

Abstract
Several ribozyme constructs have been used to dissect aspects of the group I self-splicing reaction. The Tetrahymena L-21 ScaI ribozyme, the best studied of these intron analogs, catalyzes a reaction analogous to the first step of self-splicing, in which a 5′-splice site analog (S) and guanosine (G) are converted into a 5′-exon analog (P) and GA. This ribozyme preserves the active site but lacks a short 5′-terminal segment (called the IGS extension herein) that forms dynamic helices, called the P1 extension and P10 helix. The P1 extension forms at the 5′-splice site in the first step of self-splicing and P10 forms at the 3′-splice site in the second step of self-splicing. To dissect the contributions from the IGS extension and the helices it forms we have investigated the effects of each of these elements at each reaction step. These experiments were performed with the L-16 ScaI ribozyme, which retains the IGS extension, and with 5′- and 3′-splice site analogs that differ in their ability to form the helices. The presence of the IGS extension strengthens binding of P by 40-fold, even when no new base pairs are formed. This large effect was especially surprising, as binding of S is essentially unaffected for S analogs that do not form additional base pairs with the IGS extension. Analysis of a U•U pair immediately 3′ to the cleavage site suggests that a previously identified deleterious effect from a dangling U residue on the L-21 ScaI ribozyme arises from a fortuitous active site interaction and has implications for RNA tertiary structure specificity. Comparisons of the affinities of 5′-splice site analogs that form only a subset of base pairs reveal that inclusion of the conserved G•U base pair at the cleavage site of group I introns destabilizes the P1 extension >100-fold relative to the stability of a helix with all Watson-Crick base pairs. Previous structural data with model duplexes and the recent intron structures suggest that this effect can be attributed to partial unstacking of the P1 extension at the G•U step. These results suggest a previously unrecognized role of the G•U wobble pair in self-splicing: breaking cooperativity in base pair formation between P1 and the P1 extensions. This effect may facilitate replacement of the P1 extension with P10 after the first chemical step of self-splicing and release of the ligated exons after the second step of self-splicing.
The group I intron from Tetrahymena thermophila, the first catalytic RNA to be discovered, folds into a specific structure and performs a self-splicing reaction that includes chemical steps as well as RNA conformational rearrangements. This reaction is simple, relative to pre-mRNA splicing and other RNA-mediated processes, and may serve as a tractable system to learn more about RNA catalysis and its functional conformational changes. To date much information has been garnered about the structure and catalytic function of group I introns, especially the originally discovered intron from Tetrahymena thermophila (1, 3, 8, 12–32), but much less is understood about the required conformational changes (33, 34).
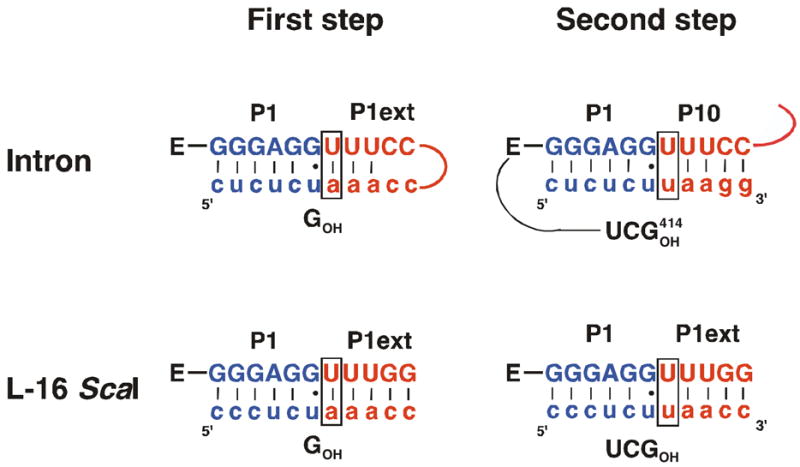
The group I self-splicing reaction has been studied in the greatest depth with shortened ribozyme constructs derived from the self-splicing intron ((1, 5, 8, 20, 21, 30, 35–37), see also: (38, 39)). The most commonly studied of these constructs is the L-21 ScaI ribozyme (Figure 1B). The 5′-end of this ribozyme positions the 5′-exon through formation of base pairs and is therefore called the internal guide sequence (IGS, Figure 1D). However, the L-21 ScaI ribozyme lacks a 5′-extension of the IGS, present in the self-splicing intron, that has the potential to form a longer helix (P1 plus P1 extension form the extended P1 duplex) during the first step of splicing (compare Figure 1A&B). Thus, the immediate environment of the cleavage site differs between the L-21 ScaI ribozyme and the self-splicing intron. Furthermore, the 5′-extension of the IGS forms another helix in the second step of self-splicing, referred to as P10. This helix is formed between the IGS extension and the 3′-exon (Figure 1A, (40–43), see also: (44)). Thus, base pairs in the P1 extension must be broken for self-splicing to proceed, and the P1 extension and P10 are dynamic secondary structure elements in self-splicing (Figure 1A).
Figure 1.

Comparison of the self-splicing RNA with the L-21 ScaI and the L-16 ScaI ribozymes. Exon sequences are shown in lower case letters, while intron or ribozyme sequences are shown in uppercase letters. The P1 duplex is shown in blue (A-D), while the P1 extension and P10 (A, C, D) are shown in red. The five 3′-terminal nucleotides that are deleted in the L-16 and the L-21 ScaI ribozymes are shown in green for the intron. The secondary structure of the remaining RNA is identical in all cases and shown below for orientation, with arrows denoting the covalent connections between the enlarged parts and the rest of the ribozyme. The reactive phosphoryl group is shown explicitly in each case as “p”. (A) Secondary structure of the region around the cleavage site in the first and second transesterification step of self-splicing. (B) Secondary structure of the L-21 ScaI ribozyme around the cleavage site. (C) Secondary structure of the L-16 ScaI ribozyme around the cleavage site. Analogs of the 5′- and 3′-splice sites are shown as substrates, with A•U and U•U pairs, respectively following the G•U wobble. Relative to the L-21 ScaI ribozyme, the L-16 ScaI ribozyme contains a five nucleotide 5′-extension (5′GGUUU3′); shown in part (A), the naturally occurring nucleotides at this position are (5′CCUUU3′). The 5′-GG residues that replace the 5′-CC in the L-16 ScaI ribozyme were included to facilitate transcription by T7 RNA polymerase. The natural 5′-exon analog contains a U five residues upstream of the cleavage site and the resulting G•U wobble pair appears to increase tertiary binding interactions with the P1 duplex (6–8). Herein, as in previous work, we have replaced this U with a C residue. Finally, to prevent binding from being too strong, we have removed the 5′-most C of the 5′-exon mimic for most of the reactions studied herein. (D) Definition of the nomenclature used herein. The additional sequence present in the self-splicing intron and the L-16 ScaI ribozyme, but absent in the L-21 ScaI ribozyme extends the IGS and is therefore termed IGS extension. Together, the IGS and the IGS extension are termed the extended IGS. Formation of base pairs with the IGS results in formation of the P1 duplex (shown in blue), which can be formed with both the L-21 and the L-16 ScaI ribozyme. Analogously, in the L-16 ScaI ribozyme formation of base pairs with the IGS extension results in formation of the P1 extension (shown in red). Together the P1 and the extended P1 duplexes are called the extended P1 duplex.
To understand the kinetic and thermodynamic contributions from the IGS extension, the P1 extension, and P10 to the self-splicing reaction we have employed an IGS-extended ribozyme. This construct, referred to as the L-16 ScaI ribozyme (Figure 1C), retains the ability to form the P1 extension and P10. Rate and equilibrium constants for individual steps in the L-16 ScaI ribozyme reaction with different bound substrates have been compared to those previously obtained with the L-21 ScaI ribozyme. This stepwise approach has allowed us to isolate the effects from the IGS extension and formation of the extended P1 or the P10 duplex on each step of the ribozyme reaction.
Materials and Methods
Materials
The L-16 ScaI ribozyme, compared to the L-21 ScaI ribozyme, has a five nucleotide extension of the internal guide sequence (IGS) at the 5′-end (5′GGUUU3′, see Figure 1 for relation of L-16 ScaI and L-21 ScaI ribozymes and the full-length intron). The naturally occurring residues at these positions are 5′CCUUU3′. The 5′-GG in the L-16 ScaI ribozyme replacing the 5′-most CC residues are included to facilitate transcription by T7 RNA polymerase. The DNA template for the L-16 ScaI ribozyme was constructed from the L-21 ScaI-containing plasmid by PCR1 mutagenesis and ligation into pUC19. Individual clones were sequenced to verify the L-16 ScaI sequence.
Conditions for transcription of the L-16 ScaI ribozyme were modified to prevent endonucleolytic cleavage, which apparently occurs at the 5′-end to yield L-21 ScaI ribozyme ((45, 46), data not shown). Transcriptions were carried out for 30 min at 30 °C in the presence of 4 mM MgCl2, 0.5 mM of each NTP, 40 mM DTT, 2 mM spermidine, 40 mM Tris, pH 8.1, 0.01% Triton X-100, 5 μg/mL ScaI-digested, purified template and T7 RNA polymerase. Reactions were stopped by the addition of excess EDTA, ethanol precipitated, resuspended, and purified over an RNeasy column (Qiagen) as described previously (5, 47). Analytical 5′-32P-phosphorylation and comparison with authentic L-21 ScaI confirmed that full-length L-16 ScaI ribozyme was obtained (~95%). This conclusion is further supported by the functional data that show that the L-16 ScaI ribozyme binds stronger to fully base-paired oligonucleotides than to oligonucleotides in which the 5′-most residues on the ribozyme cannot form base pairs (Table 1 below).
Table 1.
Binding and Dissociation Rate Constants for 5′-Splice Site Analog Substrates
| IGS-sequence: 3′GGAGGUUUGG5′ | koff (min−1) | Kd (nM) | Δ ΔG (kcal/mol) |
|---|---|---|---|
| CCUCdTMe | 1.5a | 15b | −0.2 |
| CCUCdTA | 1.9b | 19c | (0) |
| CCUCdTAAACC | 2.5•10−4 | 0.0025 | −5.4 |
| CCUCdTAAAAA | 0.17 | 1.7 | −1.5 |
Dissociation rate constants from the E•S complex at 30 °C, pH 6.0. Equilibrium dissociation constants were calculated from the measured dissociation rate constants and the association rate constant of 1•108 M−1min−1 for CCUCUAAACC and CCUCUAAAAA (data not shown). This association rate constant reflects the formation of the ribozyme-substrate duplex and is the same, within error, as that determined previously for the L-21 ScaI ribozyme (1, 2, 5). Deoxyribose substitution at position –1 (Chart 1) has no effect on the association rate constant (57). Values are averages of two or more independent experiments giving rate constants that were the same within 3-fold.
