death after DNA damage by γ-irradiation. Thus, they may acquire mutations that may result in transformation. This could be the reason behind the observed increased incidence of radiation or chemotherapy-related myeloid leukemia in patients with the NQO1 polymorphism. To test whether NQO1-/- mice will recapitulate the radiation-related leukemia in patients homozygous for the NQO1 polymorphism, wild type and NQO1-/- mice were exposed to 0, 1 and 3 grays of γ-radiation. Mice were housed in a clean facility and fed autoclaved food and water to avoid infection complications in case of radiation induced immune deficiency. Mice were observed twice a week for general health and signs of disease. One year after exposure, mice were analyzed for signs of leukemia and myeloproliferation. Blood analysis and total necropsy were performed for all the mice. Blood samples were analyzed with CBCs and flow cytometry for granulocytes. Wright stained blood smears were prepared for morphological examination of white blood cells. Bone marrow and spleen were analyzed with Flow cytometry analysis for myeloid cells. Spleen, femur, stratum, lymph nodes, thymus, liver, lung, kidney, urinary bladder and skin samples were collected for each mouse for histological analysis. Histological analysis did not reveal any sign of infection in control or γ-radiation-exposed mice. Bone marrow from a few mice in each group was also analyzed for chromosomal aberrations. Mice were diagnosed according to standardized guidelines (28). The results of blood and gross anatomy analysis are summarized in Table 1 and representative results shown in Fig. 3A and 3B. Histology of spleen and bone marrow are shown in Fig. 3C and 3D, respectively. Results on chromosomal aberrations are summarized in Table 2.
Table 1.
Frequency of myeloproliferation, lymphoma and other malignancies
| Myeloproliferation | Lymphoma | Other malignancies* | Total | Myeloid hyperplasia | |
|---|---|---|---|---|---|
| WT Control | 0/20(0) | 0/20(0) | 0/20(0) | 0/20(0) | 0/20(0) |
| NQO1-/- Control | 0/19(0) | 0/19(0) | 3/19(16) | 3/19(16) | 15/19(79) |
| WT 1gy | 0/20(0) | 0/20(0) | 0/20(0) | 0/20(0) | 0/20(0) |
| NQO1-/- 1gy | 4/18(22) | 3/18(17) | 9/18(50) | 13/18(72) | 16/18(89) |
| WT 3gy | 0/19(0) | 2/19(11) | 0/19(0) | 2/19(11) | 4/19(21) |
| NQO1-/- 3gy | 14/19(74) | 6/19(32) | 16/19(84) | 18/19(95) | 19/19(100) |
Most other malignancies were lung adenocarcinoma.
Numbers in parenthesis show the percentage of mice with myeloproliferation, lymphoma, other malignancies, total malignancies and myeloidhyperplasia.
Figure 3. Blood, lymph node, spleen, and bone marrow analysis of wild type and NQO1-/- mice one year after γ-irradiation.
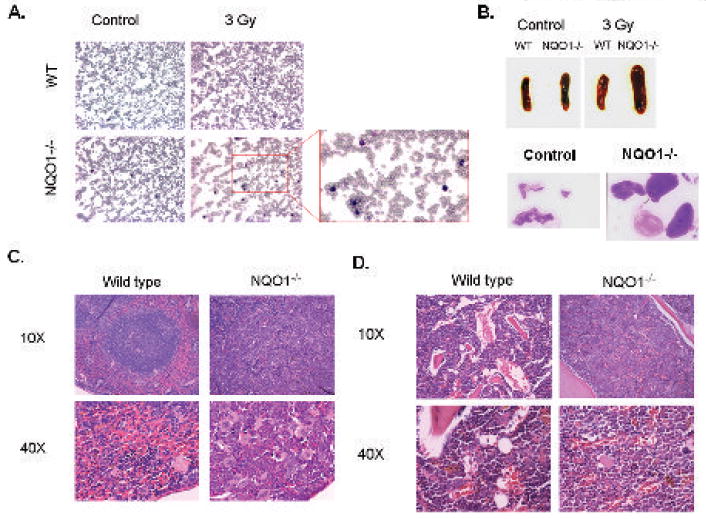
A). Blood analysis. Blood were collected from wild type and NQO1-/- mice one year after irradiation with 3 grays of γ-radiation. Blood smears were prepared and stained with Wright-Giemsa nuclear stain. Enlarged section of the blood smear of irradiated NQO1-/- mouse shows large number of mature granulocytes (polymorph-nuclear). B). Gross anatomy. Pictures of the spleen of wild type and NQO1-/- mice with and without 3 grays of γ-radiation shows enlarged NQO1-/- spleen after irradiation. Lymph nodes from NQO1-/- control (unirradiated) and irradiated mice are also shown. Wild type mice more or less showed no enlargement of lymph nodes in irradiated mice and are not shown. C). Histology of Spleen. Mice were irradiated with 3 grays of γ-radiation. One year later spleen samples were collected, fixed in 10% buffered formalin, and embedded in paraffin. Sections were cut and stained with H&E. Spleen of NQO1-/- mice, one year after irradiation, shows loss of follicular structure due to invasion of granulocytes and megakaryocytes. D). Histology of Bone. Mice were irradiated with 3 grays of γ-radiation. One year later femurs were obtained, fixed in 10% buffered formalin, decalcified, and embedded in paraffin. Sections were cut and stained with H&E. The gaps indicate adipose tissue that is lost during slide processing. The larger gaps indicate less cellular bone marrow. NQO1-/-bone marrow is hypercellular as compared to wild type mice one year after γ-irradiation.
