

Abstract
In the Buchwald-Hartwig reaction between HIPTBr (HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3 = HexaIsoPropylTerphenyl) and (H2NCH2CH2)3N it is possible to obtain a 65% isolated yield of (HIPTNHCH2CH2)2NCH2CH2NH2. A second coupling then can be carried out to yield a variety of “Hybrid” ligands, (HIPTNHCH2CH2)2NCH2CH2NHAr, where Ar = 3,5-Me2C6H3, 3,5-(CF3)2C6H3, 3,5-(MeO)2C6H3, 3,5-Me2NC5H3, 3,5-Ph2NC5H3, 2,4,6-i-Pr3C6H2, or 2,4,6-Me3C6H2). The hybrid ligands may be attached to Mo to yield [Hybrid]MoCl species. From the monochloride species a variety of other species such as [Hybrid]MoN, {[Hybrid]MoN2}Na, and {[Hybrid]Mo(NH3)}+ can be prepared. [Hybrid]MoN2 species were prepared through oxidation of {[Hybrid]MoN2}Na species with ZnCl2, but could not be isolated. [Hybrid]Mo=N-NH species could be observed as a consequence of protonation of {[Hybrid]MoN2}− species, but they too could not be isolated as a consequence of a facile decomposition to yield dihydrogen and [Hybrid]MoN2 species. Attempts to reduce dinitrogen catalytically led to little or no ammonia being formed from dinitrogen. The fact that no ammonia was formed from dinitrogen in the case of Ar = 3,5-Me2C6H3, 3,5-(CF3)2C6H3, or 3,5-(MeO)2C6H3 could be ascribed to a rapid decomposition of intermediate [Hybrid]Mo=N-NH species in the catalytic reaction, a decomposition that was shown in separate studies to be accelerated dramatically by 2,6-lutidine, the conjugate base of the acid employed in the attempted catalytic reduction. X-ray structures of [(HIPTNHCH2CH2)2NCH2CH2N{3,5-(CF3)2C6H3}]MoCl and [(HIPTNHCH2CH2)2NCH2CH2N(3,5-Me2C6H3)]MoN2}Na(THF)2 are reported.
INTRODUCTION
In the last 10 years we have been exploring early transition metal complexes that contain a triamidoamine ligand, [(RNCH2CH2)3N]3− ([RN3N]3−).1 These trianionic ligands bind to an early transition metal in a relatively high oxidation state (~3+ or higher) in a tetradentate fashion, leaving three orbitals for binding substrates in the trigonally symmetric “pocket” surrounded by the three amido substituents. Two of these orbitals have π symmetry (dxz and dyz) and one has σ symmetry (~dz2). These orbitals can be employed in three combinations (2π/1σ, 1π/2σ, or 3σ) in order to bind substrates in the trigonal pocket. Distortion of the pseudotrigonal symmetry to yield pseudooctahedral species,2 or even seven-coordinate species3 is possible in cases where tungsten is the central metal and strongly bound substrates such as isocyanides or acetylenes are involved.
Recently we have been most interested in the chemistry of Mo complexes that contain the [HIPTN3N]3− ligand, where HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3 (HexaIsoPropylTerphenyl; see Figure 1),4 and in particular in the reduction of dinitrogen at a single metal center. The HIPT-substituted ligand was designed to prevent formation of relatively stable and unreactive [ArN3N]Mo-N=N-Mo[ArN3N] complexes,1b maximize steric protection of a metal coordination site in a monometallic species, and provide increased solubility in nonpolar solvents. We showed that several [HIPTN3N]Mo complexes could be prepared that contain dinitrogen or some dinitrogen reduction product.5 Examples include paramagnetic MoN2 (where Mo = [HIPTN3N]Mo), diamagnetic [MoN2] −, diamagnetic Mo-N=N-H, diamagnetic [Mo=N-NH2]BAr′4 (Ar′ = 3,5-(CF3)2C6H3), diamagnetic Mo≡N, diamagnetic [Mo=NH]BAr′, paramagnetic [Mo(NH3)]BAr′4, and paramagnetic Mo(NH3). Several of these species were successful for the catalytic reduction of dinitrogen to ammonia with protons and electrons.4a,b Dinitrogen is reduced through a combination of reduction and protonation at a single molybdenum in a trigonally symmetric coordination pocket that is sterically protected by three hexaisopropyl terphenyl substituents. Some combination of the frontier (two π and one σ) orbitals is sufficient to bind all of the intermediates in a proposed Chart-like reduction cycle.
Figure 1.
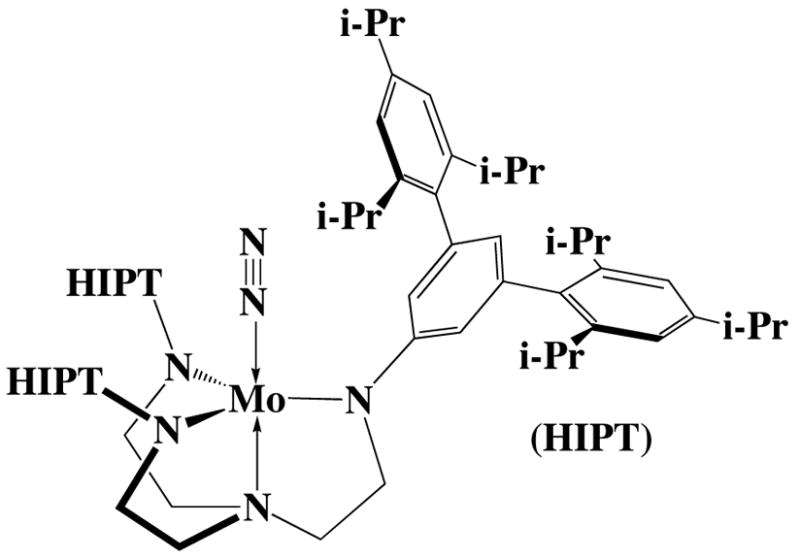
A drawing of [HIPTN3N]MoN2 = MoN2.
“Symmetric” variations of the [HIPTN3N]3− ligand that have been prepared and attached to molybdenum include the hexa-t-butylterphenyl ([HTBTN3N]3−) ligand, the hexamethylterphenyl ([HMTN3N]3−) ligand, and a variation of the [HIPTN3N]3− ligand in which a bromide is present in the para position of the central phenyl ring, [pBrHIPTN3N]3−.6 It became clear from various stoichiometric reactions involving the [HTBTN3N]3− system that proton and electron transfer was much slower than in the parent [HIPTN3N]3− system, perhaps by an order of magnitude or more. (The rate of conversion of [HTBTN3N]Mo(NH3) into [HTBTN3N]Mo(N2) has now been confirmed to be extremely slow, with a half-life of ~30 h compared to ~2 h for the parent system under a given set of conditions.7) We believe it is for this reason that [HTBTN3N]Mo≡N produces no significant ammonia from dinitrogen (0.06 equiv); the nitride is reduced to ammonia, but the system does not turn over to any significant extent. An attempted catalytic reduction with [HMTN3N]Mo≡N as the catalyst precursor was barely catalytic (0.49 equiv). The hexamethylterphenyl system was not explored in detail because of the low yields involved in synthesis of the ligand. Therefore the reason for poor catalytic activity has not been established in the hexamethylterphenyl system. Various [pBrHIPTN3N]Mo compounds were successful catalysts for reduction of dinitrogen; 6.4–7.0 equivalents of total ammonia were formed, which is not quite as high as the amount formed when [HIPTN3N]Mo species are employed. Since electronic differences (redox potentials and values for νNN) between the three variations and the parent system are relatively insignificant,6 steric differences appear to play a major role in the success or failure of the HTBT- and HMT-substituted ligand complexes for the catalytic reduction of dinitrogen.
During studies concerning the [HTBTN3N]3− system we discovered that in the arylation reaction between three equivalents of HTBTBr and (H2NCH2CH2)3N the third equivalent of HTBTBr reacts significantly more slowly than the second equivalent. Therefore by adjusting the stoichiometry (HTBTNHCH2CH2)2NCH2CH2NH2 could be isolated in good yield.6 It then became possible to add a different aryl group to the third “arm” of the ligand to yield what we call a “hybrid” (HTBTNHCH2CH2)2NCH2CH2NHAr species. We found that the same is true for (HIPTNHCH2CH2)2NCH2CH2NHAr species (Figure 2). An advantage of [(HIPTNCH2CH2)2NCH2CH2NAr]3− species is that steric protection in the coordination pocket can be tuned more finely. In this paper we report the synthesis of “hybrid” ligands and a selection of molybdenum compounds that contain some of them. We also report attempts to reduce dinitrogen catalytically with compounds that contain hybrid ligands. No Mo compound that contains one of the hybrid ligands is as successful as a catalyst as a compound that contains the parent [HIPTN3N]3− ligand, and some fail completely to produce ammonia from gaseous dinitrogen. Although the hybrid catalysts are poor catalysts, or not catalysts at all, these studies have helped us to understand what may be some of the key problems in the catalytic cycle.
Figure 2.

Labeling scheme for ligands and metal complexes described in this paper.
RESULTS AND DISCUSSION
If 2.1 equivalents of HIPTBr are employed in the Buchwald-Hartwig reaction8 between HIPTBr and (H2NCH2CH2)3N, then a mixture results that consists largely of (HIPTNHCH2CH2)2NCH2CH2NH2 mixed with a small amount of (HIPTNHCH2CH2)3N. These two products can be separated readily using a silica gel column pretreated with triethylamine, from which (HIPTNHCH2CH2)3N (11% yield) elutes readily with toluene. Flushing the column with a 1:1 mixture of toluene and tetrahydrofuran then yields (HIPTNHCH2CH2)2NCH2CH2NH2 (H41, 65% yield on a ~50 g scale; equation 1). The (HIPTNHCH2CH2)3N ligand can be accumulated and employed in other studies.