The dissociation rate constant for CCUCdTMe is too fast to be measured accurately and was therefore calculated from the measured dissociation rate constant for CCCUCUMe, with the effect from the deletion of –6C (Chart 1), and the deoxyribose substitution at position –1 accounted for by comparing the dissociation rate constants for CCUCdTAAAAA and CCCUCdTAAAAA (for –6C) and CCCUCdTA and CCCUCUA (for the –1 deoxyribose).
The dissociation rate constant for CCUCdTA is too fast to be measured accurately and was therefore calculated from the dissociation constant for CCCUCdTA accounting for the effect from the –6 base pair as described in footnote a.
RNA oligonucleotides were purchased from Dharmacon Research, Inc. (Lafayette, CO), and DNA oligonucleotides were purchased from Operon Technologies Inc. (Alameda, CA). RNA and DNA oligonucleotides were 5′-end-labeled with γ32P-ATP and purified with non-denaturing gels using standard procedures (48). Unlabeled oligonucleotides were purified by anion exchange HPLC and desalted on Sep-Pak C-18 columns (Waters, Franklin, MA) as described (5).
General Kinetic Methods
All reactions were single turnover with ribozyme in excess of 5′-32P-labeled S or P and, unless otherwise noted, were carried out at 30 °C in 50 mM Na-MES, pH 6.0, and 10 mM MgCl2. Prior to reaction, ribozyme was preincubated for 30 min at 50 °C and pH 6.0 with 10 mM MgCl2 to allow folding to the active state (1, 49). Control experiments performed by incubating radiolabeled L-16 ScaI ribozyme and monitoring its length on denaturing polyacrylamide gels confirmed that no significant processing of the L-16 ScaI ribozyme occurred during folding (data not shown). Reactions were initiated by addition of ribozyme and aliquots were removed at specified times to be quenched with two volumes of a solution containing 20 mM EDTA and 85% formamide. Radiolabeled oligonucleotides were separated by denaturing gel electrophoresis (7 M urea, 20% acrylamide) and quantitated using Phosphorimager analysis (Molecular Dynamics) with Image Quant software. For slow reactions, rate constants were obtained from initial rates with endpoints of 95% assumed. All other reactions were followed to completion, and good first order fits to the data with endpoints of ≥90% were obtained in all cases (R2 ≥ 0.98) (Kaleidagraph, Synergy Software, Reading, PA).
Dissociation Rate Constants for Substrates and Product
Dissociation rate constants for *S (see Chart 1 for oligonucleotides used) in the absence of guanosine and for P in the presence and absence of UCGAAACC 2 were determined in pulse-chase gel-shift experiments essentially as described previously (5, 36). Oligonucleotides with a 2′-deoxyribose substitution at the cleavage site were used to prevent reaction during the dissociation reaction. Trace *S (or *P) was bound to saturating amounts of ribozyme (pulse) and a large excess of unlabeled S (chase) was then added. Control experiments in which the chase was preincubated with the labeled oligonucleotide verified that the chase was effective in preventing rebinding of dissociated *S (or *P). At specified times aliquots were loaded onto a running native gel in THEM buffer (33 mM Tris, 67 mM HEPES, 1 mM EDTA, 10 mM MgCl2) to separate bound and free *S (or *P). Dissociation rate constants were obtained by fitting the fraction of free *S (or *P) to a double exponential. Double exponential fitting was necessary because a small fraction of ribozyme (~8%) released *S or *P considerably faster than the bulk of the population, consistent with release from the L-21 ScaI ribozyme. Presumably these ribozymes had lost their IGS extension through self-processing (44–46). Control experiments monitoring the length of radioactively labeled L-16 ScaI confirmed that L-16 ScaI is not substantially processed during the dissociation reactions (≥80% remaining, data not shown; for reference, processing of 50% in parallel with dissociation, to give an apparent dissociation rate constant that is approximately the sum of the rate constants for dissociation and processing, would result in only a two-fold deviation of the observed dissociation rate constant from the actual rate constant).
Chart 1.
| Abbreviation | Oligonucleotide | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IGS sequenceb: | 3′rG | rG | rG | rA | rG | rG | rU | rU | rU | rG | rG5′ |
| Position: | −6 | −5 | −3 | −1 | +1 | +3 | +5 | ||||
| −1d, ΔrSA3C2 | rC | rC | rU | rC | dT | rA | rA | rA | rC | rC | |
| −1d, ΔrSUA2C2 | rC | rC | rU | rC | dT | rU | rA | rA | rC | rC | |
| −1d, ΔrSA5 | rC | rC | rU | rC | dT | rA | rA | rA | rA | rA | |
| −1d, Δ ΔrSA5 | rC | rU | rC | dT | rA | rA | rA | rA | rA | ||
| −1d,rSA5 | rC | rC | rC | rU | rC | dT | rA | rA | rA | rA | rA |
| −1r,dSA5 | dC | dC | dC | dU | dC | rU | dA | dA | dA | dA | dA |
| dSA5 | dC | dC | dC | dU | dC | dT | dA | dA | dA | dA | dA |
| −1d, ΔrSA | rC | rC | rU | rC | dT | rA | |||||
| −1d, rSA | rC | rC | rC | rU | rC | dT | rA | ||||
| rSA | rC | rC | rC | rU | rC | rU | rA | ||||
| rSMe | rC | rC | rC | rU | rC | rU | CH3 | ||||
| −3m, −1d, ΔrSA3C2 | rC | rC | mU | rC | dT | rA | rA | rA | rC | rC | |
| −3m, −1d, rSA | rC | rC | rC | mU | rC | dT | rA | ||||
| −1d, ΔP | rC | rC | rU | rC | dT | ||||||
| d ΔP | dC | dC | dT | dC | dT | ||||||
r = 2′-OH; d = 2′-H, m = 2′-OCH3
The IGS sequence is listed in the 3′ to 5′ direction to show complementarity to the listed oligonucleotides.
The dissociation rate constant of *S from E•*S•G (or *P from E•*P•UCGAAACC) was obtained from experiments that measured the partitioning of molecules in the E•*S•G (or E•*P•UCGAAACC) complex between dissociation of *S ( or for dissociation of *P) and reaction of the ternary complex to form E•*P + GA (kc, or k−c for reaction of E•*P•UCGAAACC to form E•*S+UCG). To accurately determine the rate constant for dissociation in these experiments, the values of and kc must be similar. To determine the partitioning, radiolabeled *S (or *P) was added to the ribozyme (50 nM final concentration) to allow formation of E•*S (or E•*P). Reaction was initiated by addition of G ([G] = 2 mM; ; or UCGAAACC, [UCGAAACC] = 10 μM; ) to rapidly form the E•*S•G (or E•*P•UCGAACC) complex, along with a large excess (470 nM) of unlabeled S, to prevent rebinding of dissociated *S or *P. The observed rate constant for formation of *P from *S (or *S from *P) is the sum of the rate constants for the chemical step and dissociation. Thus, by independently measuring the rate constant for the chemical step in reactions with the chase omitted, the dissociation constant can be calculated (Eq. 1A).
| (1A) |
| (1B) |
Alternatively, the dissociation rate constant can be obtained from the fraction of *S that reacted relative to reactions in which no chase was added (fracreacted) and the observed rate constant of the reaction in the presence of the chase (kobs, Eq. 1A&B). The results from the two methods agreed within two-fold. The dissociation rate constant for –1d,ΔP (Chart 1) in the presence of UCGAAACC obtained in partitioning experiments and pulse-chase gel-shift experiments (see above) agreed within two-fold.
Association Rate Constants for S
Association rate constants for S in the absence of G were determined in pulse-chase gel-shift experiments (5, 36). Varying concentrations of ribozyme (2–10 nM) were added to trace *S to initiate the binding reaction (“pulse”). After specified times t1, a large excess of unlabeled S (340 nM) was added to prevent any additional binding of *S (“chase”). Control experiments confirmed that the chase effectively prevented subsequent binding of *S. Plotting the fraction of bound *S against t1 yields a first-order rate constant, which varies with ribozyme concentration. Plotting the first-order rate constants against the ribozyme concentration at which they were obtained gives the second-order association rate constant.
Previous work with the L-21 ScaI ribozyme has shown that in the presence of saturating concentrations of G the rate of reaction of rS is limited by the association of S (1, 50) so that (kcat/KM)S values represent the association rate constants for S in the presence of G. Moreover, (kcat/KM)S values represent the association rate constant if koff ≪ kc (51). Because these conditions hold for the L-16 ScaI ribozyme as well, (kcat/KM)S values were measured to obtain association rate constants in the presence of G. The similarity of (kcat/KM)S values, which represent the formation of the P1 duplex, for the L-21 ScaI and the L-16 ScaI ribozyme (1.5•108 and 1.0•108 M−1 min−1) further supports the notion that (kcat/KM)S values describe the association rate constants for S. To obtain (kcat/KM)S values the reaction rate constant was measured using varying concentrations of ribozyme (2–10 nM). The observed rate constants were plotted as a function of ribozyme concentration to yield the apparent second order rate constant (kcat/KM)S.
Measurement of the Equilibrium Constant for Docking
S and P bind to the ribozyme in two steps: base pairing to form the P1 duplex between S (or P) and the ribozyme [(E•S)open], followed by docking of this duplex into tertiary interactions at the active site ((E•S)open, Eq. 2A, e.g., (8, 35, 52)).
| (2A) |
| (2B) |
2′-Hydroxyl residues on S (and the IGS) as well as the exocyclic amino group of G22 of the ribozyme’s IGS stabilize docking of the ribozyme/substrate duplex (2, 7, 22, 53, 54). Modification of the 2′-hydroxyl group at position –3 to a 2′-methoxy group destabilizes the docked ribozyme/substrate duplex conformation while not affecting the stability of the P1 duplex (7, 35, 55). Thus, substrate analogs with this modification approximate the affinity of the duplex between substrate and IGS (for Ktert < 1, , Eq.2B). Comparing the observed affinity of substrates S* that have been undocked in this way and are thus held only by base pairing interactions ( ) to the affinity of wild type substrate ( ) allows the evaluation of tertiary interactions that stabilize the docked conformation for the wild type substrate (Eq. 2C obtained from Eq. 2B for the wild type substrate, (5, 22, 35, 56).
| (2C) |
Measurement of Affinities for G and GAAACC-analogs
To determine the affinities of ribozyme complexes for G or UCG the rate of reaction of *S was determined with 0–2000 μM G (or 0–1000 μM UCG). A substrate with a 2′-deoxyribose residue at position –1 was used in these experiments to ensure that the chemical step was rate-limiting (23, 57). The observed rate constant for cleavage of *S was plotted as a function of G (or UCG) concentration and fit to Eq. 3.
| (3) |
Non-linearity of ribozyme concentration dependences was observed at low ribozyme concentrations (≤1 nM) suggesting nonspecific losses of ribozyme to tube walls. To avoid these losses ribozyme concentrations ≥2 nM were used in all experiments herein. This meant, however, that S was bound to the ribozyme under all equilibrium conditions. To obtain the affinity of G to free ribozyme, we used a 5′-shortened oligonucleotide CUCdTAAAAA. The presence of the 5′-terminal nucleotides affects binding of oligonucleotides via formation of a base pair, but docking of the resulting P1 duplex is not affected (58). At 2 nM L-16 ScaI ribozyme, most CUCdTAAAAA is unbound (Kd ~ 100 nM, data not shown), as suggested from the rate of reaction compared to that for a substrate that is fully bound to ribozyme. Binding of G and UCG to the open complex was determined with 5′-splice site analogs with a 2′-methoxyribose substitution at position –3 (-3m,rSA5, Chart 1) or with 2′-deoxyribose substitutions at all positions other than the cleavage site (-1r,dSA5, Chart 1, (4, 35)).