Table 2.
Chromosomal aberrations
| 1 | WT male Control | 40, XY |
| 2 | NQO1-/- male Control | 40, XY |
| 3 | WT male 3 Gy | t(2;?) (one cell); 13q+ (one cell) |
| 4 | WT female 3 Gy | 9q+ (one cell); break in chr. 11 (one cell) |
| 5 | NQO1-/- male 3 Gy | t(5;13) (one cell); t-(5;19) (one cell); br in chr. 13 (one cell) |
| 6 | NQO1-/- female 3Gy | t(2;X) (six cells); del X (one cell) |
15 metaphases were analyzed in each case.
Note: Chromosomal aberrations were not observed in control (unirradiated) wild type and NQO1-/- mice.
Histological analysis of wild type and NQO1-/- mice demonstrated the following. Wright stained blood smears showed a larger number of mature polymorphonuclear neutrophils, which indicate a myeloproliferative disease and not acute myeloid leukemia (Fig. 3A). The spleen and lymph nodes of irradiated NQO1-/- mice were significantly enlarged (Fig. 3B). Histological section in the spleen on NQO1-/- mice showed loss of follicular structure (white/red pulp) due to invasion of granulocytes and megakaryocytes (Fig. 3C). Histological section of decalcified femur from NQO1-/- mice showed hypercellular bone marrow (Fig. 3D). Analysis of all the mice revealed that NQO1-/- mice are significantly more susceptibility to develop radiation-induced myeloproliferative disease (Table 1, P>0.001). About 74% of NQO1-/- mice exposed to 3 grays of γ-irradiation developed myeloproliferative disease (Table 1). 79% of unirradiated and 100% of irradiated NQO1-/- mice exposed to 3 grays of γ-radiation developed myeloid hyperplasia. It is also noteworthy to mention that few of the radiation-exposed wild type and a greater number of NQO1-/- mice also developed lymphomas (Table 1). In addition, the NQO1-/- mice, but not wild type, also developed blood unrelated tumors one year after γ-irradiation. These included lung adenomas and squamous cell carcinomas in the head and groin area. Incidence of blood unrelated tumors were 0% for wild type and 84% for NQO1-/- mice exposed to 3 grays of radiation. These data indicate that NQO1-/- mice develop myeloid hyperplasia and are highly susceptible to develop myeloproliferative disease after exposure to DNA damaging agents like γ-radiation. Analysis of chromosomal aberrations revealed significantly higher frequency of translocations in NQO1-/- mice, as compared to wild type mice (Table 2). Chromosomal aberrations were absent in control (unirradiated) wild type and NQO1-/- mice (Data not shown). Chromosome 5 translocations were frequent in male NQO1-/- mice exposed to γ-radiation (Table 2). Interestingly, chromosome 2 translocation in radiation-exposed female NQO1-/- mice appeared clonally selected since six out of fifteen cells demonstrated translocation from chromosome 2 to chromosome X (Table 2).