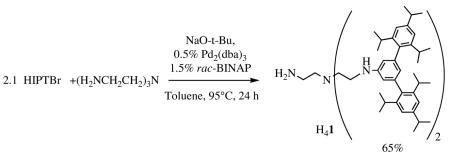 |
(1) |
A second Buchwald-Hartwig coupling can be carried out on the terminal amine in (HIPTNHCH2CH2)2NCH2CH2NH2 to yield (HIPTNHCH2CH2)2NCH2CH2NHAr species H32a-H32g (Equation 2, Figure 2). The final arylation can be complicated by overarylation of one or more secondary amines. However, careful optimization of the conditions allowed the pure ligands to be isolated and purified through column chromatography. These ligands all exhibit features in their NMR spectra that are entirely consistent with their Cs symmetry.
 |
(2) |
The hybrid ligands were attached to Mo in the same way as the [HIPTN3N]3− ligands, i.e., the parent ligands were added to a solution of MoCl4(THF)2 and the mixture was then treated with three equivalents of LiN(TMS)2 (Figure 3). The resulting bright orange paramagnetic [Hybrid]MoCl compounds 3a–g (Figure 2) could be isolated in good yields, even though they tend to be significantly more soluble than the already highly soluble [HIPTN3N]MoCl species.4 The proton NMR spectra of the [Hybrid]MoCl compounds reveal paramagnetically shifted methylene resonances similar to those observed for [HIPTN3N]MoCl, with separate methylene resonances for the HIPT and Ar-substituted arms. In some cases separate “inner” and “outer” (diastereotopic) methylene proton resonances can be observed in the HIPT-substituted arms of the ligands, consistent with the overall Cs symmetry of the hybrid complex. It should be noted that symmetric (ArNHCH2CH2)3N ligands cannot be placed onto Mo using the method described above when Ar is 2,4,6-trimethylphenyl,1b presumably for steric reasons. One mesityl or TRIP group is tolerated in 3f or 3g as a consequence of the two HIPT groups having no substituents in the ring’s ortho positions.
Figure 3.

Synthesis of molybdenum complexes that contain “hybrid” ligands.
An X-ray structure of 3c was carried out (Figure 4, Tables 1 and 2). The main difference between 3c and [HIPTN3N]3− compounds is a disruption of the trigonally symmetric pattern of steric protection around the apical position on the Mo, i.e., an opening up of one face of the ligand binding pocket. Bond distances and angles around the metal are similar to those found in symmetric [HIPTN3N]Mo, [HTBTN3N]Mo, and [pBrHIPTN3N]Mo systems.
Figure 4.

X-ray crystal structure of 3c. Hydrogen atoms have been removed for clarity.
Table 1.
Selected Bond Lengths and Angles of 3c vs. HTBTMoCl.6
| Bond | 3c | HTBTMoCl |
|---|---|---|
| Mo-Cl | 2.331(2) Å | 2.33 Å |
| Mo-N1 (amide) | 1.978(7) Å | 1.99 Å |
| Mo-N4 (amine) | 2.199(6) Å | 2.34 Å |
| N4-Mo-N1 | 100.20(18)° | 99° |
| N4-Mo-Cl | 176.10(17)° | 179° |
Table 2.
Selected Bond Lengths and Angles of 5d vs. [HIPTMoN2][MgBr(THF)3].17
| Bond | 5d | [HIPTMoN2][MgBr(THF)3] |
|---|---|---|
| N5-N6 (Nitrogen) | 1.171(6) A | 1.15 A |
| Mo-N5 (Dinitrogen) | 1.882(4) A | 1.86 A |
| Mo-N4 (Amine) | 2.209(4) A | 2.24 A |
| Mo-N3 (Amide) | 2.022(4) A | 2.02 A |
| N4-Mo-N5 | 174.09(17)° | 178° |
| N4-Mo-N1 | 81.25(16)° | 80° |
| Mo-N5-N6 | 174.2(4)° | 178° |
| N5-N6-Na | 110.7(4)° |
One of the least air-sensitive [HIPTN3N]Mo compounds that can be employed as a catalyst precursor in dinitrogen reduction is the nitride, [HIPTN3N]MoN. The bright yellow, diamagnetic [Hybrid]MoN compounds 4a–g (Figure 3) were prepared in reactions between [Hybrid]MoCl and trimethylsilylazide in benzene at elevated temperatures. NMR spectra of the diamagnetic [Hybrid]MoN species are fully consistent with their Cs symmetry. The [Hybrid]MoN species also are relatively crystalline and readily isolated. All have been analyzed and fully characterized.
Reduction of [Hybrid]MoCl compounds with sodium sand or 0.5% sodium/mercury amalgam yields diamagnetic anionic complexes that can be described as either Mo(II) dinitrogen complexes or, alternatively, as deprotonated Mo(IV) diazenido species, {[Hybrid]MoN2}Na(THF)2. Although all {[Hybrid]MoN2}Na(THF)2 complexes could be identified in solution, we attempted to isolate only 5c and 5d. Unfortunately, neither 5c nor 5d could be isolated in a sufficiently pure state to pass elemental analyses. Fortunately, however, an x-ray crystal structure was completed on a suitable crystal of 5d (Figure 5). As in 3c, the presence of an unsymmetric arm does not alter the metrical parameters around molybdenum to any significant degree (Table 2). The most obvious and significant feature is the off-axis coordination of the sodium atom (Na-N6-N6 = 110°) to the 3,5-dimethylphenyl group. Coordination of the sodium ion to the two nitrogens in the diazenido ligand decreases the Mo-N=N angle slightly to 174°.
Figure 5.

X-Ray crystal structure of 5d. Hydrogen atoms and isopropyl groups have been removed for clarity.
The {[Hybrid]MoN2}Na(THF)2 compounds 5a–g can be oxidized with ZnCl2 to yield [Hybrid]MoN2 species, 6a–g. The extreme solubility of 6a–g and the presence of 5–10% of free ligand, (HIPTNHCH2CH2)2NCH2CH2NHAr, so far have thwarted attempts to isolate these species. Therefore 6a–g could be characterized only through IR and NMR techniques. In 1H NMR spectra of these species, which are characteristic of a single Mo compound (plus free ligand), multiple resonances are observed for backbone methylene protons, as in many of the paramagnetic [Hybrid]MoCl compounds. IR spectra reveal a strong νNN absorption near 1990 cm−1, as previously observed for MoN2 compounds that contain “symmetric” ligands such as [HIPTN3N]MoN2.4 These νNN stretches are listed in Table 3. The νNN stretches in 6a–g respond to the slightly different electronic characteristics of the hybrid ligands. In the 3,5-substituted compounds, the energy of the νNN stretch moves to lower energies as the hybrid ligand’s substituents become more electron-donating, i.e., νNN = 1992 cm−1 for 6c versus νNN = 1984 cm−1 for 6e. The exception to this trend is 6g, where νNN is found at 2007 cm−1. We propose that this relatively high value results from steric crowding at the metal by the ortho isopropyl substituents, which lengthens the Mo-Nz bond slightly and therefore reduces the amount of backbonding to dinitrogen. As we shall see, however, the νNN stretches, which in any case vary relatively little, do not correlate with the success or failure of a catalytic reduction of dinitrogen, at least in the series of complexes prepared here.
Table 3.
Dinitrogen stretching modes for LMoN2 compounds.
| Compound | νN-N in cm−1 |
|---|---|
| [HIPTN3N]MoN2 | 1990 |
| [pBrHIPTN3N]MoN2 | 1992 |
| [HTBTN3N]MoN2 | 1990 |
| 6a | 1991 |
| 6b | 1988 |
| 6c | 1992 |
| 6d | 1987 |
| 6e | 1984 |
| 6f | 1986 |
| 6g | 2007 |
Two cationic ammonia complexes, {[Hybrid]MoNH3}BAr4′ species (7c and 7e) were prepared in reactions between 3c and 3e, respectively, and ammonia in dichloromethane in the presence of NaBAr′ (Ar′ = 3,5-(CF3)2C6H3). Compound 7e could be crystallized cleanly and was analyzed successfully. Compound 7c could not be freed completely from NaBAr4′, and so could be characterized only via NMR. Both 7c and 7e were utilized in studies involving exchange of ammonia for dinitrogen discussed below.
Attempts to reduce dinitrogen catalytically were carried out in a manner as described for the parent system, which produced 7–8 equivalents of ammonia per equivalent of Mo.4 Briefly, a heptane slurry containing 48 equivalents of [2,6-lutidinium][BAr4′] and 4a–g were placed in the reaction vessel and 36 equivalents of decamethylchromocene in 10 mL of heptane were added with a syringe pump.4a (See the experimental section for a representative experiment.) The reducing agent was added over a period of 6 hours, and the mixture was stirred for an additional hour. The volatiles were then vacuum transferred onto 1 M HCl in ether. The remaining solids were treated with NaO-t-Bu in a 2:1 mixture of methanol and tetrahydrofuran and the volatiles again were transferred onto 1 M HCl in ether. The ether and HCl were removed in vacuo and the resulting solid was analyzed for ammonia using the indophenol method. The results are summarized in Table 4.
Table 4.
Equivalents of ammonia obtained when LMoN is the precatalyst.
| Compound | Equiv NH3 from N2 |
|---|---|
| 4a | 0.6 |
| 4b | 0.3 |
| 4c | 0 |
| 4d | 0 |
| 4e | 0 |
| 4f | 0.2 |
| 4g | 2.0 |
None of the compounds that contains one of the new hybrid ligands is as efficient a catalyst for the reduction of dinitrogen as the [HIPTN3N]Mo analog. However, 4a, 4b, 4f, and 4g are catalytic to a small degree (0.6, 0.3, 0.2, and 2 equivalents of ammonia from dinitrogen, respectively; Table 4). At least one equivalent of ammonia is formed in all runs, i.e., the nitride is reduced to ammonia under the conditions employed. Compounds 4a and 4b are not directly comparable to the others as a consequence of the presence of the pyridyl ring, which could be protonated in several intermediates. In fact all that we can say at this point is that the [HIPTN3N]Mo system is not unique, although successful catalytic reduction appears to be extraordinarily sensitive to steric effects. It is important to know that one run in which 6c was employed yielded 0.7 equivalents of ammonia. This is an important result that we will revisit later.