To determine the affinity of UCGA and its analog UCGAAACC to E•P the rate of formation of *S from *P was followed as a function of UCGA or analog concentration, as described above ([UCGA] = 10–4000 μM; [UCGAAACC] = 0.02–2 μM). In these experiments caution was taken to ensure that UCGAAACC remained in excess of E by lowering the ribozyme concentration to 10 nM when necessary. 5′-Exon analog (P) with a 2′-deoxyribose substitution at position –1 was used to ensure that the chemical step was rate-limiting in these experiments. To measure the affinity of UCGA or UCGAAACC to free L-16 ScaI ribozyme, the shortened all-deoxyribose 5′-exon analog dΔP (Chart 1) was used (5). dΔP binds weakly to the ribozyme so that at 10 nM ribozyme most of the dΔP is unbound (Kd ~ 20 nM, data not shown). The dissociation constant obtained in this way is the weighted average of binding to E and E•P, and the reported Kd values have been adjusted to account for the fraction of E that has P bound. In addition, due to coupled binding of P and UCGAAACC, binding of UCGAAACC increases the affinity for P, so that at high UCGAAACC concentrations most E will have P bound as well UCGAAACC (see Results). The dissociation constant in Figure 2D is therefore a limit. The rate of reaction was determined as a function of UCGA or UCGAAACC concentration as described above ([UCGA] = 200–8000 μM; [UCGAAACC] = 0.1–10 μM). In the case of binding of UCGA inhibition was observed at higher oligonucleotide concentrations. This inhibition precluded determination of a reliable value for the Kd so that only a lower limit is reported in Figure 2C.
Figure 2.

Kinetic and thermodynamic framework for the ribozyme reaction. (A) Definition of rate and equilibrium constants used herein. Association rate constants (k1, k4, k7 and k8) are reported in M-1 min−1; all other rate constants are in units of min−1. The internal and external equilibrium constants are unitless and describe conversion of substrates to products. Frameworks are defined at 30 °C, 10 mM MgCl2 and pH 6.2. (B) Kinetic and thermodynamic framework for the L-21 ScaI ribozyme with CCUCdTA bound. This framework was adapted from (5). Rate constants for dissociation of CCUCdTA and CCUCdT were determined herein for direct comparison with the L-16 ScaI reactions. (C) Kinetic and thermodynamic framework for the L-16 ScaI ribozyme with CCUCdTA bound. (D) Kinetic and thermodynamic framework for the L-16 ScaI ribozyme with CCUCdTAAACC bound.
Measurement of the Rate Constant for the Chemical Step and the Internal Equilibrium
The rate constant for the chemical step in the forward (and reverse) direction was measured as described previously (5). In these reactions the rate constant for conversion of the tertiary complex E•*S•UCG to E•*P + UCGAAACC (or E•*P•UCGAAACC to E•*S + UCG) was followed ([E] = 50 nM, ; [UCG] = 100 μM; ; [UCGAAACC] = 2 μM, ). Control experiments in which the concentrations of UCG (or UCGAAACC) were varied indicated that the ternary complexes were quantitatively formed. S and P with a 2′-deoxyribose substitution at position –1 were used to ensure that the chemical step was rate-limiting (57). This conclusion was confirmed by the observation of a log-linear pH dependence between pH 6.2 and 8.1 (data not shown, (5, 59, 60)).
The internal equilibrium constant, Kint, which describes the equilibrium between the ternary complexes E•S•UCG and E•P•UCGAAACC, was obtained from the rate constant for the chemical step in the forward and reverse direction (kc and k−c, Eq. 4).
| (4) |
Measurement of the External Equilibrium
The external equilibrium constant Kext describes the equilibrium between unbound substrates and products (Eq. 5A&B). The external equilibrium constant was determined as described (5), except that the cleavage site also contained a -1-deoxyribose-substitution (dSA5 and dP) to relate it to the internal equilibrium obtained with -1-deoxyribose substituted oligonucleotides. Briefly, L-21 ScaI ribozyme was used to catalyze the equilibration of all deoxyribose S and P at subsaturating concentrations of UCG and UCGd(AAAAA). At low ribozyme concentrations most S and P is unbound (20–50 nM, Kd ≈ 85 and 150 nM for binding of dSA5 and dP to the L-21 ScaI ribozyme (2, 5)), so that the external equilibrium is observed (5) and data not shown). Previous control experiments showed that the presence of the deoxyribose residues at positions other than the cleavage site and the length of the “tail” 3′ to the cleavage site have no effect on the equilibrium constant, so that external equilibrium constants obtained with d(CCCTCTAAAAA) can be compared to internal equilibrium constants obtained with r(CCCUC)dTr(AAACC) (5).
| (5A) |
| (5B) |
| (5C) |
Reactions were carried out with 20–50 nM E, 1–5 μM UCG and 5–40 μM UCGdA5. *S or *P was used in trace so that UCG and UCGd(AAAAA) concentrations were essentially unchanged throughout the course of reaction. The value of Kext was calculated from the fraction of *P formed from *S at different ratios of UCGd(AAAAA)/UCG according to Eq. 5C, which was derived from Eq. 5B. Control experiments in which the ribozyme concentration or the concentrations of UCG and UCGd(AAAAA) were varied while maintaining a constant ratio [UCGd(AAAAA)]/[UCG], gave faster reaction but no change within 8% in the observed extent of reaction, indicating that substrates and products were subsaturating. The same value of Kext, within 15%, was obtained from reactions initiated with *P.
Measurement of RNA Duplex Stabilities
Melting temperatures were measured using a Varian Cary 1 UV-VIS spectrophotometer equipped with a thermo programmer. RNA samples at various concentrations were prepared in 1 M NaCl, 50 mM sodium phosphate buffer, and 10 mM EDTA at pH 7.0 and were transferred to a quartz cuvette and covered with a Teflon-cap. To anneal duplexes samples were heated to 95 °C and then slowly cooled to 10 °C. Duplex stability was measured subsequently by heating the sample at a rate of 1 °C/min while recording the absorbance at 260 nm every 1 °C. MeltWin v. 3.0 (61) was used to fit the resulting absorbance data in order to obtain the melting temperatures of each sample. The thermodynamic constants ΔH and ΔS were determined by linear fitting of the reciprocal of the melting temperature (Tm) against the logarithm of concentrations (C) using Eq. 6A. The value of ΔG at 30 °C was calculated from ΔH and ΔS using Eq. 6B.
| (Eq. 6A) |
| (Eq. 6B) |
Δ ΔG values represent the stabilization of an A•U and a G•C pair, respectively, relative to a G•U pair and were calculated by subtracting the stability of the latter from the stability of the former (Eq. 6C).
| (Eq. 6C) |
Results and Discussion
Several ribozymes have been constructed to learn more about group I intron self-splicing (1, 36–39, 62). The most-studied construct, the L-21 ScaI ribozyme, lacks 21 nucleotides at the 5′-end of the intron and five nucleotides at the 3′-end (corresponding to a ScaI restriction site in the encoding plasmid, Figure 1A&B, (62)). Previous investigations added back the 3′-sequence to assess the effect and importance of the P9.0 helix and the 3′-terminal guanosine residue (36, 37). Here we take an analogous stepwise approach, adding five 5′-terminal nucleotides to give the L-16 ScaI ribozyme. This ribozyme allows formation of the P1 extension, base pairing that is analogous to base pairing between the 5′-most portion of the intron and the extended IGS (Figure 1A&C, see below for a description of the nomenclature). This construct also allows formation of P10, a helix that forms in the second step of self-splicing between the extended IGS and the 3′-exon (Figure 1A&C).
We have used pre-steady-state kinetics to first dissect the contributions arising from the presence of the IGS extension and then from formation of base pairs with this extended sequence. This dissection is accomplished by comparing kinetic and thermodynamic reaction frameworks for the L-16 ScaI ribozyme reaction with appropriate substrates to that of the L-21 ScaI ribozyme reaction (Figure 2). The second section of the Results and Discussion addresses the effects from the presence or absence of a base pair immediately 3′ to the cleavage site, as this difference reflects the scenario in the first and second chemical step of self-splicing, respectively (Figure 1A&C).
Nomenclature
Compared to the L-21 ScaI ribozyme the L-16 ScaI ribozyme has a five-nucleotide 5′-extension. Because this sequence extends the IGS (internal guide sequence) it is called the IGS extension herein (Figure 1D). Together the IGS and the IGS extension (IGSext) are called the extended IGS. While binding of S (or P) to the L-21 ScaI results in formation of P1 only (blue in Figure 1D), the IGS extension in the L-16 ScaI ribozyme allows formation of another helix, termed the P1 extension (P1ext) herein (red in Figure 1D). By analogy to the extended IGS above, the P1 and P1 extension helices together are called the extended P1 duplex.
Kinetic and Thermodynamic Frameworks for the L-16 ScaI Ribozyme Reaction
We first describe practical considerations for obtaining the kinetic and thermodynamic frameworks. The details of the pre-steady-state kinetic methods employed are given in the Materials and Methods and follow directly from previous studies (1, 5, 6, 23, 35, 36). We then present and compare the frameworks. The rate and equilibrium constants are defined in Figure 2A and the data presented below are summarized in Figure 2B-D.
Equilibrium binding constants for UCG, UCGA and UCGAAACC were determined from the concentration dependences of the single turnover reaction rate constants as described in the Materials and Methods. The affinity for S and P could not be determined directly by varying ribozyme concentration. This is because these oligonucleotides bind very strongly to the ribozyme (Table 1 and Figure 2) and nonspecific losses to the tube walls occur at very low ribozyme concentrations. Instead, we measured association and dissociation rate constants and calculated affinity constants from these (5, 26, 36). Because the dissociation rate constant of the fully base paired 5′-splice site analog CCCUCdTAAACC is too slow to be measured accurately at 30 °C we instead used 5′-splice site analogs that lacked the 5′-terminal C residue (-6CΔ; Chart 1). Previous work has suggested that this residue contributes to binding solely via base pairing interactions (50) and is therefore not expected to affect the comparisons herein. This conclusion is supported by comparisons at 50 °C with full-length 5′-splice site analogs (i.e., containing residue –6C), which gave similar effects to those obtained with –6CΔ 5′-splice site analogs at 30 °C (Table S1 in the Supplementary Materials).3
Effects from Extending the IGS on Individual Steps in the Ribozyme Reaction
Effects from lengthening the IGS without formation of any new base pairing interactions have been dissected through comparison of the framework for reaction of CCUCdTA with the L-21 ScaI ribozyme and the L-16 Sca I ribozyme (Figure 2B&C, Scheme 1A, see also: (5)). The residue A(+1) of CCUCdTA can in principle form a base pair with the L-16 ScaI ribozyme, but results described below suggest that this base pair does not form. Reactions with this oligonucleotide can therefore be used to determine the effects on the reaction kinetics and thermodynamics that arise from the IGS extension that is present in the L-16 ScaI ribozyme but absent in the L-21 ScaI ribozyme (Scheme 1A).