Lower/lack of induction of apoptotic and myeloid cell differentiation factors in NQO1-/- mice
The above results raised intriguing questions regarding the mechanism of the role of NQO1 in protection against radiation-induced myeloproliferative diseases. NQO1 has been shown to protect tumor suppressor p53 against 20S proteasome degradation (29). In addition, lack of induction of p53 and reduced apoptosis were shown to contribute to benzo(a)pyrene-induced skin carcinogenesis (25). Therefore, we performed Western analysis to test the hypothesis that NQO1 regulates stability of myeloid cell apoptosis and differentiation factors. Western blots of bone marrow lysates from wild type and NQO1-/- mice unirradiated and irradiated with 3 grays γ-radiation were probed with antibodies against p53 and p53 downstream genes p21 and Bax and myeloid differentiation factors Pu.1 and C/EBPα. The results are shown in Fig. 4. Bone marrow from unirradiated NQO1-/- mice showed lower levels of p53, p21 and Bax and higher levels of C/EBPα and Pu.1 as compared to wild type mice (Fig. 4). Western analysis of the bone marrow of wild type and NQO1-/- mice 12 hours after exposure to 3 grays of γ-radiation showed significant induction of p53, p21, Bax, C/EBPα and Pu.1. Interestingly, NQO1-/- mice demonstrated lower to lack of induction of p53, p21, Bax, C/EBPα and Pu.1 upon exposure to 3 grays of γ-radiation. Interestingly, NQO1 was induced by γ-irradiation in bone marrow cells from the wild type mice, probably as a result of oxidative stress from the reactive oxygen species (ROS) generated after exposure to ionizing irradiation (Fig. 4).
Figure 4. Tumor suppressor p53 and myeloid differentiation factors in wild type and NQO1-/- bone marrow with or without γ-irradiation.
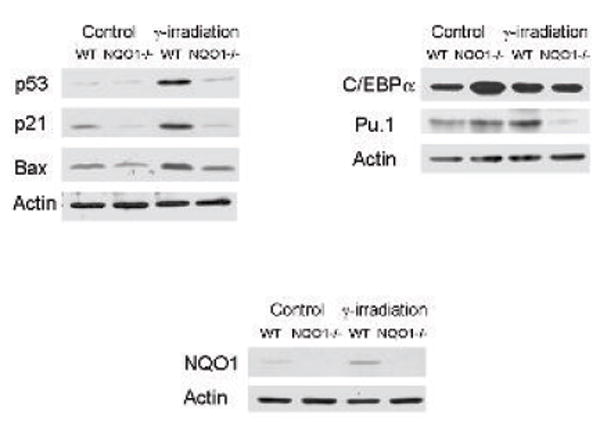
Wild type and NQO1-/- mice were irradiated with 3 grays of γ-radiation. Twelve hours later, mice were euthanized and femurs were surgically removed. Bone marrow were flushed and lysed with cold buffer containing 50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.5% Triton X-100, and protease inhibitor cocktail (Roche). One hundred micrograms of each bone marrow lysate was loaded and separated on 12 % polyacrylamide gels, blotted on the ECL membrane and probed with antibodies against p21, Bax, p53, NQO1, C/EBPα, Pu.1, and actin.
Discussion
Disruption of NQO1 gene in mice leads to myeloid cell hyperplasia as evident from the increase in myeloid cells in bone marrow and granulocytes in peripheral blood (present and previously published reports) (21). Exposure of NQO1-/- mice to γ-radiation showed myeloproliferative disease that included significant increase in blood granulocytes and bone marrow myeloid cells, enlarged lymph nodes and spleen, loss of spleen follicular structure due to invasion of granulocytes and megakaryocytes, and bone marrow hypercellularity. These results provide direct evidence that NQO1-deficient mice are highly susceptible to develop myeloproliferative disease. This conclusion is also supported by previous observations of high frequency of P187S polymorphism in NQO1 protein in AML patients after chemotherapy and radiation treatment (14, 16). NQO1 P187S protein is known to rapidly degrade and individuals carrying homozygous polymorphism have zero and heterozygous have only half of the NQO1 protein and activity (7).
Interestingly, the NQO1-/- mice did not develop AML even though the P187S mutant polymorphism in human NQO1 has been linked to therapy-related AML, especially after chemo and radiation therapy (14). Instead, NQO1-/- mice were more susceptible to develop myeloproliferative disease after exposure to γ-radiation. However, the high incidence of myeloproliferative disease in NQO1-/- mice indicates that the NQO1-/- mouse model partially recapitulates the high incidence of therapy-related AML in patients homozygous for the null polymorphism in human NQO1 gene. It is also possible that additional factors contribute to development of AML in absence of NQO1 and remains to be investigated.