A crucial step in dinitrogen reduction is conversion of a Mo(III) ammonia complex into a Mo(III) dinitrogen complex. Two of the LMoNH3 complexes were prepared in situ through reduction of 7c and 7e with CrCp*2. The resulting LMoNH3 complexes (8c and 8e) could not be isolated as a consequence of their high solubility and their ready conversion under dinitrogen into LMoN2 complexes, as observed through growth of LMoN2 in IR studies in aliquots of the solution of LMoNH3 over a period of several hours. The rate of formation of LMoN2 was shown to be approximately a first-order process in Mo through at least two half-lives, as found in the parent system.7 For 8c t1/2 = 180 min, while for 8e t1/2 < 45 min. It should be stressed that these results are only semi-quantitative, since the ammonia that is released is in equilibrium with the newly formed LMoN2 is not immediately released into the gas phase. In the parent system conversion of the ammonia complex into the dinitrogen complex was shown to be first order in dinitrogen with t1/2 ≈120 min. Exchange was estimated to actually be approximately ten times faster than observed in “bulk” exchange reactions.7 However, data for this “first-order” reaction of hybrid complexes are useful when compared with analogous data obtained for the parent system.7 In short, the rate of exchange appears to be slightly slower when CF3 groups are present (t1/2 = 180 min) and faster when methoxy groups are present (t1/2 < 45 min). Although we have not shown that the exchange rates in the hybrid systems depend upon dinitrogen pressure, as in the parent system,7 we believe that this is likely to be the case. The exchange rate is faster when the metal is slightly more electron-rich, consistent with more efficient backbonding into dinitrogen when it displaces ammonia in a bimolecular reaction. The position of the equilibria in the hybrid ligand systems are not known. In any case, the main point is that slow exchange cannot be the reason why these catalysts fail, since the exchange rates for the CF3- and OMe-substituted hybrid ligands bracket that observed in the successful HIPT system. We also were able to observe free NH3 in runs where two NH3 collections (initial volatiles and post base workup) were separated and quantified. When 4c is used as the precatalyst 0.3 out of the 0.97 equivalents of the ammonia observed was found in the initial volatiles, indicating that NH3 is indeed released during catalysis and that approximately 1/3 of the ammonia is present as NH3 rather than NH4+.
If exchange of ammonia for dinitrogen in the hybrid systems is not the problem with 8c and 8e as catalyst precursors, then what is the problem? We focused on the next step in the catalytic reaction, formation of the neutral diazenide (M-N=NH) species. As opposed to M-N=NR species (e.g., R = alkyl), M-N=NH species are rare in the literature, although some have been claimed.9 However, to our knowledge only the molybdenum and tungsten compounds in the symmetric HIPT system have been confirmed to have a proton on the β nitrogen in 15N-labeled compounds. Fortunately, the [ArN3N]Mo-N=NH species are diamagnetic, so the presence of a proton on Nβ can be established through 15N labeling studies. Due to its relative ease of synthesis and the availability of an 19F NMR handle, we focused on the [ArN3N]Mo-N=NH compound containing the 3,5-trifluoromethylphenyl-substituted arm (9c). Although the results for this compound have been reported recently,7 a summary is reported below.
Treatment of 5c (both 15N and 14N) with a proton source (initially H(OEt2)2BAr4′) resulted in formation of 9c. This species was identified through observation of Mo-N=NH at 8.6 ppm, with JNβH = 54.5 Hz and JNαH = 8 Hz (absolute values) in the 15N labeled compound (Figure 6). These values are nearly identical to those obtained for [HIPTN3N]Mo-N=NH.4b Compound 9c decomposes in a manner that is first order in Mo to form 6c quantitatively (and H2) with a t1/2of 17 ± 2 h. In contrast, pure [HIPTN3N]Mo-N=NH decomposes only very slowly to [HIPTN3N]MoH (at 60 °C t1/2 = 90 h4b). Interestingly, during some of these experiments we also were able to observe conversion of Mo-15N=15NH to Mo-14N=14NH (t1/2 of 4.5 h; Figure 7). The mechanism of this exchange (which also can be observed in [HIPTN3N]Mo-15N=15NH, albeit with a much slower t1/2 of approximately 150 h) is under further study. We suspect that nitrogen exchange is dependent upon nitrogen pressure, i.e., it does not involve β hydride elimination to yield [HIPTN3N]MoH followed by “insertion” of dinitrogen into the Mo-H bond. It should be noted that 9c is prepared in situ. Therefore we do not know whether the pure compound (if it somehow could be purified) would decompose at the same rate as that prepared in situ.
Figure 6.

Synthesis and decomposition of hybrid diazenide 9c.
Figure 7.

14N2 exchange into the 15N2-labeled diazenide 9c.
When [2,6-lutidinium]BAr4′ was used to form 9c the decomposition rate was enhanced by an order of magnitude, resulting in a t1/2 of 1.1 h. When 9c was prepared with H(OEt2)2BAr4′, and 4 equivalents of 2,6-lutidine, 2,4,6-trimethylpyridine (collidine), or Et3N were added subsequently, 9c could not be observed 5 minutes after addition of the base; only 6c and H2 were observed as the major products. The ability of Et3N to decompose 9c catalytically suggests that reaction is unlikely to be involve redox chemistry, as it might when some lutidine is the base. These results suggest that a lutidine-catalyzed shunt that produces hydrogen is operative in the less sterically shielded compounds (Figure 8). This finding could explain the inability of 4c to catalyze dinitrogen reduction. Clearly much more work needs to be done to understand this base-catalyzed chemistry. It is especially important to understand if dihydrogen formation is bimolecular or unimolecular in Mo. Regardless of the precise mechanism it is nevertheless ironic that one method of forming dihydrogen takes place in a cycle that involves the dinitrogen complex as the catalyst! Of course there are other ways of forming dihydrogen, the most direct consisting of reduction of the acid by the chromocene reducing agent.
Figure 8.

Hydrogenase shunt that limits catalysis in less sterically demanding systems.
It is interesting and important to note that when 6c is employed in a catalytic run, 0.7 equivalents of NH3 were found. The fact that some ammonia is formed suggests that the reaction can proceed through the diazenido stage early in the reaction, but no additional ammonia is then formed from free dinitrogen. Apparently any diazenido species that is formed upon addition of one electron and one proton to MoN2 is not completely decomposed by 2,6-lutidine, since only one equivalent is present at this stage. Later in the cycle, however, more 2,6-lutidine is present and the reaction then does not proceed beyond the diazenido stage.
CONCLUSIONS AND COMMENTS
A number of compounds that contain unsymmetric “hybrid” ligands have been synthesized that help expand our knowledge of molybdenum-centered catalytic dinitrogen reduction. While none of the compounds catalyzes dinitrogen reaction as successfully as compounds that contain the parent [HIPTN3N]3− ligand, and several fail completely, the study of several observable intermediates in dinitrogen reduction have helped us understand what is necessary for successful catalytic reduction. The primary requirement is that the binding pocket be sterically protected to a dramatic degree, not only to prevent bimetallic reactions, but also, we have discovered, to prevent decomposition of unstable dinitrogen reduction intermediates, especially the diazenido complex, through as yet an unknown mechanism. We also have shown that electron-donating ligands may speed the rate of ammonia for dinitrogen exchange. However, increasing the rate of exchange of ammonia for dinitrogen was not sufficient to avoid the shunt that prevents catalytic turnover. Clearly many other studies are required in order to understand important details of the shunt that produces dihydrogen.
The larger question remains whether abiological studies of the type described here suggest that dinitrogen is reduced to ammonia at Mo in the FeMo nitrogenase.10,11 Since we have (I believe) proven that dinitrogen can be reduced with protons and electrons catalytically to a mixture of ammonia and hydrogen at a single Mo center in systems of the type described here, and since no other abiological system will accomplish this feat,12 we are biased toward molybdenum being the metal that will reduce dinitrogen most easily, and the site of reduction in the FeMo nitrogenase. It is possible that in “alternative” FeV and all Fe nitrogenases13 dinitrogen is reduced at V or Fe instead of Mo in a similar core structure, with V and Fe being progressively less selective toward forming ammonia instead of dihydrogen. The dramatic sensitivity of catalytic dinitrogen reduction toward small changes in the nature of the triamidoamine ligand, as we have demonstrated here, further emphasizes that dinitrogen reduction is an extremely complex and sensitive catalytic reaction, with many points where it can fail. We hope to continue to improve our understanding of the complex process of catalytic reduction of dinitrogen at Mo and believe that an abiological vanadium-based catalytic reduction of dinitrogen is a goal that can be reached with the right ligand system. Catalytic reduction by V catalysts remains to be achieved, as does the catalytic reduction of dinitrogen with Fe-based systems.14
EXPERIMENTAL SECTION
General
Air and moisture sensitive compounds were manipulated utilizing standard Schlenk and dry-box techniques under an atmosphere of dinitrogen. All glassware used was oven and/or flame dried immediately prior to use. Pentane, diethyl ether, toluene, and benzene were purged with dinitrogen and passed through activated alumina columns. Benzene additionally was passed through a Q5 column.15 THF and benzene-d6 were dried over sodium/benzophenone ketyl and vacuum transferred prior to use. All other solvents mentioned were freeze-pump-thaw degassed 3 times prior to use. All dried and deoxygenated solvents were stored in a dinitrogen-filled glove box over molecular sieves or in Teflon-sealed glass solvent bombs. 1,3,5-Triisopropylbenzene (Aldrich), 2,4,6-tribromoaniline (Lancaster), N-bromo-succinimide (Aldrich), NaO-t-Bu (Aldrich), Pd2(dba)3 (Strem), rac-BINAP (Strem), and tris(2-aminoethyl)amine (Aldrich) were used as received, unless indicated otherwise. HexaIsoPropylTerphenyl bromide (HIPTBr),16,17 2,4,6-trimethoxy-1-iodobenzene, and 4-bromo-2,6-dimethylpyridine18 were prepared according to published procedures or with slight modifications. Proton and carbon NMR spectra were recorded on a Varian Mercury 300. NMR spectra are referenced to internal residual solvent peaks (1H and 13C NMR) or to external C6H5F (δ = −113.15 ppm in the 19F NMR spectrum). All stated coupling constants should be considered as absolute values.