Scheme 1.

Binding of the 5′-Exon Analog P Is Strengthened by the IGS Extension
Binding of S to free ribozyme is unaffected or only slightly affected by the IGS extension ( and 10 nM for binding of CCUCdTA to the L-16 or the L-21 ScaI ribozyme, respectively, Figures 2B&C). CCUCdTMe and CCUCdTA bound to the L-16 ScaI ribozyme with the same affinity, within error ( and 19 nM, Table 1). This similarity suggested that the base pair between A(+1) and U(21) in the IGS extension (Scheme 1) is not formed; CCUCdTA was used subsequently to dissect effects that arise from the presence of the IGS extension without formation of the P1 extension.
The 5′-exon analog (P) binds about 40-fold stronger to the free L-16 ScaI ribozyme than to the L-21 ScaI ribozyme ( and 600 pM, respectively, Figures 2B&C). This large additional energetic contribution to binding of P was surprising, especially as there was essentially no change in binding of S. Previous work with the L-21 ScaI ribozyme has shown that binding of P is stronger than binding of S, and a metal ion that coordinates the 3′-OH of P has been suggested to be responsible for this effect (5, 25, 63). It is possible that this metal ion interaction is further optimized on the L-16 ScaI ribozyme. Alternatively, the IGS extension could be forming fortuitous interactions with the core when P is bound. To account for the effect on binding of P but not S, these fortuitous interactions would have to be sensitive to the presence of the reactive phosphoryl group, such that they would be prevented from forming with the phosphoryl group present. Regardless, these results underscore the high sensitivity of RNA interactions to local variations and rearrangements.
Binding of G Is Similar With and Without the IGS Extension
Binding of G to the L-16 ScaI and L-21 ScaI ribozymes with substrate bound in the open complex is similar ( and 802 ± 67 μM for binding to L-16 ScaIopen and L-21 ScaIopen, respectively; Table 2). With bound S the binding of G to the L-16 ScaI ribozyme is about two-fold stronger than binding to the L-21 ScaI ribozyme (2.1- and 2.2-fold stronger binding to the L-16 ScaI ribozyme with CCUCdTA and CCUCdTAAACC bound, respectively; Table 2).
Table 2.
G and UCG Binding to the L-21 and the L-16 ScaI Ribozymes
| Kd (μM)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| E•Sopen |
E•SA
|
E•SA3C2 |
|||||||
| L-21 | L-16 | Ratio | L-21 | L-16 | Ratio | L-21 | L-16 | Ratio | |
| G | 930 ± 54 | 802 ± 67 | 1.1 | 124 ± 4 | 59 ± 9 | 2.1 | 76 ± 16 | 35 ± 3 | 2.2 |
| UCG | 33 ± 2 | 31 ± 7 | 1.1 | 5 ± 2 | 7 ± 1 | 0.7 | 28 ± 7 | 11 ± 2 | 2.5 |
| ratio | 28 | 26 | 25 | 8 | 2.7 | 3.2 | |||
All experiments were performed at 30 °C, pH 7.2 and in the presence of 10 mM MgCl2. Comparisons between L-21 ScaI and L-16 ScaI ribozymes were performed side-by-side and repeated two or more times. No significant difference was detected for binding of UCG to E•Sopen with –1d,-3m,rSA or –1r,dSA5 bound ( and 85 μM, respectively).
Formation of P9.0
The P9.0 duplex is formed between the residues directly 5′ of the 3′-terminal G residue of the intron and single stranded residues of the intron adjacent to the G-binding site. Comparison of the affinity of UCG, which can form P9.0, and G for the L-16 ScaI ribozyme with CCCUCdTA bound in the open complex shows that UCG binding is stronger than G binding, suggesting that P9.0 is formed on the L-16 ScaI ribozyme akin to its formation on the L-21 ScaI ribozyme (47, 64) ( and for binding to L-21 ScaI ribozyme and and for binding to L-16 ScaI ribozyme each with –1d,rSA bound in the open complex; Table 2). Furthermore, the energetic contribution from formation of P9.0 is similar for the L-21 ScaI and the L-16 ScaI ribozymes (28- and 26-fold stronger binding to L-21 ScaI and L-16 ScaI ribozyme, respectively; Table 2).
Previous work has shown that interference between P9.0 and the residue at position A(+2) of the 3′-tail of the 5′-splice site analog weakens binding of UCG ((47) and Table 2). The observation that the contribution from P9.0 is also reduced when the longer substrates are used on the L-16 ScaI ribozyme suggests that formation of a duplex with the +2 residue does not eliminate the interference (only 2.7- and 3.2-fold stronger binding of UCG vs. G to E•CCUCdTAAACC with the L-21 ScaI and the L-16 ScaI ribozymes, respectively; Table 2). Furthermore, the reduced contribution from P9.0 on the L-16 ScaI ribozyme with CCUCdTA bound suggests that an analogous interference with P9.0 can arise from the IGS extension, which includes a residue complementary to the +2 residue (25- and 8-fold stronger binding of UCG vs. G to the L-21 ScaI and the L-16 ScaI ribozymes, respectively, both with CCUCdTA bound in the closed complex; Table 2).
In the recent structure of the Azoarcus intron with P10 formed the distance between P9.0 and P10 (which contains both the IGS extension and the substrate-tail) exceeds 8 Å, too far to explain steric interference via a direct effect (65). It is possible that these distances are different in the Tetrahymena and Azoarcus RNAs or in different conformations of the ribozyme. Alternatively, the interference may not be direct but mediated by other parts of the ribozyme. Residues of P9 come within less than 4 Å of P9.0, making it the closest neighbor of P9.0 and thereby a candidate for mediating indirect interactions.
Binding of UCGA
Above we have shown that CCUCdTA can be used to isolate effects from the IGS extension only, as the A(+1) residues does not base pair with U21 of the extended IGS. Analogously, UCGA can be used to dissect the effects from the IGS extension on the reverse reaction if the corresponding base pair with U21 is not formed. To investigate this potential effect we compared the contribution of the base pairs with the IGS extension when formed with CCUCdTAAACC or UCGAAACC relative to CCUCdTA or UCGA, respectively. If the base pair between the IGS extension and UCGA were formed, the increase in stability for UCGAAACC relative to UCGA would be expected to be larger than the increase in stability of CCUCdTAAACC relative to CCUCdTA. This is because for CCUCdTAAACC part of the energy from formation of the base pairs in the P1 extension must pay for an energetic penalty to allow formation of the first A•U pair (see below). If this A•U pair were preformed in UCGA, formation of the additional base pairs in the P1 extension would not incur this energetic penalty for helix initiation and thus would be predicted to provide additional stabilization. However, the increased affinity for CCUCdTAAACC relative to CCUCdTA is the same, within error, as the increased affinity for UCGAAACC relative to UCGA (Table 3). These results, although indirect, suggest that the 3′-A of UCGA does not form its potential base pair with the IGS extension. UCGA was therefore used to probe the reverse of the L-16 ScaI ribozyme reaction without base pairs to the IGS extension formed.4
Table 3.
Thermodynamic Contribution from the P1 Extension
| Oligonucleotide | Kd (μM) | Δ ΔG (kcal/mol) |
|---|---|---|
| CCUCdTA | 3.0•10−3 | |
| CCUCdTAAACC | 0.4•10−6 a | − 5.4 |
| UCGA | 1050 | |
| UCGAAACC | 0.11 | − 5.5 |
Dissociation constants from ternary complexes E•S•G and E•P•UCGA(AACC) were obtained at 30 °C. S and P had 2′-deoxyribose substitutions at position -1.
Calculated from the dissociation rate constant of S from E•S and the observation of 6-fold cooperativity of S and G binding with this oligonucleotide substrate (Table 2). Direct measurement was not possible because dissociation was slower than reaction to form P.
UCGA binds to the L-16 ScaI ribozyme with P bound in the closed complex with a dissociation constant of 1050 μM, 12-fold weaker than binding to the L-21 ScaI ribozyme (Figure 2B and C). This destabilization could arise from interference, direct or indirect, between P9.0 and the IGS extension, as suggested previously and above for binding of UCG (47). With the L-21 ScaI ribozyme UCGAAAAA binding is three-fold weaker than UCGA binding ( and for binding to E•P, data not shown), consistent with an interfering effect with the extended IGS. Alternatively, it is possible that the weaker binding of UCGA to the L-16 ScaI ribozyme is due to an increased contribution from an unfavorable interaction with the reactive phosphoryl group that was previously suggested for the L-21 ScaI ribozyme 5 (5).
Effects of Formation of the Extended P1 Duplex on Individual Steps in the Ribozyme Reaction
In the sections above we have shown that extending the IGS strengthens binding of G and P about two- and 40-fold, respectively, while binding of UCGA is weakened 12-fold. We next present results for the L-16 ScaI ribozyme with CCUCdTAAACC bound (Figure 2D). These data suggest that the P1 extension is formed but with a decreased stability relative to a continuously stacked duplex. Furthermore, formation of the P1 extension has no significant effect on the kinetics or equilibria of subsequent steps.
Effects from the formation of the P1 extension were isolated through comparison of the frameworks of the L-16 ScaI ribozyme reactions with CCUCUdTA and CCUCdTAAACC bound (Scheme 1B). This was possible as the IGS extension is present in both cases but only CCUCdTAAACC, not CCUCdTA, forms the base pairs of the P1 extension.
Formation of the Extended P1 Duplex
To test formation of the base pairs with the IGS extension, we compared the affinity of a series of 5′-splice site analogs (Table 1). The 104-fold stronger binding of CCUCdTAAACC (nucleotides with complementarity to the IGS extension of the L-16 ScaI ribozyme are underlined in this section) relative to CCUCdTMe suggests that at least some of the potential base pairs are formed. We next compared the affinities of 5′-splice site analogs that could only form subsets of the additional P1 base pairs. The 600-fold stronger binding of CCUCdTAAACC compared to CCUCUdTAAAAA suggests that the 3′-terminal C•G base pairs are formed. The stronger affinity of CCUCUdTAAAAA than CCUCdTMe suggests that at least some of the three A•U base pairs are formed. Nevertheless, addition of these potential three base pairs only gave a 10-fold increase in affinity, whereas an effect of ~1000-fold, ~100-fold more, would be expected for this addition to a Watson-Crick duplex (9). This difference is consistent with the absence of increased binding with a single A(+1) relative to a methyl group in this position as discussed above. To test whether the A(+1) formed a base pair in the longer duplex we changed the base pairing at this position from a A•U to a U•U, using CCUCdTUAACC. This mismatched oligonucleotide base pair resembles the ligated exon product of the second step of self-splicing (Scheme 2).
