

Abstract
Delineating the mechanisms of survival pathways that exist in neurons will provide important insight into how neurons utilize intracellular proteins as neuroprotectants against the causes of acute neurodegeneration. We have employed cultured rat cerebellar granule cells as a model for determining the mechanisms of these intraneuronal survival pathways. Glutamate has long been known to kill neurons by an N-methyl-d-aspartate (NMDA) receptor-mediated mechanism. Paradoxically, subtoxic concentrations of NMDA protect neurons against glutamate-mediated excitotoxicity. Because NMDA protects neurons in physiologic concentrations of glucose and oxygen, we refer to this phenomenon as physiologic preconditioning. One of the major mechanisms of NMDA neuroprotection involves the activation of NMDA receptors leading to the rapid release of brain-derived neurotrophic factor (BDNF). BDNF then binds to and activates its cognate receptor, receptor tyrosine kinase B (TrkB). The efficient utilization of these two receptors confers remarkable resistance against millimolar concentrations of glutamate that kill more than eighty percent of the neurons in the absence of preconditioning the neurons with a subtoxic concentration of NMDA. Exactly how the neurons mediate neuroprotection by activation of both receptors is just beginning to be understood. Both NMDA and TrkB receptors activate nuclear factor kappaB (NF-kB), a transcription factor known to be involved in protecting neurons against many different kinds of toxic insults. By converging on survival transcription factors, such as NF-kB, NMDA and TrkB receptors protect neurons. Thus, crosstalk between these very different receptors provides a rapid means of neuronal communication to upregulate survival proteins through release and transcriptional activation of messenger RNA.
Keywords: N-methyl-d-aspartate, TrkB, receptor activation, cerebellar granular cells, survival pathways, neurons
PRECONDITIONING AND NEUROPROTECTION
Preconditioning in vivo has been long been known to reduce infarct size. The initial study was conducted in gerbil brain where two-minute priming insults were performed followed by five minutes of test challenge at intervals of one to four days. Determination of surviving neurons in the CA1 region of the hippocampus was conducted seven days after recirculation.1,2 Similar paradigms performed in rats have also demonstrated tolerance.3 Protection in brain regions other than the hippocampus has been demonstrated in the gerbil model.4 Importantly, significant protection has been observed in the gerbil hippocampus CA1 neurons where the priming insult was repeated over successive days2 or after single insults.5 Other models, such as spreading depression6-10 and global ischemia,11 have been found to be neuroprotective against a subsequent ischemic insult. Although the mechanism(s) of tolerance have not been delineated, it seems clear that ischemia induced in gerbils result in the release of the excitatory amino acid glutamate as well as other amino acids.12
The role of NMDA receptors in glutamate-mediated excitotoxicity has been studied in rat cerebellar granule cells in vitro. Overactivation of NMDA receptors by excitotoxic concentrations of either glutamate (100μM) or NMDA (1mM) results in neuronal cell death.13 NMDA is about twenty-fold less potent than glutamate in mediating neuronal cell death via NMDA receptors.13
Paradoxically, subtoxic concentrations of glutamate or NMDA protect vulnerable neurons against the excitotoxic effects of glutamate acting on NMDA receptors in cultured rat cerebellar granule cells.13 The neuroprotective effect was mediated by NMDA receptors and was time and concentration dependent.14 Since our initial report, other laboratories have demonstrated neuroprotection in neuronal culture models including pretreating with glutamate,15 through the release of glutamate in magnesium-free medium16 or in oxygen-glucose deprivation models.17,18
NMDA PROTECTS CULTURED NEURONS VIA A BDNF AUTOCRINE LOOP
Although in vivo protection models require further characterization, it seems clear that neuronal vulnerability can be reduced by seemingly fundamental and lasting changes in animal and in neuronal culture model paradigms. We employed cultured cerebellar granule cells to further characterize the neuroprotective effect of NMDA because: (1) the neurons are relatively homogenous (about 95% neurons); (2) the neurons express all of the glutamate receptor subtypes; and (3) the neurons are responsive to various neurotrophic factors and in particular to the neurotrophins,19,20 a family of trophic factors related by primary amino acid sequence homology, whose members include brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), neurotrophin-3 (NT-3), and NT-4/5.21-25 The biological activity of neurotrophins depends upon the activation of high-affinity receptors (Trk), a family of structurally related receptors having a similar intrinsic protein-tyrosine kinase activity26,27 but different ligand binding properties. TrkA, TrkB, and TrkC are, respectively, the receptors for NGF, BDNF, and NT-3.28-30