(HIPTNHCH2CH2)2NCH2CH2NH2
The procedure is similar to previous syntheses of symmetric ligands,4 but utilizes 2.1 equivalents of HIPTBr instead of 3.1 equivalents. A 100 mL toluene solution of Pd2(dba)3 (0.91 g, 0.9 mmol) and rac-BINAP (1.85 g, 2.9 mmol) were stirred with mild heating to pre-form the active catalyst. The catalyst solution was then filtered through Celite into a 500 mL toluene solution containing HIPTBr (78.21 g, 139 mmol), tris(2-aminoethyl)amine (10.09 g, 67 mmol), and NaO-t-Bu (20.4g, 212 mmol). This mixture was then heated at ~100°C for 2 days. After 2 days the organic layer was combined with 500 mL of water and the mixture was extracted with ether. The organic layers were combined and dried over magnesium sulfate. This ether solution was then filtered and the ether was removed in vacuo. Column chromatography was then carried out on a silica gel column. Byproducts were eluted with toluene (including 11 g (10%) of the symmetric ligand) and the product was eluted with a 1:1 mixture of toluene and THF. (Note that it is important to pretreat the silica gel with triethylamine in order to obtain acceptable separations.) Upon removal of solvent from the column fractions in vacuo, 47.3 g of a pale yellow solid was isolated (64%): 1H NMR (C6D6, 20°C) δ 7.22 (s, 8H, 3,5,3″,5″ -H), 6.50 (s, 6H, 2′,4′,6′-H), 4.92 (t, JHH = 4.9 Hz, 2H, NH), 3.17 (septet, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.94 (overlapped septet, JHH = 6.6 Hz, 4H, 4,4″-CHMe2), 2.85 (overlapped multiplet, 4H, NH2CH2CH2 and NH2CH2CH2), 2.74 (br q, JHH = 5.2 Hz, 4H, NHCH2CH2), 2.03 (br t, JHH = 5.2 Hz, 4H, NHCH2CH2), 1.32 (d, JHH = 7.1 Hz, 24H, 4,4″-CH(CH3)2), 1.26 (br d, JHH = 6.9 Hz, 48H, 2,6,2″,6″-CH(CH3)2). 0.44 (br s, 2H, NH2); 13C NMR (C6D6, 20°C) δ 148.77, 148.40, 147.19, 142.48, 138.69, 121.38, 121.03, 112.84, 55.91, 52.97, 41.64, 40.19, 31.18, 25.36, 25.05, 24.91. MS (ESI) 1107.9119 ([M+H]+; calcd 1107.9116).
4-bromo-2,6-diphenyl-pyridene
This compound was synthesized via a method similar to that of Talik et. al.18 Briefly, 2,6-diphenyl-pyridin-4-ylamine (3.75 g, 15.4 mmol) and copper bromide (11.5 g, 80.3 mmol) were added to 150 mL of 48% hydrobromic acid and cooled to < 5°C in an ice bath. Sodium nitrite (22.5 g, 326 mmol) in 100 mL water was slowly added, ensuring that the temperature did not rise above 5°C. After addition the mixture was allowed to stir overnight, at which time it was basified with 150 g of sodium hydroxide in 300 mL water. The product was extracted with ether, which was dried over magnesium sulfate, filtered, and dried in vacua, yielding a bright orange oil. This was triturated with methanol to yield 1.55 g of a bright orange powder, (34%) which is sparingly soluble in most solvents. 1H NMR (C6D6, 20°C) δ 7.95 (dd, 4H, , 2,6,2″,6″-H), 7.42 (s, 2H, 3′,5′-H) 7.22 (m, 6H, 3,4,5,3″,4″,5″-H). MS (ESI) 309.0146 ([M+H]+; calcd 309.0148).
H3[LutHIPT2N3N] (2a)
Pd2(dba)3 (0.054g, 0.058mmol) and rac-BINAP (0.11g, 0.18 mmol) were stirred with mild heating in 25 mL toluene to preform the bright orange catalyst. This solution was then filtered through Celite into a 300 mL toluene solution containing 1 (4.41 g, 3.98 mmol), 4-bromo-2,6-dimethyl-pyridine (0.80 g, 4.30 mmol), and NaO-t-Bu (0.76 g, 7.91 mmol). This solution was then heated at 95°C for 2 days, after which time it was filtered through Celite and concentrated in vacuo to dryness. The resulting solid was dissolved in pentane and loaded onto a silica column which had been pretreated with triethylamine. The side products were eluted with toluene, while the product was eluted with a 1:1 mixture of toluene and THF; yield 3.66 g of a foamy, tan solid (76%): 1H NMR (C6D6, 20°C) δ 7.22 (s, 8H, 3,5,3″,5″ –H), 6.53 (br t, JHH = 1.4 Hz, 2H, 4′-H), 6.48 (d, JHH = 1.3 Hz, 4H, 2′,6′-H), 6.02 (s, 2H, Lut-3,5-H), 3.93 (t, JHH = 4.9 Hz, 1H, Lut-NH), 3.71 (t, JHH = 4.8 Hz, 2H, HIPT-NH), 3.16 (septet, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.92 (septet, JHH = 6.9 Hz, 4H, 4,4″-CHMe2), 2.80 (br q, JHH = 5.4 Hz, 4H, HIPT-NHCH2CH2), 2.63 (br q, JHH = 5.9 Hz, 2H, Lut-NHCH2CH2), 2.39 (s, 6H, Lut-CH3), 2.16 (br t, JHH = 5.7 Hz, 4H, HIPT-NHCH2CH2), 2.11 (br t, JHH = 6.8 Hz, 2H, Lut-NHCH2CH2), 1.32 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.28 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2). 1.25 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2); 13C NMR (C6D6, 20°C) δ 158.72, 154.58, 148.55, 148.4, 147.15, 142.68, 138.44, 121.96, 121.09, 112.84, 104.75, 52.78, 52.15, 41.48, 40.51, 35.26, 31.21, 30.94, 25.30, 25.10, 24.93, 24.87. MS (ESI) 1212.9662 ([M+H]+; calcd 1212.9695).
H3[PhLutHIPT2N3N] (2b)
A solution of Pd2(dba)3 (0.045g, 0.049mmol) and rac-BINAP (0.094g, 0.15 mmol) in 25 mL toluene was stirred with mild heating until the orange catalyst was formed. The orange solution was then filtered through Celite into a 300 mL toluene solution containing 1 (3.75 g, 3.39 mmol), 4-bromo-2,6-diphenylpyridine (1.55 g, 5.02 mmol), and NaO-t-Bu (0.64 g, 6.66 mmol). This mixture was then heated at 95°C for 2 days, at which time it was filtered through Celite and concentrated in vacuo to dryness. The resulting solid was dissolved in pentane and the product was isolated through column chromatography on silica column as described in earlier preparations; yield 2.07 g of the product as a light yellow foamy solid (46%): 1H NMR (C6D6, 20°C) δ 8.32 (m, 4H, PhLut-2,6,2″,6″-H), 7.33 (m, 6H, PhLut-3,4,5,3″,4″,5″-H), 7.22 (s, 8H, 3,5,3″,5″ -H), 6.75 (s, 2H, PhLut-3′,5′-H), 6.57 (br t, JHH = 1.3 Hz, 2H, 4′-H), 6.51 (d, JHH = 1.4 Hz, 4H, 2′,6′-H), 3.98 (t, JHH = 5.2 Hz, 1H, PhLut-NH), 3.60 (br s, 2H, HIPT-NH, 3.17 (septet, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.89 (m, 8H, 4,4″-CHMe2 and HIPTNHCH2CH2 overlapping), 2.75 (br q, JHH = 5.8 Hz, 2H, PhLutNHCH2CH2, 2.24 (m, 6H, HIPTNHCH2CH2 and PhLutNHCH2CH2 overlapping), 1.31 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.29 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2). 1.28 (br d, JHH = 6.9 Hz, 36H, 2,6,2″,6″-CH(CH3)2); 13CNMR(C6D6, 20°C) δ 158.41, 155.34, 148.61, 148.28, 147.16, 142.75, 141.27, 138.38, 129.14, 127.88, 122.10, 121.12, 112.82, 103.64, 52.87, 41.42, 35.24, 31.22, 25.31, 25.03, 24.83. MS (ESI) 1337.0025 ([M+H]+; calcd 1337.0008).
H3[3,5-Bis(CF3)HIPT2N3N] (2c)
This compound was prepared in a manner similar to other hybrid ligand systems. Briefly, a toluene solution of 0.050 g (55 mmol) of Pd2(dba)3 and 0.101 g (162 mmol) of rac-BINAP was heated until orange. This solution was filtered through Celite into 100 mL of toluene containing 4 g (3.6 mmol) of 1, 1.05 g (3.6 mmol) of 1-bromo-3,5-(bis)trifluoromethylbenzene, and 0.69 g (7.2 mmol) of NaO-t-Bu. This solution was then heated at 105°C for 2 days and filtered. The volatiles were removed in vacuo and the residue was then extracted into pentane. The pentane insoluble material was removed via filtration, and the resulting solution was subject to column chromatography on silica gel. The product was eluted with toluene to yield 0.91 g (20%) of the final product as a light yellow solid: 1H NMR (C6D6, 20°C) δ 7.25 (s, 1H, CF3 arm 4-H), 7.22 (s, 8H, HIPT 3,5,3″,5″-H), 6.64 (s, 2H, CF3 arm 2,6-H), 6.57 (s, 2H, HIPT 4′-H), 6.48 (s, 4H, HIPT-2′,6′-H), 3.88 (t, JHH = 5.2 Hz, 1H, CF3 arm NH), 3.51 (br s, 2H, HIPT-NH), 3.15 (septet, 8H, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.90 (septet, JHH = 6.9 Hz, 4H, 4,4″-CHMe2), 2.82 (br t, JHH = 5.8 Hz, 4H, HIPT-NCH2CH2), 2.40 (br q, JHH = 5.2 Hz, 2H, CF3 arm-NCH2CH2), 2.17 (br t, JHH = 5.8 Hz, 4H, HIPT-NHCH2CH2), 2.10 (br t, JHH = 5.8 Hz, 2H, CF3 arm-NCH2CH2) 1.32 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.28 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2), 1.25 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2); 13C NMR (C6D6, 20°C) δ 149.41, 148.66, 148.25, 147.13, 142.79, 138.31, 133.33, 132.91, 122.28, 121.12, 122.89, 112.30, 110.36, 52.95, 52.21, 41.55, 40.91, 35.25, 31.21, 25.28, 24.99, 24.83; 19F NMR (C6D6, 20°C) δ −62.74. HRMS (ESI) 1319.9156 ([M+H]+; calcd 1319.9177).