Tumor suppressor p53 is associated with apoptosis (30). Increase or decrease in p53 is directly related to similar alterations in apoptosis. C/EBPα and Pu.1 are myeloid differentiation factors. C/EBPα (CCAAT/enhancer-binding protein alpha) is the main lineage determination regulator that favors myeloid vs. lymphoid lineage commitment. C/EBPα knockout mice did not have any neutrophils in the blood, which indicates the essential role of C/EBPα in myeloid differentiation (31). Pu.1 knockout mice also lack blood neutrophils (32). C/EBPα and Pu.1 regulate and induce myeloid cell differentiation into mature granulocytes (33). Differentiation stops proliferation, and thus prevents myeloproliferative disease and myeloid leukemia. Lower levels and mutations in C/EBPα and Pu.1 have been associated with AML development and prognosis (34, 35).
The present studies revealed that decreased apoptosis in myeloid cells and increased myeloid cell differentiation led to myeloid hyperplasia in unirradiated NQO1-/- mice. Lower p53 and Bax presumably led to decreased apoptosis in NQO1-/- mice. The increase in myeloid differentiation factors C/EBPα and Pu.1 most likely contributed to myeloid hyperplasia in NQO1-/- mice. In other words, increase in differentiation factors resulted in greater number of myeloid cell differentiation from its progenitors that led to myeloid hyperplasia in NQO1-null mice. Exposure to γ-radiation induced accumulation of high levels of p53/Bax and myeloid differentiation factors C/EBPα and Pu.1 in the bone marrow of wild type mice. Induction of these factors resulted in the induction of apoptosis and differentiation of myeloid cells in wild type bone marrow, leading to the prevention of mutagenesis and transformation. In NQO1-/- mice, γ-irradiation did not significantly induce p53/Bax and apoptosis. Lack of induction of p53/Bax and apoptosis after γ-irradiation presumably allowed cells with DNA damage to continue to proliferate. The lack of induction of C/EBPα and Pu.1 after irradiation also limited the differentiation of myeloid cells in γ-radiation-exposed NQO1-/- mice. Therefore, the reduced apoptosis combined with lack of differentiation resulted in increased incidence of myeloproliferative disease in NQO1-/- mice.
One of the intriguing questions is, how does NQO1 regulate apoptosis/differentiation and associated factors? Recently, we and others have shown a role of NQO1 in protection against 20S proteasomal degradation of cellular factors including p53 (29, 36). The loss of NQO1 in NQO1-/- mice leads to degradation of p53 that contributes to benzo(a)pyrene-induced skin cancer (22). Therefore, it is possible that NQO1 also protects apoptotic factor p53 and differentiation factors C/EBPα and Pu.1 against 20S proteasomal degradation in γ-radiation-exposed bone marrow from wild type mice. This leads to protection by eliminating the radiation-damaged cells and promoting differentiation to myeloid cells. However, lack of NQO1 in NQO1-/- mice leads to rapid degradation of p53, C/EBPα and Pu.1. This results in significantly lower levels of these factors in radiation exposed NQO1-/- mice leading to myeloproliferative disease. This however does not explain the increase in C/EBPα and Pu.1 in unirradiated NQO1-/- mice bone marrow. It is possible that mechanisms that regulate expression of C/EBPα and Pu.1 are altered in NQO1-/- mice and remains to be determined.
Mouse chromosome 2 aberrations have been linked to development of radiation-induced AML (37-39). Interestingly, mouse chromosome 2 contains clustered tumor suppressor gene loci and Pu.1 gene (40). In addition, loss of a whole chromosome 5 or a deletion of the long arm, del(5q), is a recurring abnormality in myelodysplastic syndromes (MDSs) and AML (41-42). Intriguingly, exposure of NQO1-/- mice to 3 grays of γ-radiation induced chromosome 5 and 2 translocations. The chromosome 2 translocation appeared clonally selected. It is possible that chromosome 2 and 5 translocations contributed to radiation-induced myeloproliferative diseases in NQO1-/-. It is also possible that lack of induction of p53 in NQO1-/- mice in response to γ-radiation led to chromosome 2 and 5 translocations, which resulted in myeloproliferative disease and remains to be determined.