Scheme 2.
Changing the +1A to a +1U in the context of the base-paired oligonucleotide (CCUCdTAAACC and CCUCdTUAACC) decreases the affinity for the oligonucleotide 100-fold (Δ ΔG = 2.4 kcal/mol6, Table 4). These data suggest that in the context of a duplex the +1 A•U base pair is formed. In contrast, a single A•U pair at that position is not formed, as described above. These observations suggest that the G•U wobble pair that precedes the A•U pair does not behave as a Watson-Crick base pair for which formation of a subsequent A•U pair would be favored.
Table 4.
Binding of the +1 U•U Mismatched Substrate
| CCUCdT- | Δ ΔGbp (kcal/mol)
|
kc (min−1) | Ktert | |
|---|---|---|---|---|
| −G | +G | |||
| -AAACC | −5.4a | −5.4 | 0.039 | 23 |
| -UAACC | −3.9 | −3.0 | 0.041 | 230 |
All experiments were performed at 30 °C and pH 7.2. All substrates contained a 2′-deoxyribose substitution at the cleavage site (position –1) to prevent cleavage. Ktert values were obtained by comparison of docked and undocked substrates as described in the Materials and Methods, with oligonucleotide substrates undocked by the introduction of a 2′-methoxyribose substitution at position –3 (26, 35, 55). Δ ΔGbp values were obtained by comparison of the affinity of the CCUCdTAAACC or CCUCdTUAACC with CCUCdTA.
Value from Table 1 included for comparison.
The observation that the G•U wobble pair at the cleavage site differs from Watson-Crick base pairs is consistent with observations from structural work on model duplexes containing a G•U wobble pair. This work shows that stacking of the G•U wobble pair with the 3′-helix (relative to the G, in this case the P1 duplex) is continuous, while stacking with the 5′-helix (relative to G, in this case the P1 extension) is altered and involves only the G and the opposite strand of the helix (the substrate “3′-tail”), with the U and its opposite strand (the IGS extension) remaining unstacked on the 5′-side ((66–69), see also: (70)).
To evaluate the ribozyme results, we compared the destabilization arising from an internal G•U pair (relative to a G•C or a A•U pair) to that of a terminal G•U pair in model duplexes (Table 5). The destabilization was calculated by comparing the stabilities of model duplexes with internal and terminal G•U, A•U and G•C pairs. Duplex stabilities were measured in thermal denaturation experiments as described in the Materials and Methods. The destabilization from an internal G•U pair relative to a A•U or a G•C pair is 0.7 and 1.2 kcal/mol, respectively, while a terminal G•U is stabilized relative to an A•U by 0.4 kcal/mol and destabilized relative to a G•C by only 0.6 kcal/mol (Table 5). This suggests that there is an added destabilizing effect from internally located G•U pairs that amounts to 0.6–1.1 kcal/mol (Δ Δ ΔGAU = 1.1 kcal/mol and Δ Δ ΔGGC = 0.6 kcal/mol). Thus, this energetic analysis suggests that internal G•U wobble pairs behave differently from terminal G•U pairs, in that they have an additional detrimental effect that is not explained simply by the change in base pairing. These results are consistent with the structural studies of duplexes with internal G•U pairs described above ((66–69), see also: (70)).
Table 5.
Effect from Introduction of a G•U Pair in Model Duplexes
| Internal
|
Terminal
|
|||||
|---|---|---|---|---|---|---|
| GU | AU | GC | GU | AU | GC | |
| ΔG (kcal/mol) | −14.6 | −15.3 | −15.8 | −9.6 | −9.2 | −10.2 |
| Δ ΔG (kcal/mol) | (0) | −0.7 | −1.2 | (0) | +0.4 | −0.6 |
Duplex stabilities ΔG were measured in thermal denaturation experiments in 1 M NaCl, 10 mM EDTA and 50 mM sodium phosphate, pH 7.0. The sequence of the duplexes was as follows (in the 5′ to 3′ direction, with the base pair being varied underlined): GCCUCUAAAC/GUUUGGAGGC, GCCUCUAAAC/GUUUAGAGGC and GCCUCCAAAC/GUUUGGAGGC for duplexes containing internal GU, AU and GC pairs and GCCUCU/GGAGGC, GCCUCU/AGAGGC and GCCUCC/GGAGGC for duplexes with terminal GU, AU and GC pairs. Δ ΔG values represent stabilities for AU and GC pairs relative to GU pairs. Δ Δ ΔG values represent the additional destabilization of internal GU pairs relative to terminal GU pairs and are calculated by subtracting the Δ ΔG values for terminal GU pairs from the Δ ΔG values for internal GU pairs.
The data on model duplexes show that internal G•U duplexes are intrinsically destabilized. This intrinsic destabilization appears to be exacerbated when the extended P1 duplex is bound to the L-16 ribozyme, where the destabilization amounts to >2.8 kcal/mol7 instead of 0.6–1.1 kcal/mol. This difference suggests that the ribozyme may make use of features in the duplex architecture to further destabilize the duplex with an internal G•U wobble pair demarcating the cleavage site G•U.
Consistent with these biochemical and structural results on model duplexes and the extended P1 duplex in the Tetrahymena ribozyme the recent X-ray crystal structures of the group I intron from Azoarcus also shows disrupted helical stacking at the G•U wobble pair (65, 71). The potential importance of partial unstacking of the extended P1 duplex for self-splicing is discussed in the Summary and Implications section.
Accommodation of the Chemical Transition State Within a Helix
In the L-21 ScaI ribozyme the cleavage site is located at the end of a helical segment, whereas for self-splicing and for the L-16 ScaI ribozyme with CCUCdTAAACC bound, the cleavage site is embedded in a duplex (Figure 1). This constraint within a duplex could increase or decrease the rate of the chemical step if the helical geometry were preferred or not accommodated in the transition state, respectively. We therefore compared the rate constants for the forward and reverse chemical steps (k5 and k−5) with a fully base paired substrate (CCUCUdTAAACC) and a substrate that does not form a base pair in the P1 extension (CCUCdTA). Log-linear pH dependences with a slope of one between pH 6.2 and 8.1 suggest that the chemical step is rate limiting (59, 60) in these experiments (data not shown). The rate constants for reaction of the base paired and non-base paired substrates are similar (k5 = 0.04 and 0.14 min−1 and k−5 = 0.04 and 0.07 min−1 for CCUCdTAAACC and CCUCdTA, respectively, Figure 2C&D).
The similar rate constants suggest that the constraint on the helical conformation by the extended P1 duplex has at most a small effect on adoption of the transition state structure. This effect could be small because the transition state conformation can be reached within the helix or because the extended P1 duplex is not highly constrained due to the limited stacking following the G•U wobble pair at the cleavage site.
Cooperativity in Binding of Reactants
Previous work with the L-21 ScaI ribozyme showed that binding of S strengthened binding of G (or UCG) and vice versa (5, 23, 28). This thermodynamic coupling of substrates is also observed for the L-16 ScaI ribozyme with CCUCdTA or CCUCdTAAACC bound (Figure 2C&D). The ~10-fold coupling effect with the L-16 ScaI is similar to the 6-fold effect with the L-21 ScaI ribozyme ((5) and Figure 2B-D). Thus, neither the IGS extension nor the base pairs formed with the IGS extension to give the extended P1 duplex substantially affect the thermodynamic coupling of substrates.
Binding of the 5′-exon analog (P) also strengthens binding of UCGA to the L-21 ScaI ribozyme ~5-fold ((5) and Figure 2B). In the presence of the extended IGS alone weak binding of UCGA prevented determination of the extent of product coupling. When the P1 extension is formed cooperative binding of products is maintained with a coupling effect of at least 6-fold (Figure 2D).
No Significant Contribution to 5′-Splice Site Analog (S) Binding From Tertiary Interactions with the P1 Extension
A large body of previous work has shown that binding of S or P to the ribozyme occurs in two steps: formation of the P1 duplex and subsequent docking of this duplex into a binding site at the active site (1, 8, 52, 53). In the “docked” state specific tertiary interactions between the ribozyme and the P1 duplex contribute to binding of S and P (1, 7, 54, 72–74). The strength of these tertiary interactions is measured as Ktert, the equilibrium constant for binding in tertiary interactions. To determine whether there were additional tertiary interactions from the P1 extension, we compared the Ktert for the L-21 ScaI and the L-16 ScaI ribozymes.
Single-molecule FRET measurements at 22 °C have shown that Ktert for the L-21 ScaI and the L-16 ScaI (with all base pairs in the P1 extension formed) are similar with values of 22 ± 5 and 32 ± 4, respectively (55). These data are consistent with bulk measurements using modified substrate analogs at 30 °C (Ktert = 15 and 23 for binding to the L-21 ScaI and the L-16 ScaI ribozyme, respectively, data not shown). The simplest interpretation from these data is that there are no significant interactions between the P1 extension and the ribozyme. The recent crystal structure of the Azoarcus intron suggests the formation of a single hydrogen bond between the 2′-OH group of the last residue in J4/5 (G116 in Tetrahymena) and the 2′-OH group of the first residue of the IGS extension (G21 in Tetrahymena) (65). A previous measurement of the effect of removing tertiary interactions involving 2′-OH groups (by modification to 2′-H) gave effects of about 0.5 kcal/substitution, corresponding to an effect of about two-fold (75). Such a small effect is consistent with the similar tertiary stabilization observed for ribozymes with and without extended P1 duplexes.
Probing the Effect of a U•U Mismatch After the Cleavage Site
As described above, there is a change in base pairing between the first and second chemical steps in self-splicing (Figure 3 and Scheme 2) that originates from the replacement of the 5′-end of the intron with the 3′-exon. These segments differ in the identity of the +1 residue, A or U, resulting in formation of an A•U or a U•U base pair in the first and second step, respectively. Previous work with the L-21 ScaI ribozyme has shown that a U(+1)-containing substrate weakened G binding (76). These results were surprising and could have arisen from fortuitous, non-productive interactions of the ligated exon analog that interfered with catalysis. Alternatively, it was possible that a previously unrecognized mechanism used to render self-splicing irreversible had been uncovered. To distinguish between these possibilities we further investigated the effect from the U(+1) substitution using the L-16 ScaI ribozyme.
Figure 3.