H3[3,5-DimethylHIPT2N3N] (2d)
This compound was prepared in a manner similar to other hybrid ligand systems. Briefly, 0.037 g (4 mmol) of Pd2(dba)3 and 0.075 g (12 mmol) of rac-BINAP were stirred in toluene to form the orange catalyst. This solution was then filtered through Celite into a toluene mixture containing 3 g (2.8 mmol) of 1, 0.5 g (2,7 mmol) of 1-bromo-3,5-dimethylbenzene, and 0.52 g (5.4 mmol) of NaO-t-Bu. This solution was then heated at 85°C overnight, with the temperature then being raised to 95°C for one day. The mixture was then filtered through Celite and the solvent removed in vacuo. The solid was taken up in pentane and the solution was filtered again through Celite. The product was isolated from a silica gel column. The compound was eluted with a 9:1 mixture of toluene and THF to yield 0.78 g (24%) of the product as a light yellow solid: 1H NMR (C6D6, 20°C) δ 7.22 (s, 8H, HIPT 3,5,3″,5″-H), 6.52 (br t, JHH = 1.1 Hz, 2H, HIPT 4′-H), 6.46 (d, JHH = 1.1 Hz, 4H, HIPT-2′,6′-H), 6.37 (s, 1H, dimethyl-4-H), 6.26 (s, 2H, dimethyl-2,6-H), 3.94 (br s, 2H, HIPT-NH), 3.75 (br s, 1H, dimethyl-NH, 3.16 (septet, JHH = 6.9 Hz, 8H, HIPT-2,6,2″,6″-CHMe2), 2.95 (septet JHH = 6.9 Hz, 4H, HIPT-4,4″-CHMe2), 2.79 (m, 8H, contains HIPT-NCH2CH2 and dimethyl-NCH2CH2), 2.17 (m, 8H, contains HIPT-NCH2CH2 and dimethyl-NCH2CH2), 2.08 (s, 6H, dimethyl-CH3), 1.46 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.38 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2), 1.27 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2); 13C NMR (C6D6, 20°C) δ 148.79, 148.49, 148.44, 147.17, 142.58, 139.28, 138.58, 121.66, 121.03, 120.74, 112.71, 111.71, 52.59, 52.29, 41.73, 41.41, 35.27, 31.19, 30.84, 25.32, 25.06, 24.89, 21.93. HRMS (ESI) 1211.9734 ([M+H]+; calcd 1211.9742).
H3[3,5-DimethoxyHIPT2N3N] (2e)
This compound was prepared in a manner similar to other hybrid ligand systems. Briefly, 0.037 g (0.04 mmol) of Pd2(dba)3 and 0.075 g (0.12 mmol) of rac-BINAP were heated in toluene. This solution was filtered through Celite into a solution containing 3g (2.7 mmol) of 1, 0.56 g (2.5 mmol) of 3,5-dimethoxy bromobenzene, and 0.52 g (5.4 mmol) of NaO-t-Bu in 75 mL of toluene. This container was then sealed and heated at 90°C for 2 days. The resulting brown solution was filtered and the solvent was removed in vacuo. A column was employed to isolate the product, with pentane and toluene as the eluents; yield 2.2 g (65%) of product as a light yellow powder: 1H NMR (C6D6, 20°C) δ 6.46 (s, 4H, 3,5,3″,5″ HIPT-H), 6.42 (s, 4H, 3,5,3″,5″ HIPT-H), 6.40 (d, JHH = 2 Hz, 2H, dimethoxy 2,6-H), 6.13 (t, JHH = 2 Hz, 1H, dimethoxy 4-H), 5.94 (t, JHH = 1.9 Hz, 2H HIPT 4′-H), 5.88 (d, JHH = 1.9 Hz, 4H HIPT 2′6′-H), 3.94 (br s, 1H, dimethoxy N-H), 3.77 (br s, 2H, HIPT N-H), 3.27 (s, 6H, dimethoxy-OCH3), 3.11 (septet, JHH = 6.8 Hz, 8H HIPT-2,6,2″,6″-CHMe2), 2.85 (septet, JHH = 6.8 Hz, 4H HIPT-4,4″-CHMe2), 2.75 (m, 10H, contains HIPT-NCH2CH2 and dimethoxy-NCH2CH2 and dimethoxy NCH2CH2), 2.14 (br t, 4H, HIPT-NCH2CH2), 1.26 (d, JHH = 6.8 Hz, 24H HIPT-4,4″ –CH(CH3)2), 1.22 (d, JHH = 6.8 Hz, 24H HIPT-2,6,2″,6″ –CH(CH3)2), 1.19 (d, JHH = 6.8 Hz, 24H HIPT-2,6,2″,6″ –CH(CH3)2); 13C NMR (C6D6, 20°C) δ 162.9, 162.5, 150.8, 148.4, 147.2, 142.6, 138.6, 138.2, 129.7, 126.0, 121.0, 112.8, 92.9, 54.9, 52.8, 41.5, 35.3, 31.2, 25.3, 25.0, 24.9. MS (ESI) 1243.9694([M+H]+; calcd 1243.9641).
H3[MesHIPT2N3N] (2f)
This compound was prepared in a manner similar to other hybrid ligand systems. Briefly, 0.037 g (40 mmol) of Pd2(dba)3 and 0.075 g (12 mmol) of rac-BINAP were heated in toluene and the orange solution was then filtered through Celite into a toluene mixture containing 3 g (2.7 mmol) of 1, 0.59 g (29.8 mmol) of 1-bromo-2,4,6-trimethylbenzene, and 0.52 g (5.4 mmol) of NaO-t-Bu. The vessel was then sealed and heated at 110°C for 2 days. The product mixture was worked up in a manner similar to that described for previous ligands and the product was isolated via column chromatography with an 8:1 mixture of toluene and THF as the eluent; yield 2 g (60%) of a pale yellow solid: 1H NMR (C6D6, 20°C) δ 7.22 (s, 8H, HIPT 3,5,3″,5″-H), 6.77 (s, 2H, Mes-3,5-H), 6.54 (s, 2H, HIPT 4′-H), 6.48 (s, 4H, HIPT-2′,6′-H), 3.79 (br s, 2H, HIPT-NH), 3.18 (septet, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.88 (m, 8H, containing 4,4″-CHMe2 and HIPT-NCH2CH2), 2.78 (br t, JHH = 5.8 Hz, 2H, Mes-NCH2CH2), 2.2 (m, 15H, containing Mes-CH3, Mes-NCH2CH2, and HIPT-NCH2CH2), 1.32 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.28 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2) 1.26 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2); 13C NMR (C6D6, 20°C) δ 162.80, 148.48, 147318, 144.65, 142.59, 138.54, 131.03, 130.23, 129.14, 121.83, 121.07, 112.77, 55.29, 53.30, 46.83, 41.56, 35.26, 31.19, 25.32, 25.12, 24.86, 21.06, 19.22. HRMS (ESI) 1225.9906 ([M+H]+; calcd 1225.9899).
H3[TripHIPT2N3N] (2g)
This compound was prepared in a manner similar to other hybrid ligand systems. Briefly, 0.037 g (40 mmol) of Pd2(dba)3 and 0.077 g (162 mmol) of X-Phos were heated mildly in toluene to pre-form the active orange catalyst species. This was then filtered into a toluene mixture containing 3 g (2.7 mmol) of 1, 0.92 g (3.2 mmol) of 1-bromo,2,4,6-triisopropylbenzene, and 0.39 g (40.5 mmol) of NaO-t-Bu. This mixture was then heated at 120°C for 2 days. The solvent was removed in vacuo, the resulting solid was taken up in pentane, and the mixture was filtered in preparation for column chromatography on silica gel. Elution with a 8:1 mixture of toluene and THF mixture yielded a mixture of (HIPTNHCH2CH2)2NCH2CH2NH2 and the desired product from which the solvent was removed in vacuo. The resulting solid was taken up in toluene and passed through a silica plug, yielding 0.35 g (10%) of the desired product as a light yellow solid: 1H NMR (C6D6, 20°C) δ 7.21 (s, 8H, HIPT 3,5,3″,5″-H), 7.06 (s, 2H, Trip-3,5-H), 6.54 (br t, JHH = 1.1 Hz, 2H, HIPT 4′-H), 6.51 (d, JHH = 1.1 Hz, 4H, HIPT-2′,6′-H), 3.88 (br s, 2H, HIPT-NH), 3.38 (septet, JHH =6.9 Hz, 2H, Trip-2,6-CHMe2), 3.17 (septet, JHH = 6.9 Hz, 8H, HIPT-2,6,2″,6″-CHMe2), 2.89 (m, 12H, containing HIPT-NCH2CH2, Trip-NCH2CH2, HIPT-4,4″-CH(CH3)2, and Trip-4-CH(CH3)2), 2.44 (br t, JHH = 5.5 Hz, 2H, Trip-NCH2CH2), 2.33 (br t, JHH = 5.5 Hz, 4H, HIPT-NCH2CH2), 1.32 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.25 (m, 60H, 2,6,2″,6″-CH(CH3)2); 13C NMR (C6D6, 20°C) δ 148.58, 148.47, 147.17, 144.52, 142.77, 142.10, 138.51, 121.98, 121.91, 121.05, 113.00, 55.60, 53.70, 50.16, 42.13, 35.26, 31.17, 30.84, 28.56, 25.35, 25.01, 24.92, 24.96, 24.87. HRMS (ESI) 1310.0833 ([M+H]+; calcd 1310.0838).
[LutHIPT2N3N]MoCl (3a)
The procedure followed was nearly identical to published procedures.4,6 Briefly, H32a (0.6 g, 0.50 mmol) and MoCl4(THF)2 (0.19 g, 0.50 mmol) were dissolved in THF (40 mL). This solution was stirred for one hour and (Me3Si)2NLi (0.258 g, 0.1.54 mmol) was then added slowly. The solution was stirred for 2 h and the solvent was removed in vacuo with mild heating. The solid residue was extracted with pentane (2×10 mL) followed by benzene (3 × 20 mL) and all extracts were passed through Celite. The filtrate was then evaporated to dryness in vacuo. The residue was recrystallized from pentane to yield 0.42 g of an orange solid in multiple crops (63%): 1H NMR (C6D6, 20°C) δ 11.8 (br s), 7.3 (m), 3.1 (br s), 3.0 (m), 2.7 (br s), 1.6 (br s), 1.4 (br s), −16.7 (br s), −23.8 (br s), −65.0 (br s), −69.4 (br s). Anal Calcd for C85H118ClMoN5: C, 76.11; H, 8.87; N, 5.22. Found: C, 76.01; H, 9.06, N, 5.16.