In summary, the results suggested that the loss of NQO1 leads to decrease in p53 protein and apoptosis and increase in differentiation factors C/EBPα and PU.1, which leads to myeloid hyperplasia in NQO1-/- mice. In addition, lower/lack of induction of apoptotic and differentiation factors led to radiation-induced myeloproliferative disease in NQO1-/- mice. These results led to the conclusion that NQO1 acts as an endogenous factor in protection against myeloid hyperplasia and radiation-induced myeloproliferation. This conclusion is highly significant for the 4% human individuals who are homozygous for a polymorphism P187S allele of NQO1 and lack NQO1 protein and activity. The conclusion is also significant for the 20-25% of individuals carrying one polymorphic NQO1 P187S allele and half of the wild type NQO1 protein and activity.
Acknowledgments
We are thankful to Dr. Dorothy Lewis, Baylor College of Medicine, Houston, TX for help in Flow cytometric analysis. We are also thankful to Dr. Brad Patrick, University of Maryland School of Medicine, Baltimore for critically reading the manuscript. This investigation was supported by NCI grant RO1 CA81057 and NIEHS grant RO1 ES07943.
Abbreviations
- NQO1
NAD(P)H:quinone oxidoreductase 1
- WT
Wild Type
- NQO1-/-
NQO1 deficient
References
- 1.Radjendirane V, Joseph P, Jaiswal AK. Gene expression of DT-diaphorase (NQO1) in cancer cells. In: Forman HJ, Cadenas E, editors. Oxidative Stress and Signal Transduction. New York: Chapman & Hall; 1997. pp. p441–75. [Google Scholar]
- 2.Riley RJ, Workman P. DT-diaphorase and cancer chemotherapy. Biochem Pharm. 1992;43:1657–69. doi: 10.1016/0006-2952(92)90694-e. [DOI] [PubMed] [Google Scholar]
- 3.Lind C, Hochstein P, Ernster L. DT-diaphorase: Properties, reaction mechanism, metabolic function. A progress report. In: King TE, Mason HS, Morrison M, editors. Oxidases and Related Redox Systems. Oxford: Pergamon Press; 1982. pp. p321–47. [Google Scholar]
- 4.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 5.Jaiswal AK, Bell DW, Radjendirane V, Testa JR. Localization of human NQO1 gene to chromosome 16q22 and NQO2-6p25 and associated polymorphism. Pharmacogenetics. 1999;9:413–8. doi: 10.1097/00008571-199906000-00020. [DOI] [PubMed] [Google Scholar]
- 6.Traver RD, Horikoshi T, Danenberg KD, et al. NAD(P)H:Quinone Oxidoreductase gene expression in human colon carcinoma cells: Characterization of a mutation which modulates DT-diaphorase activity and mitomycin sensitivity. Cancer Res. 1992;52:797–802. [PubMed] [Google Scholar]
- 7.Siegel D, Anwar A, Winski SL, Kepa JK, Zolman KL, Ross D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol Pharmacol. 2001;59:263–8. doi: 10.1124/mol.59.2.263. [DOI] [PubMed] [Google Scholar]
- 8.Siegel D, McGuinessess SM, Winski SL, Ross D. Genotype-phenotype relationships in studies of a polymorphism in NAD(P)H:quinone oxidoreductase 1. Pharmacogenetics. 1999;9:113–21. doi: 10.1097/00008571-199902000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Kelsey KT, Ross D, Traver RD, et al. Ethnic variation in the prevalance of a common NAD(P)H:quinone oxidoreductase polymorphism and its implications for anti-cancer chemotherapy. Br J Cancer. 1997;76:852–4. doi: 10.1038/bjc.1997.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosvold EA, McGlynn KA, Lustbader ED, Buetow KH. Identification of an NAD(P)H:quinone oxidoreductase polymorphism and its association with lung cancer and smoking. Pharmacogenetics. 1995;5:199–206. doi: 10.1097/00008571-199508000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Schulz WA, Krummeck A, Rosinger I, Schmitz-Drager BJ, Sies H. Predisposition towards urolithiasis associated with the NQO1 null-allele. Pharmacogenetics. 1998;8:453–4. doi: 10.1097/00008571-199810000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Weincke JK, Spitz MR, McMillan A, Kelsey KT. Lung Cancer in Mexican-Americans and African Americans is associated with the wild-type genotype of the NAD(P)H:quinone oxidoreductase polymorphism. Cancer Epidemiol Biomark Prev. 1997;6:87–92. [PubMed] [Google Scholar]