Cartoon model of the self-splicing reaction. The P1 helix is shown in blue and the P1 extension and P10 are shown in red. In the first chemical step of self-splicing (transition from I to II) an exogenous G (shown in orange) attacks at the 5′-splice site. Note that in II the extended P1 duplex is nicked. In a subsequent conformational step (transition from II to III), the G, now covalently linked to the P1 extension, leaves the G binding site. Next, G414, the last nucleotide of the intron (shown in cyan), enters the G binding site and the 3′-exon is aligned in P10 for ligation (III to IV). In the second chemical step (IV to V) exons are ligated to be released in a last step (V to VI).
Comparison of the contribution of tertiary interactions towards binding of CCUCdTUAACC and CCUCdTAAACC shows that in the absence of G the U(+1) containing substrate docks more strongly into the active site than the A(+1) containing substrate (Ktert = 230 and 23 for CCUCdTUAACC and CCUCdTAAACC, respectively, Table 4) providing evidence for tertiary interactions with U(+1). Further, the tertiary binding energy of CCUCdTUAACC is 4.0 kcal/mol in the absence of bound G, 1 kcal/mol more than observed in the presence of bound G (Table 4). The stronger binding energy without G present suggests that G binding disrupts the tertiary interactions with CCUCdTUAACC.
Comparison of the rate of reaction from the L-16 ScaI E•S•G complex with U(+1) and A(+1) containing substrates shows that both reactions proceed with the same rate constant (kc = 0.039 and 0.041 min−1 for reaction of CCUCdTAAACC and CCUCdTUAACC, respectively, Table 4). This observation suggests that the presence of guanosine restores interactions that are important for catalysis, supporting the notion that the effect from the U(+1) is fortuitous and non-productive.
The previous results and the analysis of the +1U effect suggest that the interactions of the U(+1) containing substrate with the L-21 ScaI are fortuitous, providing another example of RNA’s conformational promiscuity (e.g., (30, 55, 77, 78). The promiscuous interaction can be overcome by occupying other portions of the active site in their cognate interactions with G and by immersing the U•U pair within a duplex, effects that have implications for understanding the origins of tertiary structure specificity in RNA (58).
Summary and Implications
Summary of Effects from the IGS Extension and the P1 Extension
A stepwise approach was taken to dissect the contribution from individual elements within the group I intron towards the self-splicing reaction. Starting from the well-characterized L-21 ScaI ribozyme, which lacks 21 nucleotides at the 5′-terminus and five nucleotides at the 3′-terminus, we added back sequence elements at the 5′-end and determined their effect on individual steps of the ribozyme reaction. Figure 4 summarizes effects from the IGS extension (dark columns) and from formation of the P1 extension (light columns). These effects are summarized briefly below.
Figure 4.

Summary of effects on equilibrium constants from IGS extension (dark columns) and the formation of the P1 extension (light columns) on the ribozyme reaction. Equilibrium constants are defined in Figure 2A. refers to binding of UCGA (dark columns) or UCGAAACC (light columns) in the presence of bound P. Similarly, refers to binding of –1d,rSA (dark columns) or –1d,rSA3C2 (light bars) in the presence of bound UCG. All values are relative to the L-21 ScaI reaction and were calculated from Figure 2. Effects from the IGS extension were determined from comparison of reactions with the L-16 ScaI ribozyme that has CCUCdTA bound. The combined effects from the IGS extension and formation of base pairs with that extension were determined from reactions of the L-16 ScaI ribozyme with CCUCdTAAACC bound.
The presence of the IGS extension (dark columns in Figure 4) strengthens binding of G and P and weakens binding of UCGA (second and third set of columns in Figure 4, respectively). Further addition of the base pairs with the IGS extension (light columns in Figure 4) only affects reaction steps in which the base pairs directly tether the ligand to the ribozyme (first and fourth set of columns in Figure 4). These data suggest that the IGS extension modulates kinetics and thermodynamics in self-splicing beyond its role in positioning the 3′-exon for ligation in the second step of self-splicing. P bound to the L-16 ScaI ribozyme 40-fold stronger than to the L-21 ScaI ribozyme (Figure 2B&C). This large effect from the IGS extension was especially surprising as binding of the 5′-splice site analog lacking the ability to form the P1 extension was essentially unaffected.
Lastly, the effects from the IGS extension on the product side of the framework are larger than the effects on the substrate side, with 2–3-fold stronger binding of (UC)G and two-fold weaker binding of S, compared to 40-fold stronger binding of P and 12-fold weaker binding of UCGA (Figure 2B&C). This asymmetry suggests that the communication between the ribozyme extension and the active site is different when S and UCG or P and UCA are bound. The origin of these differences is not understood.
The Role of the G•U Pair in Self-splicing
The P1 extension is a dynamic secondary structure element. During the first step of self-splicing, three A•U pairs are formed in the P1 extension. These base pairs have to be broken to allow positioning of the 3′-splice site, which occurs through base pairing of the 3′-exon with the IGS extension to form the P10 helix (Figure 3, (40–43)). A recent mutational analysis showed that a modest stabilization of the P1 extension, which is expected to slow dissociation of this duplex, results in splicing defects (79). It thus appears possible that disruption of the three A•U pairs could be rate-limiting or nearly rate-limiting for the first step of self-splicing. These considerations lead to the questions: How are the base pairs in the P1 extension preferentially broken while those in P1 remain intact to prevent dissociation of the 5′-exon before the second step of self-splicing? And how is rapid dissociation of the ligated exons, the final product, ensured?
As noted above, previous structural data with model duplexes suggested that stacking is disrupted by an internal G•U wobble pair, so that stacking between the P1 and the P1 extension helices might not be expected to be continuous (66–68, 70). This structural picture is supported by the recent x-ray structures of the group I intron (65, 71). Furthermore, this structural picture of internal G•U pairs is reflected in their energetics; substitution of a Watson Crick pair with a G•U pair in a model duplex has a larger effect within a helix than at the end (see Formation of the Extended P1 Duplex above) and an even larger effect of >2.8 kcal/mol measured on the ribozyme.
The discontinuity in stacking between P1 and the P1 extension presumably causes the observed loss in cooperativity of base pair formation at the junction of these helices such that a lone base pair beyond the G•U pair is not formed. In self-splicing this interruption of cooperativity might facilitate release of the ligated exons after the second step of self-splicing. The discontinuity in stacking could also facilitate exchange of the P1 extension with the 3′-exon after the first chemical step in splicing (Figure 3, II-IV), while holding onto the 5′-exon in P1 for exon ligation. For exchange of the P1 extension with P10 to be facilitated by the G•U pair, it is required that the G•U pair exerts its destabilizing effect even in the case of a nicked duplex (as found after the first chemical step of self-splicing (Figure 3, II). While it is possible that the disruptive effect from the G•U wobble pair is dependent on a covalent linkage, and would then disappear with a nick, the data in Table 3 show that the contribution from the P1 extension is independent of covalent linkage to P1, suggesting that the G•U pair also disrupts the noncovalently linked, nicked helix. This notion is also consistent with the crystal structures on the group I intron in the intermediate form (65, 71). Thus, the G•U pair may serve a previously unappreciated role in self-splicing. Besides helping to define the position of the cleavage site through tertiary interactions (2, 73), the G•U wobble pair uncouples the stability of the P1 and P1 extension helix, allowing coordination of conformational transitions needed to accomplish the two transesterification steps in self-splicing.
Proline is an amino acid that disrupts α-helices or β-sheets in proteins. This disruption arises because proline does not have a free backbone amide for hydrogen bonding with carbonyl groups within helices or sheets. We suggest that G•U wobble pairs in RNA molecules can act analogously. Internal G•U wobble pairs reduce the stacking interactions between two helical segments, thereby reducing or removing the cooperativity between these segments so that they become quasi-independent, rigid modules.
Supplementary Material
Footnotes
This work was supported by NIH grant GM49243. K.K. was supported in part by a predoctoral fellowship from the Boehringer Ingelheim Fonds.
Abbreviations: S refers to the 5′-splice site analog without specifying the identity of the 2′-substituents, which can be a hydrogen atom or a hydroxyl or methoxy group, and without specifying the length of the ‘tail’ 3′ of the cleavage site (see Chart 1 for the specific oligonucleotides used); P refers to the 5′-exon analog (see Chart 1) without specifying the identity of 2′-substituents as for S; *S, radioactively labeled S; *P, radioactively labeled P; E, L-16 ScaI or L-21 ScaI ribozyme; PCR, polymerase chain reaction; HPLC, high pressure liquid chromatography; IGS, internal guide sequence, sequence of the ribozyme that is complementary to the 5′-exon (see Figure 1D); Me, methyl group (CH3); HEPES, 4-(2-hydroxy-ethyl)-piperazine-1-ethane-sulfonic acid; MES, 2-(N-morpholino)-ethane-sulfonic acid; MOPS, 3-(N-morpholino)-propanesulfonic acid; EDTA, ethylenediaminetetraacetic acid.
The guanosine residue that binds in the guanosine binding site is underlined throughout for clarity.
The 5′-splice site analogs used herein and in previous work (e.g. (1–5)) also replace a G•U wobble pair five base pairs upstream of the cleavage site with a G•C pair. It appears that this wobble pair contributes to tertiary binding of the P1 helix (6–8).
If an A•U base pair were formed, the observed 12-fold weaker binding of UCGA due to the IGS extension would be an underestimate.
The IGS extension adds five nucleotides to give the L-16 ScaI ribozyme. Therefore, the 5′-terminal triphosphate group, which is present from transcription, is farther from the active site. The proximity of the triphosphate to the cleavage site, its large negative charge, and its capacity to bind a divalent metal ion, raised the possibility that the differences between the L-16 and the L-21 ScaI ribozyme originated from displacement of the triphosphate group rather than the added nucleotides. To test this possibility, we prepared L-21 ScaI ribozyme with a 5′-terminal hydroxyl group (L-21 ScaIOH) by transcription with a 10-fold excess of G over GTP. The presence of predominantly (≥85%) 5′-OH-containing ribozyme was confirmed via incorporation of 32P-ATP at the 5′-end without preincubation with phosphatase to remove the 5′-phosphate (data not shown). Binding of G and P to L-21 ScaIOH and to L-21 ScaI was tested as these equilibria showed the largest difference between L-21 ScaI and the L-16 ScaI ribozyme. The dissociation constants for G and for P from L-21 ScaIOH and L-21 ScaI were the same within error ( and 93 μM, and 600 pM for binding to L-21 ScaIOH and L-21 ScaI, respectively, Figure S1A and Figure S1B) and differed significantly from those for the L-16 ScaI ribozyme ( , Figure 2C and Table 2), suggesting that the presence of the additional 5′-nucleotides rather than the absence of the nearby triphosphate group is responsible for the observed effects.
Because in the absence of G the +1U-containing substrate makes non-productive tertiary interactions that contribute ~10-fold to its affinity, the affinities of CCUCdTUAACC and CCUCdTAAACC are compared in the presence of G, which disrupts these alternative interactions (see Probing the Effect of a U•U Mismatch After the Cleavage Site). This analysis isolates the contribution of the A•U base pair to stability of the reactive complex.