[PhLutHIPT2N3N]MoCl (3b)
The procedure followed was nearly identical to published procedures.4,6 Briefly, H32b (1 g, 0.75 mmol) and MoCl4(THF)2 (0.286 g, 0.75 mmol) were dissolved in THF (75mL). This solution was stirred for one hour and (Me3Si)2NLi (0.388 g, 2.3 mmol) was then added slowly. After 2 hours the solvent was removed in vacuo. The solid residue was extracted with pentane (2×10 mL) and benzene (3 × 20 mL) and all filtrates were passed through Celite. The filtrates were then reduced to dryness in vacuo. The product was crystallized from pentane to yield 0.9 g of an orange-brown solid in multiple crops (82%): 1H NMR (C6D6, 20°C) δ 14.4 (br s), 8.3 (br s), 7.50 (br m), 7.2 (s), 3.1 (br s), 2.9 (br s), 2.5 (br s), 1.3 (br s), −21.5 (br s), −62.2 (br s), −81.1 (br s), −86.7 (br s). Anal. Calcd for C95H122ClMoN5: C, 77.86; H, 8.39; N, 4.78. Found: C, 77.73; H, 8.46; N, 4.83.
[3,5-Bis(CF3)HIPT2N3N]MoCl (3c)
This compound was synthesized similarly to other compounds of this type described above. Briefly, 0.75 g (0.57 mmol) of H32c as added to 50 mL THF. MoCl4(THF)2 (0.24 g; 0.62 mmol) was then added slowly and the mixture was stirred for one hour, during which time the solution darkened to a deep red. To this red solution 0.30 g (1.8 mmol) of (Me3Si)2NLi was added and the reaction was stirred for 2 h. The solvent was removed in vacuo and the resulting solid was extracted into pentane. The mixture was filtered through Celite and the solvent removed in vacuo. Recrystallization of the residue from pentane yielded 0.45 g (55%) of a deep orange powder: 1H NMR (C6D6, 20°C) δ 11.1 (br s), 7.27 (s), 7.19 (s), 3.18 (br s), 2.95 (m), 1.62 (br s), 1.49 (br s), 1.34 (br s), −10.3 (br s), −13.8 (br s), −29.7 (br s), −69.5 (br s), −82.7 (br s), −86.9 (br s); 19F NMR (C6D6, 20°C) δ −57.9 ppm. Anal. Calcd for C86H113ClF6MoN4: C, 71.32; H, 7.86; N, 3.87. Found: C, 71.25; H, 7.76; N, 3.94.
[3,5-DimethylHIPT2N3N]MoCl (3d)
This compound was synthesized similarly to other compounds of this type described above. Briefly, 0.76 g (0.63 mmol) of H32d was added to 0.26 g (0.68 mmol) of MoCl4(THF)2 in 50 mL THF. This mixture was stirred for one hour and 0.33 g (1.9 mmol) of (Me3Si)2NLi was added. After two hours the solvent was removed in vacuo and the resulting solid was extracted into pentane and the extract was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized from pentane to yield 0.47 g (56%) of a deep orange solid: 1H NMR (C6D6, 20°C) d 9.68 (br s), 7.26 (s), 7.24 (s), 3.18 (br s), 2.97 (m), 2.05 (s), 1.54 (br s), 1.44 (s), 1.36 (br s), −11.25 (br s), −13.80 (br s), −23.5 (br s), −66.5 (br s), −77.5 (br s), −84.0 (br s). Anal. Calcd for C86H119ClMoN4: C, 77.07; H, 8.95; N, 4.18. Found: C, 76.87; H, 9.06; N, 4.11.
[3,5-DimethoxyHIPT2N3N]MoCl (3e)
This compound was synthesized similarly to other compounds of this type described above. Briefly, 0.41 g (0.3 mmol) of H32e was added to 0.13 g (0.3 mmol) of MoCl4(THF)2 in 30 mL of THF. Upon addition of the molybdenum, the solution’s color immediately darkened to deep red. The solution was stirred for one hour and 0.17 g (1 mmol) of (Me3Si)2NLi was then added. The solution slowly turned orange-red. The solvent was removed in vacuo and the solid was extracted into pentane and the extract was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized from pentane yielding 0.32 g (70%) of the compound as a deep orange powder: 1H NMR (C6D6, 20°C) δ 11.6 (br s), 10 (br s), 7.27 (s), 3.96 (s), 3.18 (s), 2.95 (s), 2.63 (s), 1.34 (s), −14.5 (br s), −20.4 (br s), −69.6 (br s), −79.6 (br s). Anal. Calcd for C86H119ClMoN4O2: C, 75.27; H, 8.74; N, 4.08. Found: C, 75.06; H, 8.65; N, 4.08.
[MesHIPT2N3N]MoCl (3f)
This compound was synthesized similarly to other compounds of this type described above. Briefly, 1.0g (0.81 mmol) H32f was added to 70 mL of THF. MoCl4(THF)2 (0.32 g, 1.0 mmol) was added slowly with stirring. After 2 h 0.42 g (2.6 mmol) of (Me3Si)2NLi was added. After 1 hour the solvent was evaporated in vacuo and the resulting solid was dissolved into pentane and the extract was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized from pentane to yield 0.74 g (67%) of a deep orange crystalline solid: 1H NMR (C6D6, 20°C) δ 17.73 (br s), 7.28 (s), 7.22 (s), 6.42 (br s), 3.83 (s), 3.5 (br s), 3.17 (br s), 2.97 (m), 1.52 (br s), 1.41 (br s), −20 (br s), −23.5 (br s), −74.68 (br s), −97.50 (br s). Anal. Calcd for C87H121ClMoN4: C, 77.16; H, 9.01; N, 4.14. Found: C, 77.04; H, 9.04; N 4.11.
[TripHIPT2N3N]MoCl (3g)
This compound was synthesized similarly to other compounds of this type described above. Briefly, 0.35 g (0.25 mmol) of H32g was dissolved in 50 mL of THF. MoCl4(THF)2 (0.112 g (0.29 mmol) was added and the solution was stirred for 2 hours. The color changed from orange to bright red. To this solution 0.138 g (0.83 mmol) of (Me3Si)2NLi was added slowly and the solution was stirred for an additional hour. The solvent was removed in vacuo and the resulting solid was extracted into pentane and the mixture was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized to yield 0.22 g (56%) of a bright orange powder in multiple crops: 1H NMR (C6D6, 20°C) δ 18.14 (br s), 9.66 (br s), 7.30 (s), 7.20 (s), 4.09 (br s), 3.53 (br s), 3.20 (br s), 2.99 (m), 1.40 (br s), 1.26 (s), −11.52 (br s), −16.97 (br s), −23.75 (br s), −61.65 (br s), −83.70 (br s), −96.15 (br s). Anal Calcd for C93H1133ClMoN4: C, 77.65; H, 9.32; N, 3.89. Found: C, 77.42; H, 9.38; N, 3.77.
[LutHIPT2N3N]MoN (4a)
This compound could not be isolated in pure form, as the reaction between (3a) and Me3SiN3 is low yielding and the final product consequently was contaminated with H32a, even after multiple recrystallizations. A sample containing ~10% H32a (according to 1H NMR) was utilized in test catalytic runs.
[PhLutHIPT2N3N]MoN (4b)
Me3SiN3 (0.044 g, 0.38 mmol) and 3b (0.138 g, 0.092 mmol) were added to 25 mL of benzene and the mixture was heated overnight at 90°C. The resulting yellow solution was stripped to dryness in vacuo. The residue was taken up in pentane, the mixture was filtered through Celite, and filtrate volume was reduced to 0.5 ml in vacuo. Upon cooling, 50 mg of a bright, canary yellow solid was obtained (37%): 1H NMR (C6D6, 20°C) δ 8.35 (d, 4H, JHH=1.3 Hz, PhLut, 2,6,2″,6″-H), 8.14 (s, 2H, PhLut 3′,5′-H), 7.79 (d, 4H, JHH = 1.1 Hz, HIPT 3′,5′-H), 7.40 (m, 6H, PhLut 3,4,5,3″,4″,5″-H), 7.17 (m, 8H, HIPT 3,5,3″,5″-H), 6.73 (br t, 2H, JHH = 1.1 Hz, HIPT 4′-H), 3.59 (br t, 4H, JHH = 5.1 Hz, HIPT-NHCH2CH2), 3.20 (br t, 2H, JHH = 4.2 Hz, PhLut-NCH2CH2), 3.11 (septet, 8H, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.89 (septet, JHH = 6.9 Hz, 4H, 4,4″-CHMe2), 2.08 (br t, 2H, JHH= 3.4 Hz, PhLut-NCH2CH2), 2.02 (br t, 4H, JHH = 5.1 Hz, HIPT-NCH2CH2), 1.33 (dd, 24H, 4,4″-CH(CH3)2), 1.22 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2), 1.19 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2), 1.16 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2), 1.11 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2). Anal Calcd for C95H122MoN6: C, 79.02; H, 8.52; N, 5.82. Found: C, 78.88; H, 8.45; N, 5.88.
[3,5-Bis(CF3)HIPT2N3N]MoN (4c)
This compound was made in a manner similar to other compounds of its type. Briefly, 0.17 g (117 mmol) of 3c and 0.07 g (608 mmol) of Me3SiN3 was added to 50 mL of benzene in a Teflon® sealed glass bomb. This was heated to 90°C for 12 hours and then brought to dryness in vacuo. The resulting yellow solid was taken into pentane and filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized to yield 0.12 g (72%) of a bright yellow solid: 1H NMR (C6D6, 20°C) δ 7.93 (s, 2H, CF3 arm 2,6-H), 7.70 (d, JHH = 1.4 Hz, 4H, HIPT-2′,6′-H), 7.39 (s, 1H, CF3 arm 4-H), 7.19 (s, 8H, HIPT 3,5,3″,5″-H), 6.81 (br t, JHH = 1.1 Hz, 2H, HIPT 4′-H), 3.53 (br t, JHH = 4.7 Hz, 4H, HIPT-NCH2CH2), 3.15 (overlapping septets, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.90 (septet, JHH = 6.9 Hz, 4H, 4,4″-CHMe2), 2.83 (br t, JHH = 4.8 Hz, 2H, CF3 arm-NCH2CH2), 1.97 (br t, JHH = 5.1 Hz, 4H, HIPT-NHCH2CH2), 1.92 (br t, JHH = 5.0 Hz, 2H, CF3 arm-NCH2CH2), 1.34 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.26 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2), 1.22 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2), 1.20 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2), 1.11 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″ - CH(CH3)2). 19F NMR (C6D6, 20°C), d −62.4. Anal Calcd for C86H113F6MoN5: C, 72.40; H, 7.98; N, 4.91. Found: C, 72.56; H, 7.98; N, 4.81.