- 13.Rothman N, Smith MT, Hayes RB, et al. Benzene poisoning, a risk factor for hematologic malignancy, is associated with NQO1 609 C-->T mutation and rapid fractional excretion of chlorzoxazone. Cancer Res. 1997;57:2839–42. [PubMed] [Google Scholar]
- 14.Larson RA, Wang Y, Banerjee M, et al. Prevalence of the inactivating 609C-->T polymorphism in the NAD(P)H:quinone oxidoreductase (NQO1) gene in patients with primary and therapy-related myeloid leukemia. Blood. 1999;94:803–7. [PubMed] [Google Scholar]
- 15.Smith MT, Wang Y, Kane E, et al. Low NAD(P)H:quinone oxidoreductase1 activity is associated with increased risk of acute leukemia in adults. Blood. 2001;97:1422–6. doi: 10.1182/blood.v97.5.1422. [DOI] [PubMed] [Google Scholar]
- 16.Wiemels JL, Pagnamenta A, Taylor GM, Eden OB, Alexander FE, Greaves MF. A lack of a functional NAD(P)H:quinone oxidoreductase allele is selectively associated with pediatric leukemias that have MLL fusions. Cancer Res. 1999;59:4095–9. [PubMed] [Google Scholar]
- 17.Smith MT, Wang Y, Skibola CF, et al. Low NAD(P)H:quinone oxidoreductase activity is associated with increased risk of leukemia with MLL translocations in infants and children. Blood. 2002;100:4590–3. doi: 10.1182/blood-2001-12-0264. [DOI] [PubMed] [Google Scholar]
- 18.Radjendirane V, Joseph P, Lee H, et al. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J Biol Chem. 1998;273:7382–9. doi: 10.1074/jbc.273.13.7382. [DOI] [PubMed] [Google Scholar]
- 19.Gaikwad A, Long DJ, II, Stringer JL, Jaiswal AK. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J Biol Chem. 2001;276:22559–64. doi: 10.1074/jbc.M101053200. [DOI] [PubMed] [Google Scholar]
- 20.Long DJ, II, Gaikwad A, Multani A, et al. Disruption of the NAD(P)H:quinone oxidoreductase 1 (NQO1) gene in mice causes myelogenous hyperplasia. Cancer Res. 2002;62:3030–6. [PubMed] [Google Scholar]
- 21.Bauer AK, Faiola B, Abernethy DJ, et al. Genetic susceptibility to benzene-induced toxicity: role of NADPH:quinone oxidoreductase-1. Cancer Res. 2003;63:929–35. [PubMed] [Google Scholar]
- 22.Long DJ, II, Waikel RL, Wang X, Perlaky L, Roop DR, Jaiswal AK. NAD(P)H:quinone oxidoreductase 1 deficiency increases susceptibility to benzo(a)pyrene-induced mouse skin carcinogenesis. Cancer Res. 2000;60:5913–5. [PubMed] [Google Scholar]
- 23.Long DJ, Waikel RL, Wang XJ, Roop DR, Jaiswal AK. NAD(P)H:quinone oxidoreductase 1 deficiency and increased susceptibility to 7,12-dimethylbenz[a]-anthracene-induced carcinogenesis in mouse skin. J Natl Cancer Inst. 2001;93:1166–70. doi: 10.1093/jnci/93.15.1166. [DOI] [PubMed] [Google Scholar]
- 24.Iskander K, Paquet M, Brayton C, Jaiswal AK. Deficiency of NRH:quinone oxidoreductase 2 increases susceptibility to 7,12-dimethybenz(a)anthracene and benzo(a)pyrene-induced skin carcinogenesis. Cancer Res. 2004;64:5925–8. doi: 10.1158/0008-5472.CAN-04-0763. [DOI] [PubMed] [Google Scholar]
- 25.Iskander K, Gaikwad A, Paquet M, et al. Lower induction of p53 and decreased apoptosis in NQO1-null mice leads to increased sensitivity of chemical-induced skin carcinogenesis. Cancer Res. 2005;65:2054–8. doi: 10.1158/0008-5472.CAN-04-3157. [DOI] [PubMed] [Google Scholar]
- 26.Pathak S. Chromosome banding techniques. J Reprod Med. 1976;17:25–8. [PubMed] [Google Scholar]