This value is calculated from the observed 10-fold stronger binding to the L-16 ScaI ribozyme of CCUCdTAAAAA relative to CCUCdTMe compared to the >1000-fold stronger binding calculated from nearest neighbor rules for extended P1 duplexes with an A•U or the >4000-fold stronger binding calculated for extended P1 duplexes with an G•C pair replacing the cleavage site G•U pair (9). Comparison of model duplex data to nearest neighbor rules suggest that nearest neighbor estimates are accurate for these specific sequences (Table 5 and (9–11)).
References
- 1.Herschlag D, Cech TR. Catalysis of RNA Cleavage By the Tetrahymena-Thermophila Ribozyme. 1 Kinetic Description of the Reaction of an RNA Substrate Complementary to the Active-Site. Biochemistry. 1990;29:10159–10171. doi: 10.1021/bi00496a003. [DOI] [PubMed] [Google Scholar]
- 2.Knitt DS, Narlikar GJ, Herschlag D. Dissection of the Role of the Conserved G-Center-Dot-U Pair in Group-I RNA Self-Splicing. Biochemistry. 1994;33:13864–13879. doi: 10.1021/bi00250a041. [DOI] [PubMed] [Google Scholar]
- 3.Narlikar GJ, Herschlag D. Direct Demonstration of the Catalytic Role of Binding Interactions in an Enzymatic-Reaction. Biochemistry. 1998;37:9902–9911. doi: 10.1021/bi980495t. [DOI] [PubMed] [Google Scholar]
- 4.Shan S, Yoshida A, Sun SG, Piccirilli JA, Herschlag D. Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc Natl Acad Sci U S A. 1999;96:12299–12304. doi: 10.1073/pnas.96.22.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karbstein K, Carroll KS, Herschlag D. Probing the Tetrahymena Group I Reaction in Both Directions. Biochemistry. 2002;41:11171–11183. doi: 10.1021/bi0202631. [DOI] [PubMed] [Google Scholar]
- 6.Herschlag D, Cech TR. Catalysis of RNA Cleavage By the Tetrahymena-Thermophila Ribozyme. 2 Kinetic Description of the Reaction of an RNA Substrate That Forms a Mismatch At the Active-Site. Biochemistry. 1990;29:10172–10180. doi: 10.1021/bi00496a004. [DOI] [PubMed] [Google Scholar]
- 7.Narlikar GJ, Khosla M, Usman N, Herschlag D. Quantitating tertiary binding energies of 2′ OH groups on the P1 duplex of the Tetrahymena ribozyme: Intrinsic binding energy in an RNA enzyme. Biochemistry. 1997;36:2465–2477. doi: 10.1021/bi9610820. [DOI] [PubMed] [Google Scholar]
- 8.Bevilacqua PC, Kierzek R, Johnson KA, Turner DH. Dynamics of Ribozyme Binding of Substrate Revealed by Fluorescence-Detected Stopped-Flow Methods. Science. 1992;258:1355–1357. doi: 10.1126/science.1455230. [DOI] [PubMed] [Google Scholar]
- 9.Serra MJ, Turner DH. Predicting Thermodynamic Properties of RNA. Methods in Enzymology. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 10.Kierzek R, Burkard ME, Turner DH. Thermodynamics of single mismatches in RNA duplexes. Biochemistry. 1999;38:14214–14223. doi: 10.1021/bi991186l. [DOI] [PubMed] [Google Scholar]
- 11.Bevilacqua JM, Bevilacqua PC. Thermodynamic analysis of an RNA combinatorial library contained in a short hairpin. Biochemistry. 1998;37:15877–15884. doi: 10.1021/bi981732v. [DOI] [PubMed] [Google Scholar]
- 12.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 13.Michel F, Westhof E. Modeling of the 3-Dimensional Architecture of Group-I Catalytic Introns Based On Comparative Sequence-Analysis. J Mol Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- 14.Celander DW, Cech TR. Visualizing the Higher-Order Folding of a Catalytic RNA Molecule. Science. 1991;251:401–407. doi: 10.1126/science.1989074. [DOI] [PubMed] [Google Scholar]
- 15.Wang JF, Cech TR. Tertiary Structure around the Guanosine-Binding Site of the Tetrahymena Ribozyme. Science. 1992;256:526–529. doi: 10.1126/science.1315076. [DOI] [PubMed] [Google Scholar]
- 16.Lehnert V, Jaeger L, Michel F, Westhof E. New Loop-Loop Tertiary Interactions in Self-Splicing Introns of Subgroup Ic and Id : a Complete 3D Model of the Tetrahymena-Thermophila Ribozyme. Chemistry & Biology. 1996;3:993–1009. doi: 10.1016/s1074-5521(96)90166-0. [DOI] [PubMed] [Google Scholar]
- 17.Cate JH, Gooding AR, Podell E, Zhou KH, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal-Structure of a Group-I Ribozyme Domain: Principles of RNA Packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 18.Golden BL, Gooding AR, Podell ER, Cech TR. A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science. 1998;282:259–264. doi: 10.1126/science.282.5387.259. [DOI] [PubMed] [Google Scholar]
- 19.Zaug AJ, Grabowski PJ, Cech TR. Autocatalytic Cyclization of an Excised Intervening Sequence Rna Is a Cleavage Ligation Reaction. Nature. 1983;301:578–583. doi: 10.1038/301578a0. [DOI] [PubMed] [Google Scholar]
- 20.Zaug AJ, Cech TR. The Intervening Sequence RNA of Tetrahymena Is an Enzyme. Science. 1986;231:470–475. doi: 10.1126/science.3941911. [DOI] [PubMed] [Google Scholar]
- 21.Pyle AM, McSwiggen JA, Cech TR. Direct Measurement of Oligonucleotide Substrate Binding to Wild-Type and Mutant Ribozymes from Tetrahymena. Proc Natl Acad Sci U S A. 1990;87:8187–8191. doi: 10.1073/pnas.87.21.8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herschlag D, Eckstein F, Cech TR. Contributions of 2′-Hydroxyl Groups of the RNA Substrate to Binding and Catalysis By the Tetrahymena Ribozyme: an Energetic Picture of an Active-Site Composed of RNA. Biochemistry. 1993;32:8299–8311. doi: 10.1021/bi00083a034. [DOI] [PubMed] [Google Scholar]
- 23.McConnell TS, Cech TR, Herschlag D. Guanosine Binding to the Tetrahymena Ribozyme: Thermodynamic Coupling With Oligonucleotide Binding. Proc Natl Acad Sci U S A. 1993;90:8362–8366. doi: 10.1073/pnas.90.18.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bevilacqua PC, Johnson KA, Turner DH. Cooperative and Anticooperative Binding to a Ribozyme. Proc Natl Acad Sci U S A. 1993;90:8357–8361. doi: 10.1073/pnas.90.18.8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narlikar GJ, Gopalakrishnan V, McConnell TS, Usman N, Herschlag D. Use of Binding-Energy by an RNA Enzyme for Catalysis by Positioning and Substrate Destabilization. Proc Natl Acad Sci U S A. 1995;92:3668–3672. doi: 10.1073/pnas.92.9.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shan S, Herschlag D. Probing the role of metal ions in RNA catalysis: Kinetic and thermodynamic characterization of a metal ion interaction with the 2′-moiety of the guanosine nucleophile in the Tetrahymena group I ribozyme. Biochemistry. 1999;38:10958–10975. doi: 10.1021/bi990388e. [DOI] [PubMed] [Google Scholar]
- 27.Shan S, Kravchuk AV, Piccirilli JA, Herschlag D. Defining the catalytic metal ion interactions in the Tetrahymena ribozyme reaction. Biochemistry. 2001;40:5161–5171. doi: 10.1021/bi002887h. [DOI] [PubMed] [Google Scholar]
- 28.Shan S, Herschlag D. Dissection of a metal-ion-mediated conformational change in Tetrahymena ribozyme catalysis. RNA. 2002;8:861–872. doi: 10.1017/s1355838202020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szewczak AA, Ortolevadonnelly L, Ryder SP, Moncoeur E, Strobel SA. A Minor-Groove RNA Triple-Helix Within the Catalytic Core of a Group-I Intron. Nat Struct Biol. 1998;5:1037–1042. doi: 10.1038/4146. [DOI] [PubMed] [Google Scholar]
- 30.Karbstein K, Herschlag D. Extraordinarily Slow Binding of Guanosine to the Tetrahymena Group I Ribozyme: Implications for RNA Structure and Preorganization. Proc Natl Acad Sci U S A. 2003;100:2300–2305. doi: 10.1073/pnas.252749799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zarrinkar PP, Williamson JR. Kinetic intermediates in RNA folding. Science. 1994;265:918–924. doi: 10.1126/science.8052848. [DOI] [PubMed] [Google Scholar]
- 32.Zarrinkar PP, Williamson JR. The P9.1-P9.2 peripheral extension helps guide folding of the Tetrahymena ribozyme. Nucleic Acids Res. 1996;24:854–858. doi: 10.1093/nar/24.5.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaug AJ, McEvoy MM, Cech TR. Self-Splicing of the Group-I Intron From Anabaena Pre-Transfer-RNA : Requirement For Base-Pairing of the Exons in the Anticodon Stem. Biochemistry. 1993;32:7946–7953. doi: 10.1021/bi00082a016. [DOI] [PubMed] [Google Scholar]
- 34.Golden BL, Cech TR. Conformational Switches Involved in Orchestrating the Successive Steps of Group-I RNA Splicing. Biochemistry. 1996;35:3754–3763. doi: 10.1021/bi952599z. [DOI] [PubMed] [Google Scholar]
- 35.Narlikar GJ, Herschlag D. Isolation of a Local Tertiary Folding Transition in the Context of a Globally Folded RNA. Nat Struct Biol. 1996;3:701–710. doi: 10.1038/nsb0896-701. [DOI] [PubMed] [Google Scholar]
- 36.Mei R, Herschlag D. Mechanistic Investigations of a Ribozyme Derived From the Tetrahymena Group-I Intron: Insights Into Catalysis and the 2nd Step of Self-Splicing. Biochemistry. 1996;35:5796–5809. doi: 10.1021/bi9527653. [DOI] [PubMed] [Google Scholar]
- 37.Zarrinkar PP, Sullenger BA. Probing the interplay between the two steps of group I intron splicing: Competition of exogenous guanosine with omega G. Biochemistry. 1998;37:18056–18063. doi: 10.1021/bi982193x. [DOI] [PubMed] [Google Scholar]
- 38.Zaug AJ, Davilaaponte JA, Cech TR. Catalysis of RNA Cleavage By a Ribozyme Derived From the Group-I Intron of Anabaena Pre-tRNA(Leu) Biochemistry. 1994;33:14935–14947. doi: 10.1021/bi00253a033. [DOI] [PubMed] [Google Scholar]