[3,5-DimethylHIPT2N3N]MoN (4d)
This compound was made in a similar manner to other compounds of this type. Briefly, 0.15 g (0.11 mmol) of 2d and 0.064 g (0.56 mmol) of Me3SiN3 was added to 40 mL benzene and the mixture was heated in a Teflon®-sealed bomb at 100°C for one day. The volatiles were removed in vacuo and the resulting solid was taken up in pentane and the mixture was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized to yield 0.073 g (50%) of a bright yellow powder: 1H NMR (C6D6, 20°C) δ 7.86 (d, JHH = 1.1 Hz, 4H, HIPT-2′,6′-H), 7.21 (dd, JHH=1.4 Hz and 3.3 Hz, 8H, HIPT 3,5,3″,5″-H), 7.05 (s, 2H, dimethyl-2,6-H), 7.76 (br t, JHH = 1.1 Hz, 2H, HIPT 4′-H), 6.51 (s, 1H, dimethyl-4-H), 3.54 (br t, JHH = 5.2 Hz, 4H, HIPT-NCH2CH2), 3.64 (br t, JHH = 5.2 Hz, 2H, dimethyl-NCH2CH2), 3.19 (septet, JHH = 6.9 Hz, 8H, HIPT-2,6,2″,6″-CHMe2), 2.93 (septet, JHH = 6.9 Hz, 4H, HIPT-4,4″-CHMe2), 2.18 (s, 6H, dimethyl-CH3), 2.03 (m, 8H, contains HIPT-NCH2CH2 and dimethyl-NCH2CH2), 1.34 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.26 (d, JHH = 6.9 Hz, 24H, 2,2″,6,6″-CH(CH3)2), 1.21 (d, JHH=6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2), 1.16 (d, JHH=6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2). Anal Calcd for C86H119MoN5: C, 78.32; H, 9.09; N, 5.31. Found: C, 78.19; H, 8.96; N, 5.23.
[3,5-DimethoxyHIPT2N3N]MoN (4e)
This was synthesized similarly to other compounds of its type. Briefly, 0.12 g (0.08 mmol) of 3e was added to 0.05 g (0.4 mmol) of Me3SiN3 in 25 mL of benzene. This mixture was heated at ~100°C overnight. The solvent was removed in vacuo and the resulting solid taken up in pentane and the solution was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized from pentane to yield 0.07 g (55%) of a bright yellow powder in multiple crops: 1H NMR (C6D6, 20°C) δ 7.91 (d, JHH = 0.8 Hz, 4H, HIPT-2′,6′-H), 7.23 (d, JHH = 1.8 Hz, 2H, dimethoxy-2,6-H), 7.21 (s, 8H, HIPT 3,5,3″,5″-H), 6.76 (s, 2H, HIPT 4′-H), 6.30 (t, JHH = 1.8 Hz, 1H, dimethoxy-4-H), 3.55 (br t, 4H, HIPT-NCH2CH2), 3.43 (s, 6H, -OCH3), 3.31 (br t, 2H, dimethoxy-NCH2CH2), 3.20 (septet, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 2.92 (septet, JHH = 6.9 Hz, 4H, 4,4’’’’-CHMe2), 1.94 (br t, 6H, overlapped HIPTNCH2CH2 and dimethoxyNCH2CH2), 1.32 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.22 (overlapping doublets, JHH = 6.9 Hz, 24H, 2,6,2″,6″ -CH(CH3)2), 1.14 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2). Anal. Calcd for C86H119MoN5O2: C, 76.47; H, 8.88; N, 5.18. Found: C, 76.38; H, 8.85; N, 5.06.
[MesHIPT2N3N]MoN (4f)
This compound was synthesized similarly to compounds of this type. Briefly, 0.19 g (0.14 mmol) of 3f was combined with 0.1 g (0.86 mmol) of Me3SiN3 in 25 mL of benzene and heated in a Teflon® sealed glass bomb at 90°C for one day. The solvent was removed in vacuo, and the solid was extracted into pentane and the extract was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized from pentane to yield 0.11 g (59%) of a bright yellow powder: 1H NMR (C6D6, 20°C) δ 7.76 (d, JHH = 1.1 Hz, 4H, HIPT-2′,6′-H), 7.20 (s, 8H, HIPT 3,5,3″,5″-H), 6.70 (br t, JHH = 1.1 Hz, 2H, HIPT 4′-H), 6.65 (s, 2H, Mes-3,5-H), 3.47 (m, 4H, HIPT-NCH2CH2), 3.20 (m, 10H, containing 2,6,2″,6″-CHMe2 and Mes-NCH2CH2), 2.90 (septet, JHH = 6.9 Hz, 4H, 4,4″-CHMe2), 2.25 (s, 9H, Mes-CH3) 2.1 (m, 6H, containing Mes-NCH2CH2, and HIPT-NCH2CH2), 1.34 (d, JHH = 6.9 Hz, 24H, 4,4″-CH(CH3)2), 1.30 (d, JHH = 6.9 Hz, 24H, 2,6,2″,6″-CH(CH3)2), 1.20 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)22,6,2″,6″), 1.17 (d, JHH = 6.9 Hz, 12H, 2,6,2″,6″-CH(CH3)2). Anal Calcd for C87H121MoN5: C, 78.40; H, 9.15; N, 5.25. Found: C, 77.26; H, 9.17; N, 5.19.
[TripHIPT2N3N]MoN (4g)
This compound was synthesized in a manner similar to previously characterized compounds of this type. Briefly, 0.05 g (0.035 mmol) of 3g was combined with 0.028 g (0.24 mmol) in 10 mL of benzene and heated 100°C for one day. The solvent was removed in vacuo and the resulting solid was extracted into pentane and the extract was filtered through Celite. The solvent was removed in vacuo and the residue was recrystallized from pentane to yield 0.04 g (80%) of a bright yellow powder: 1H NMR (C6D6, 20°C) δ 7.77 (d, JHH = 1.1 Hz, 4H, HIPT-2′,6′-H), 7.19 (m, 8H, HIPT 3,5,3″,5″-H), 7.08 (s, 2H, Trip-3,5-H), 6.73 (br t, JHH = 1.1 Hz, 2H, HIPT 4′-H), 3.52 (m, 6H, HIPT-NCH2CH2), 3.34 (br t, JHH = 5.5 Hz, 2H, Trip-NCH2CH2), 3.17 (m, 10H, containing HIPT-2,6,2″,6″-CHMe2, and Trip-2,6- CHMe2), 2.95 (septet, JHH = 6.9 Hz, 2H, HIPT-4,4″-CHMe2), 2.81 (septet, JHH = 6.9 Hz, 1H, Trip-4-CHMe2), 2.21 (m, 8H, containing Trip-NCH2CH2 and HIPT-NCH2CH2), 1.38 (d, JHH = 6.9 Hz, 24H, HIPT 4,4″-CH(CH3)2), 1.24 (m, 36H, contains Trip-4- CH(CH3)2 and HIPT-2,6,2″,6″-CH(CH3)2), 1.13 (m, 24H, contains Trip- 2,6,2″,6″-CH(CH3)2 and HIPT- 2,6,2″,6″-CH(CH3)2). Anal Calcd for C93H133MoN5: C, 78.83; H, 9.46; N, 4.94. Found: C, 78.91; H, 9.48; N, 4.86.
{[3,5-Bis(CF3)HIPT2N3N]MoN2}Na(THF)2 (5c)
Compound 3c (0.56 g) was dissolved in 20 mL THF and 4.8 g of 0.5% Na/Hg amalgam was added. The mixture was stirred with a glass stir bar for two hours until the solution turned a deep green. The solution was decanted from the mercury and the volatiles were removed in vacuo. The resulting solids were dissolved in pentane and the mixture was filtered through Celite to yielding a deep purple solution. The product was recrystallized from pentane to yielding 0.45 g (70%) of a purple powder in multiple crops. A similar method was utilized to synthesize the 15N2 labeled species: 1H NMR (C6D6, 20°C) δ 7.68 (s, 1H, CF3 arm 4-H), 7.59 (s, 2H, HIPT 4′-H), 7.45 (d, JHH = 1.2 Hz, 2H, CF3 arm 2,6-H), 7.35 (d, JHH = 1.3 Hz, 4H, HIPT- 2′,6′-H), 7.18 (s, 4H, HIPT 3,5,3″,5″-H), 7.13 (s, 4H, HIPT 3,5,3″,5″-H), 3.76 (m, 6H, containing both HIPT-NCH2CH2 and CF3-NCH2CH2), 3.38 (overlapping septets, JHH = 6.9 Hz, 8H, 2,6,2″,6″-CHMe2), 3.22 (br m, 8H, THF O-CH2), 2.84 (overlapping septets, JHH = 6.9 Hz, 4H, 4, 4″-CHMe2), 1.94 (br m, containing both HIPT-NCH2CH2 and CF3- NCH2CH2), 1.23 (overlapping doublets, 52H containing 2,4,6,2″,4″,6″-CH(CH3)2), 1.1 (d, JHH = 6.9 Hz, 12H, 4, 4″-CH(CH3)2). 19F NMR (C6D6, 20°C), δ −61.76. IR (νNN 1801 cm−1; 15N2 labeled νNN = 1741 cm−1). Elemental analyses are variable as a consequence (it is believed) of a variable amount of THF being present depending on individual preparation and isolation procedures. Variable amounts of THF also have been observed in the parent compound.172
{LMoN2}Na(THF)2
Synthetic procedures for all other LMoN2Na(THF)2 compounds were similar to that for 5c. It was found that both Na/Hg amalgam or finely divided Na sand would reduce LMoCl similarly, so typically Na sand was used. A crystal structure was performed on 5d, but elemental analyses were not successful and reproducible for any compound of this type.