- 27.Sawyer JR, Moore MM, Hozier JC. High resolution G-banding chromosomes of the mouse. Chromosoma. 1987;95:350–8. doi: 10.1007/BF00293182. [DOI] [PubMed] [Google Scholar]
- 28.Kogan SC, Ward JM, Anver MR, et al. Hematopathology subcommittee of the mouse models of human cancers consotium. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100:238–45. doi: 10.1182/blood.v100.1.238. [DOI] [PubMed] [Google Scholar]
- 29.Gong X, Kole L, Iskander K, Jaiswal AK. NRH:quinone oxidoreductase 2 and NAD(P)H:quinone oxidoreductase 1 protect tumor suppressor p53 against 20S proteasomal degradation leading to stabilization and activation of p53. Cancer Res. 2007;67:5380–8. doi: 10.1158/0008-5472.CAN-07-0323. [DOI] [PubMed] [Google Scholar]
- 30.Pietsch EC, Humbey O, Murphy ME. Polymorphism in the p53 pathway. Oncogene. 2006;25:1602–11. doi: 10.1038/sj.onc.1209367. [DOI] [PubMed] [Google Scholar]
- 31.Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–74. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson KL, Smith KA, Conners K, McKercher SR, Maki RA, Torbett BE. Myeloid development is selectively disrupted in Pu.1 null mice. Blood. 1998;91:3702–10. [PubMed] [Google Scholar]
- 33.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific factor, PU.1. Nature Genet. 2004;36:624–30. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 33.Miranda MB, Johnson DE. Signal transduction pathways that contribute to myeloid differentiation. Leukemia. 2007;21:1363–77. doi: 10.1038/sj.leu.2404690. [DOI] [PubMed] [Google Scholar]
- 34.Kaeferstein A, Krug U, Tiesmeier J, et al. The emergence of a C/EBPalpha mutation in the clonal evolution of MDS towards secondary AML. Leukemia. 2003;17:343–9. doi: 10.1038/sj.leu.2402805. [DOI] [PubMed] [Google Scholar]
- 35.Dekoter RP, Kamath MB, Houston IB. Analysis of concentration-dependent functions of PU.1 in hematopoiesis using mouse models. Blood Cells Mol Dis. 2007;39:316–20. doi: 10.1016/j.bcmd.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asher G, Loten J, Sachs L, et al. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc Natl Acad Sci USA. 2002;99:13125–30. doi: 10.1073/pnas.202480499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jawad M, Cole C, Zanker A, Priscilla L, Fitch S, Plumb M. Evidence for clustered tumour suppressor gene loci on mouse chromosome 2 and 4 in radiation-induced acute myeloid leukemia. Int J Radiat Biol. 2006;82:383–91. doi: 10.1080/09553000600784161. [DOI] [PubMed] [Google Scholar]
- 38.Rithidech K, Dunn JJ, Roe BA, Gordon CR, Cronkite EP. Evidence for two commonly deleted regions on mouse chromosome 2 in g ray-induced acute myeloid leukemic cells. Exp Hemat. 2002;30:564–70. doi: 10.1016/s0301-472x(02)00799-3. [DOI] [PubMed] [Google Scholar]
- 39.Zimonjic DR, Pollock JL, Westervelt P, Popescu NC, Ley TJ. Acquired, nonrandom chromosomal abnormalities associated with the development of acute promyelocytic leukemia in transgenic mice. Proc Natl Acad Sci USA. 2000;97:13306–11. doi: 10.1073/pnas.97.24.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walter MJ, Park JS, Ries RE, et al. Reduced Pu.1 expression causes myeloid progenitor expansion and increased leukemia penetrance in mice expressing PML-RARa. Proc Natl Acad Sci USA. 2005;102:12513–8. doi: 10.1073/pnas.0504247102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joslin JM, Fernald AA, Tennant TR, et al. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood. 2007;110:719–26. doi: 10.1182/blood-2007-01-068809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lezon-Geyda K, Najfeld V, Johnson EM. Deletions of PURA, at 5q31, and PURB, at 7p13, in myelodysplastic syndrome and progression to acute myelogenous leukemia. Leukemia. 2001;15:954–62. doi: 10.1038/sj.leu.2402108. [DOI] [PubMed] [Google Scholar]