- 39.Kuo LY, Davidson LA, Pico S. Characterization of the Azoarcus ribozyme: tight binding to guanosine and substrate by an unusually small group I ribozyme. Biochim Biophys Acta. 1999;1489:281–292. doi: 10.1016/s0167-4781(99)00200-6. [DOI] [PubMed] [Google Scholar]
- 40.Davies RW, Waring RB, Ray JA, Brown TA, Scazzocchio C. Making ends meet: a model for RNA splicing in fungal mitochondria. Nature. 1982;300:719–724. doi: 10.1038/300719a0. [DOI] [PubMed] [Google Scholar]
- 41.Price JV, Cech TR. Determinants of the 3′ splice site for self-splicing of the Tetrahymena pre-rRNA. Genes Dev. 1988;2:1439–1447. doi: 10.1101/gad.2.11.1439. [DOI] [PubMed] [Google Scholar]
- 42.Michel F, Hanna M, Green R, Bartel DP, Szostak JW. The Guanosine Binding-Site of the Tetrahymena Ribozyme. Nature. 1989;342:391–395. doi: 10.1038/342391a0. [DOI] [PubMed] [Google Scholar]
- 43.Suh ER, Waring RB. Base pairing between the 3′ exon and an internal guide sequence increases 3′ splice site specificity in the Tetrahymena self-splicing rRNA intron. Mol Cell Biol. 1990;10:2960–2965. doi: 10.1128/mcb.10.6.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Been MD, Cech TR. Sites of circularization of the Tetrahymena rRNA IVS are determined by sequence and influenced by position and secondary structure. Nucleic Acids Res. 1985;13:8389–8408. doi: 10.1093/nar/13.23.8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaug AJ, Kent JR, Cech TR. A Labile Phosphodiester Bond at the Ligation Junction in a Circular Intervening Sequence RNA. Science. 1984;224:574–578. doi: 10.1126/science.6200938. [DOI] [PubMed] [Google Scholar]
- 46.Zaug AJ, Kent JR, Cech TR. Reactions of the Intervening Sequence of the Tetrahymena Ribosomal Ribonucleic-Acid Precursor: pH-Dependence of Cyclization and Site-Specific Hydrolysis. Biochemistry. 1985;24:6211–6218. doi: 10.1021/bi00343a027. [DOI] [PubMed] [Google Scholar]
- 47.Russell R, Herschlag D. Specificity from steric restrictions in the guanosine binding pocket of a group I ribozyme. RNA. 1999;5:158–166. doi: 10.1017/s1355838299981839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaug AJ, Grosshans CA, Cech TR. Sequence-Specific Endoribonuclease Activity of the Tetrahymena Ribozyme: Enhanced Cleavage of Certain Oligonucleotide Substrates That Form Mismatched Ribozyme Substrate Complexes. Biochemistry. 1988;27:8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- 49.Russell R, Herschlag D. New pathways in folding of the Tetrahymena group I RNA enzyme. J Mol Biol. 1999;291:1155–1167. doi: 10.1006/jmbi.1999.3026. [DOI] [PubMed] [Google Scholar]
- 50.Narlikar GJ, Bartley LE, Khosla M, Herschlag D. Characterization of a local folding event of the Tetrahymena group I ribozyme: Effects of oligonucleotide substrate length pH; and temperature on the two substrate binding steps. Biochemistry. 1999;38:14192–14204. doi: 10.1021/bi9914309. [DOI] [PubMed] [Google Scholar]
- 51.Fersht A. Enzyme Structure and Mechanism. W.H.Freeman and Company; New York: 1984. [Google Scholar]
- 52.Herschlag D. Evidence For Processivity and 2-Step Binding of the RNA Substrate From Studies of J1/2 Mutants of the Tetrahymena Ribozyme. Biochemistry. 1992;31:1386–1399. doi: 10.1021/bi00120a015. [DOI] [PubMed] [Google Scholar]
- 53.Pyle AM, Cech TR. Ribozyme Recognition of RNA by Tertiary Interactions with Specific Ribose 2′-OH Groups. Nature. 1991;350:628–631. doi: 10.1038/350628a0. [DOI] [PubMed] [Google Scholar]
- 54.Strobel SA, Cech TR. Tertiary Interactions With the Internal Guide Sequence Mediate Docking of the P1 Helix Into the Catalytic Core of the Tetrahymena Ribozyme. Biochemistry. 1993;32:13593–13604. doi: 10.1021/bi00212a027. [DOI] [PubMed] [Google Scholar]
- 55.Bartley LE, Zhuang X, Das R, Chu S, Herschlag D. Exploration of the transition state for tertiary structure formation between an RNA helix and a large structured RNA. J Mol Biol. 2003;328:1011–1026. doi: 10.1016/s0022-2836(03)00272-9. [DOI] [PubMed] [Google Scholar]
- 56.Shan S, Herschlag D. An unconventional origin of metal-ion rescue and inhibition in the Tetrahymena group I ribozyme reaction. RNA. 2000;6:795–813. doi: 10.1017/s1355838200000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herschlag D, Eckstein F, Cech TR. The Importance of Being Ribose at the Cleavage Site in the Tetrahymena Ribozyme Reaction. Biochemistry. 1993;32:8312–8321. doi: 10.1021/bi00083a035. [DOI] [PubMed] [Google Scholar]
- 58.Narlikar GJ, Bartley LE, Herschlag D. Use of duplex rigidity for stability and specificity in RNA tertiary structure. Biochemistry. 2000;39:6183–6189. doi: 10.1021/bi992858a. [DOI] [PubMed] [Google Scholar]
- 59.Herschlag D, Khosla M. Comparison of pH Dependencies of the Tetrahymena Ribozyme Reactions With RNA 2′-Substituted and Phosphorothioate Substrates Reveals a Rate-Limiting Conformational Step. Biochemistry. 1994;33:5291–5297. doi: 10.1021/bi00183a036. [DOI] [PubMed] [Google Scholar]
- 60.Knitt DS, Herschlag D. pH Dependencies of the Tetrahymena Ribozyme Reveal an Unconventional Origin of an Apparent pK(a) Biochemistry. 1996;35:1560–1570. doi: 10.1021/bi9521147. [DOI] [PubMed] [Google Scholar]
- 61.McDowell JA, Turner DH. Investigation of the structural basis for thermodynamic stabilities of tandem GU mismatches: solution structure of (rGAGGUCUC)2 by two-dimensional NMR and simulated annealing. Biochemistry. 1996;35:14077–14089. doi: 10.1021/bi9615710. [DOI] [PubMed] [Google Scholar]
- 62.Zaug AJ, Been MD, Cech TR. The Tetrahymena Ribozyme Acts Like an RNA Restriction Endonuclease. Nature. 1986;324:429–433. doi: 10.1038/324429a0. [DOI] [PubMed] [Google Scholar]
- 63.Piccirilli JA, Vyle JS, Caruthers MH, Cech TR. Metal-Ion Catalysis in the Tetrahymena Ribozyme Reaction. Nature. 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- 64.Moran S, Kierzek R, Turner DH. Binding of Guanosine and 3′ Splice Site Analogs to a Group-I Ribozyme: Interactions With Functional-Groups of Guanosine and With Additional Nucleotides. Biochemistry. 1993;32:5247–5256. doi: 10.1021/bi00070a037. [DOI] [PubMed] [Google Scholar]
- 65.Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature. 2004;430:45–50. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- 66.Allain FH, Varani G. Structure of the P1 helix from group I self-splicing introns. J Mol Biol. 1995;250:333–353. doi: 10.1006/jmbi.1995.0381. [DOI] [PubMed] [Google Scholar]
- 67.Baeyens KJ, De Bondt HL, Holbrook SR. Structure of an RNA double helix including uracil-uracil base pairs in an internal loop. Nat Struct Biol. 1995;2:56–62. doi: 10.1038/nsb0195-56. [DOI] [PubMed] [Google Scholar]
- 68.Masquida B, Sauter C, Westhof E. A sulfate pocket formed by three GoU pairs in the 0.97 A resolution X- ray structure of a nonameric RNA. RNA. 1999;5:1384–1395. doi: 10.1017/s1355838299991173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi K, Wahl M, Sundaralingam M. Crystal structure of an RNA duplex r(G GCGC CC)2 with non-adjacent G*U base pairs. Nucleic Acids Res. 1999;27:2196–2201. doi: 10.1093/nar/27.10.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Masquida B, Westhof E. On the wobble GoU and related pairs. RNA. 2000;6:9–15. doi: 10.1017/s1355838200992082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stahley MR, Strobel SA. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science. 2005;309:1587–1590. doi: 10.1126/science.1114994. [DOI] [PubMed] [Google Scholar]
- 72.Strobel SA, Cech TR. Translocation of an RNA Duplex On a Ribozyme. Nat Struct Biol. 1994;1:13–17. doi: 10.1038/nsb0194-13. [DOI] [PubMed] [Google Scholar]
- 73.Strobel SA, Cech TR. Minor-Groove Recognition of the Conserved G•U Pair At the Tetrahymena Ribozyme Reaction Site. Science. 1995;267:675–679. doi: 10.1126/science.7839142. [DOI] [PubMed] [Google Scholar]
- 74.Strobel SA, Cech TR. Exocyclic Amine of the Conserved G•U Pair At the Cleavage Site of the Tetrahymena Ribozyme Contributes to 5′-Splice Site Selection and Transition-State Stabilization. Biochemistry. 1996;35:1201–1211. doi: 10.1021/bi952244f. [DOI] [PubMed] [Google Scholar]
- 75.Silverman SK, Cech TR. Energetics and cooperativity of tertiary hydrogen bonds in RNA structure. Biochemistry. 1999;38:8691–8702. doi: 10.1021/bi9906118. [DOI] [PubMed] [Google Scholar]
- 76.Liao XM, Anjaneyulu PSR, Curley JF, Hsu M, Boehringer M, Caruthers MH, Piccirilli JA. The Tetrahymena ribozyme cleaves a 5 ′-methylene phosphonate monoester similar to 10(2)-fold faster than a normal phosphate diester: Implications for enzyme catalysis of phosphoryl transfer reactions. Biochemistry. 2001;40:10911–10926. doi: 10.1021/bi010801u. [DOI] [PubMed] [Google Scholar]
- 77.Walstrum SA, Uhlenbeck OC. The self-splicing RNA of Tetrahymena is trapped in a less active conformation by gel purification. Biochemistry. 1990;29:10573–10576. doi: 10.1021/bi00498a022. [DOI] [PubMed] [Google Scholar]
- 78.Shan S, Narlikar GJ, Herschlag D. Protonated 2′-aminoguanosine as a probe of the electrostatic environment of the active site of the Tetrahymena group I ribozyme. Biochemistry. 1999;38:10976–10988. doi: 10.1021/bi9903897. [DOI] [PubMed] [Google Scholar]
- 79.Guo F, Cech TR. In vivo selection of better self-splicing introns in Escherichia coli: The role of the P1 extension helix of the Tetrahymena intron. RNA. 2002;8:647–658. doi: 10.1017/s1355838202029011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.