LMoN2 (6a–g)
No neutral dinitrogen complexes could be isolated as pure compounds. They could be observed only via IR and NMR spectroscopy. Typically LMoCl was treated with a 0.5 % Na/Hg amalgam or sodium sand in THF. When the solution had turned from bright orange to either green (6b, 6f, or 6g) or purple (6a, 6c, 6d, or 6e) it was filtered through a Celite plug onto a mild oxidant such as ZnCl2. The resulting brown solution was then filtered through Celite and the solvent removed in vacuo. NMR and IR analysis suggested that 10–20% of the free ligand was present in the resulting solid, depending on the compound and the specific experiment. The paramagnetically shifted protons in the 1H NMR were similar to those of the symmetric compounds of this type (3 peaks at δ ~ 10 to 20ppm, 2 peaks at ~ −3 to −10 ppm, and 3 peaks at −20 to −40 ppm).
[(3,5-Bis(CF3)HIPT2N3N)MoNH3][BAr4′] (7c)
This compound has been identified via NMR from the 1:1 reaction of 3c and NaBAr4′ under 6 equivalents of ammonia in dichloromethane. After the mixture was stirred for one day, the solvent was removed in vacuo and the resulting solid was filtered through Celite using pentane as the eluent. The solvent was removed in vacuo and the residue was recrystallized from pentane to yield the product as a red solid: 1H NMR (C6D6, 20°C) δ 8.23 (s, BAr4′), 7.61 (s, BAr4′), 7.12 (br s), 2.88 (br s), 2.68 (br s), 1.29 (br s), 1.16 (br s), −12.5 (br s), −17.5 (br s), −33 (br s); 19F NMR (C6D6, 20°C) δ −61.3 (MoNH3+), −61.45 (BAr4′). We were unable to remove NaBAr4′ from the solid completely (~5% via 19F NMR).
[(3,5-DimethoxyHIPT2N3N)Mo(NH3)][BAr4′] (7e)
A mixture of 3e (100 mg) and 65 mg of Na[BAr4′] was dissolved in 10 mL of CH2Cl2 in a 50 mL Teflon® sealed vessel. Dry ammonia (200 torr, ~6 equiv) was vacuum-transferred onto this solution. The reaction mixture turned bright red after being stirred for 6 h. The volatiles were removed in vacuo and the resulting solid was extracted into pentane. The pentane extract was filtered through Celite and the solvents were removed in vacuo. The product was isolated as a red solid upon crystallization of the residue from pentane; yield 120 mg (75%): 1H NMR (C6D6, 20°C) δ 8.36 (br s, BAr4′), 7.68 (br s, BAr4′), 6.67 (br s), 6.36 (br s), 3.5 (br s), 3.2 (s), 2.7 (br m), 1.85 (br m), 1.2 (br m), −4.0 (br s), −8.5 (br s). Anal. Calcd for C118H134BF24MoN5O2: C, 63.92; H, 6.09; N, 3.16. Found: C, 64.10; H, 6.13; N, 2.97.
X-Ray Structural Studies
Low temperature diffraction data were collected on a Siemens Platform three-circle diffractometer coupled to a Bruker-AXS SMART Apex CCD detector with graphite-monochromated MoKα radiation (λ = 0.71073 Å), performing φ and ω-scans. The structures were solved by direct methods using SHELXS19 and refined against F2 on all data by full-matrix least squares with SHELXL-97.20 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were included into the model at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms they are linked to (1.5 times for methyl groups). Crystal and structural refinement data for all structures is listed in the supporting information.
The structure of 3c is strongly affected by disorder: both CFs groups and about half of all carbon atoms are found distributed over two positions. These disorders were refined with the help of similarity restraints on 1–2 and 1–3 distances and displacement parameters as well as rigid bond restraints for anisotropic displacement parameters. The relative occupancies of the disordered components were refined freely, while constraining the total occupancy of both components to unity. Probably owing to the massive disorder the crystal diffracted only to about 1.0 Å resolution and gave rise to data of only mediocre quality. To counteract the effects of the resulting low data-to-parameter ratio, rigid bond and similarity restraints were used for the displacement parameters of all atoms.
5d crystallizes with one molecule of C86H119MoN6, one sodium ion and the following solvent molecules in the asymmetric unit: two THF molecules (one of which is disordered) coordinated to the sodium ion, one sixth of a non-coordinated THF molecule (sixfold disordered about the crystallographic -3 axis), two half occupied heptane molecules, and one sixth of a pentane molecule (sixfold disordered about the crystallographic -3 axis). The pentane molecule is probably a heptane molecule where the two methyl groups are disordered in addition to the disorder described, however refinement as heptane was not stable. These disorders were refined as described above.
Supplementary Material
Crystal data and structure refinement, atomic coordinates and equivalent isotropic displacement parameters, bond lengths and angles, and anisotropic displacement parameters for 3c and 5d. This material is available free of charge via the Internet at http://pubs.acs.org (3c = 04072) and 5d = 04169).
Acknowledgments
R.R.S. is grateful to the National Institutes of Health (GM 31978) for research support.
References
- 1.(a) Schrock RR. Acc Chem Res. 1997;30:9. [Google Scholar]; (b) Greco GE, Schrock RR. Inorg Chem. 2001;40:3850. doi: 10.1021/ic001122v. [DOI] [PubMed] [Google Scholar]; (c) Greco GE, Schrock RR. Inorg Chem. 2001;40:3861. doi: 10.1021/ic001123n. [DOI] [PubMed] [Google Scholar]; (d) O’Donoghue MB, Davis WM, Schrock RR. Inorg Chem. 1998;37:5149. [Google Scholar]
- 2.Dobbs DA, Schrock RR, Davis WM. Inorg Chim Acta. 1997;36:171. [Google Scholar]
- 3.Greco GE, O’Donoghue MB, Seidel SW, Davis WM, Schrock RR. Organometallics. 2000;79:1132. [Google Scholar]
- 4.(a) Yandulov DV, Schrock RR. Science. 2003;301:76. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]; (b) Yandulov DV, Schrock RR. Inorg Chem. 2005;44:1103. doi: 10.1021/ic040095w. [DOI] [PubMed] [Google Scholar]; (c) Yandulov DV, Schrock RR. Canad J Chem. 2005;83:341. [Google Scholar]
- 5.Schrock RR. Acc Chem Res. 2005;38:955. doi: 10.1021/ar0501121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritleng V, Yandulov DV, Weare WW, Schrock RR, Hock AR, Davis WM. J Am Chem Soc. 2004;126:6150. doi: 10.1021/ja0306415. [DOI] [PubMed] [Google Scholar]
- 7.Weare WW, Dai C, Byrnes MJ, Chin J, Schrock RR. Proc Nat Acad Sci. doi: 10.1073/pnas.0602778103. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Wolfe JP, Wagaw S, Marcoux J-R, Buchwald SL. Acc Chem Res. 1998;31:805. [Google Scholar]; (b) Hartwig J. Acc Chem Res. 1998;31:852. [Google Scholar]
- 9.(a) Lehnert N, Tuczek F. Inorg Chem. 1999;38:1659. doi: 10.1021/ic980939+. [DOI] [PubMed] [Google Scholar]; (b) Lehnert N, Tuczek F. Inorg Chem. 1999;38:1671. doi: 10.1021/ic9809409. and references therein. [DOI] [PubMed] [Google Scholar]
- 10.(a) Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]; (b) Hardy RWF, Bottomley F, Burns RC. A Treatise on Dinitrogen Fixation. Wiley-Interscience; New York: 1979. [Google Scholar]; (c) Veeger C, Newton WE. Advances in Nitrogen Fixation Research. Dr. W. Junk/Martinus Nijhoff; Boston: 1984. [Google Scholar]; (d) Coughlan MP. Molybdenum and Molybdenum-containing Enzymes. Pergamon; New York: 1980. [Google Scholar]
- 11.(a) Rees DC, Howard JB. Curr Opinion Chem Biol. 2000;4:559. doi: 10.1016/s1367-5931(00)00132-0. [DOI] [PubMed] [Google Scholar]; (b) Bolin JT, Ronco AE, Morgan TV, Mortenson LE, Xuong LE. Proc Nat Acad Sci. 1993;90:1078. doi: 10.1073/pnas.90.3.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim J, Rees DC. Science. 1992;257:1677. doi: 10.1126/science.1529354. [DOI] [PubMed] [Google Scholar]; (d) Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 12.The only other known systems that will catalytically reduce dinitrogen are those discovered by Shilov (Shilov AE. Russ Chem Bull Int Ed. 2003;52:2555.). These systems require Mo and are catalytic with respect to Mo, but reduce dinitrogen to a mixture of hydrazine and ammonia (~10:1). The reaction is run in methanol in the presence of magnesium hydroxide and a strong reducing agent such as sodium amalgam. Few mechanistic details are known.
- 13.(a) Eady RR. Chem Rev. 1996;96:3013. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]; (b) Smith BE. Adv Inorg Chem. 1999;47:159. [Google Scholar]; (c) Rehder D. Coord Chem Rev. 1999;182:297. [Google Scholar]
- 14.(a) Brown SD, Peters JC. J Am Chem Soc. 2005;127:1913. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]; (b) Brown SD, Peters JC. J Am Chem Soc. 2004;126:4538. doi: 10.1021/ja0399122. [DOI] [PubMed] [Google Scholar]; (c) Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]; (d) Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]; (e) Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J Am Chem Soc. 2006;128:756. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]; (f) Holland PL. Can J Chem. 2005;83:296. [Google Scholar]; (g) Smith JM, Lachicotte RJ, Holland PL. J Am Chem Soc. 2003;125:15752. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]; (h) Smith JM, Lachicotte RJ, Holland PL. J Am Chem Soc. 2003;125:15752. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]
- 15.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Orgamometallics. 1996;15:1518. [Google Scholar]
- 16.Yandulov DV, Schrock RR. J Am Chem Soc. 2002;124:6252. doi: 10.1021/ja020186x. [DOI] [PubMed] [Google Scholar]
- 17.Yandulov DV, Schrock RR, Rheingold AL, Ceccarelli C, Davis WM. Inorg Chem. 2003;42:796. doi: 10.1021/ic020505l. [DOI] [PubMed] [Google Scholar]
- 18.Talik T, Talik Z, Ban-Oganowska H. Synthesis. 1974:293. [Google Scholar]
- 19.Sheldrick GM. Acta Cryst. 1990;A46:467. [Google Scholar]
- 20.Sheldrick GM. SHELXL 97. University of Göttingen; Germany: 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal data and structure refinement, atomic coordinates and equivalent isotropic displacement parameters, bond lengths and angles, and anisotropic displacement parameters for 3c and 5d. This material is available free of charge via the Internet at http://pubs.acs.org (3c = 04072) and 5d = 04169).