We established that BDNF, but not NGF or NT-3, is the only neurotrophin that protects cerebellar granule cells against glutamate toxicity on day eight in vitro. This is probably attributed to the fact that TrkB is the only functional neurotrophin receptor expressed in cerebellar granule cells on day eight in vitro, as also demonstrated by the ability of BDNF to induce phosphorylation of phospholipase C-γ.31 This protein interacts with activated Trk and is involved in neurotrophin signaling.32 Most importantly, we showed that NMDA elicited a time-dependent increase in BDNF in the culture medium and TrkB tyrosine phosphorylation, suggesting that one of the mechanisms underlying the ability of NMDA to activate TrkB tyrosine phosphorylation is the increased release of BDNF, which in turn activated tyrosine phosphorylation of its own receptor in an autocrine manner.33 This autocrine loop, although described for developing sensory neurons of the dorsal root ganglia,34 is novel for central nervous system neurons. The accumulation of BDNF in the culture medium was an early event in that it occurred within two minutes after exposure to a subtoxic concentration of NMDA (100μM). The accumulation was greater at five minutes and was not attributable to an increase in intracellular BDNF levels or BDNF mRNA because intracellular protein and mRNA content were unchanged. An increase in BDNF levels in the culture medium was also observed after a three-hour incubation with NMDA. By this time, the NMDA-mediated increase in BDNF in the medium was accompanied by a concomitant accumulation of BDNF mRNA, suggesting that at three hours, in addition to the release of BDNF, NMDA affected BDNF synthesis. Thus, NMDA elicited two temporally distinct responses: an early release of BDNF and a later synthesis and release of BDNF.33
NMDA ACTIVATES NUCLEAR FACTOR kB TO PROTECT NEURONS AGAINST GLUTAMATE-MEDIATED EXCITOTOXICITY
The nuclear factor kappa B (NF-kB) family of dimeric transcription factors regulates genes involved in immunologic responses, cell proliferation, growth factor regulation, and apoptosis. Also known as the Rel family of transcription factors, the transcriptionally active forms of NF-kB are made up from combinations of the five monomeric polypeptides, p50 (NF-kB1), p65 (RelA), p52 (NF-kB2), c-Rel, and RelB. Each of the monomeric proteins shares a 300-amino acid region known as the Rel homology domain (RHD).35 In addition, the RHD confers DNA binding activity of Rel family members to kB elements having the consensus sequence 5'-GGPuNNPyPyCC-3'.35 Translocation to the nucleus and DNA binding by NF-kB are inhibited through the non-covalent association of one of a family of transcription factor inhibitor proteins, called I-kB (I-kBα, I-kBβ, I-kBγ, or I-kBε).36 A number of signal transduction cascades converge and result in the phosphorylation of I-kB allowing the NF-kB dimer to translocate into the nucleus and to bind to consensus sequences of DNA indicated above for transcriptional activation of specific mRNAs.37 Previous work has demonstrated that glutamate activates NF-kB in neurons.38,39
Four promoters of the rat BDNF gene are known to be active in brain.40 The primary DNA sequences of these promoter regions are highly conserved between rats and humans and appear to be differentially transcribed in the brain, at least in the rat.41 The rat BDNF gene is composed of four 5'- untranslated exons (1-4), and each BDNF transcript is initiated separately within the 5' flanking region from each of these untranslated exons. The primary transcripts are differentially spliced to a single 3'- coding region, exon 5, which encodes the entire sequence for the mature polypeptide. At least one functional element has been described that controls levels of exon3-specific BDNF mRNAs. This sequence (5'-TCACGTCA-3') is located 35bp from a known exon 3 transcription start site40 and was shown to mediate L-type voltage gated calcium-dependent BDNF gene expression via CREB or some other related transcription factor in primary cortical neurons.42 We observed that one of these segments, from the 5'- flanking region of exon 3, contained an NF-kB candidate sequence. Because BDNF is one of the proteins involved in mediating the neuroprotective effect of NMDA and because the rat BDNF gene has potential binding sites for activated NF-kB, we examined if NF-kB played a role in the neuroprotective activity of NMDA.
NEUROPROTECTIVE CONCENTRATIONS OF NMDA ACTIVATE NF-kB
Exposure of cultured cerebellar granule cells to a maximum neuroprotective concentration of NMDA (100μM) induced a distinct retardation band of a DNA-protein binding activity within 40min using the double-stranded rat BDNF gene-derived NF-kB target DNA in an electrophoretic mobility shift assay (see Figure 1). Specificity of the NF-kB-DNA association was shown by incubating the nuclear extracts with a 100-fold molar excess unlabeled double-stranded rat BDNF gene-derived oligonucleotide, which resulted in the disappearance of the band (Fig. 1). Furthermore, using specific antibodies specific to either p50 or p65 of NF-kB demonstrated that both subunits were part of the DNA-protein complex.14
FIGURE 1.

N-Methyl-d-aspartate activates NF-kB in nuclear extracts prepared from rat cerebellar granule cells. Autoradiograph shows that NMDA (100 μM) activates a specific NF-kB DNA-protein binding complex within 40 minutes in cultured rat cerebellar granule cells on day eight in vitro. No specific complex was observed in untreated neurons (no treatment). The addition of an unlabeled competitor DNA resulted in the disappearance of the specific band (NMDA + cold). No bands were detected in the probe (probe only).
BDNF ACTIVATES NF-kB BINDING ACTIVITY
We showed previously that one of the major molecular mechanisms of NMDA neuroprotection is the release of BDNF, which in turn binds to and activates TrkB receptors through an autocrine loop.33 Because NF-kB has been shown to be neuroprotective in many systems, we hypothesized that exogenous BDNF activates TrkB receptors leading to the activation of NF-kB. Incubation of cultured neurons with BDNF (100ng/mL) induced two distinct retardation bands using the double-stranded rat BDNF gene-derived NF-kB target DNA (see Figure 2). Specificity of the NF-kB-DNA interaction was shown by incubating the nuclear extracts with a 100-fold molar excess of unlabeled double-stranded target DNA, which resulted in the disappearance of the specific bands (Fig. 2). Because BDNF activates NMDA receptors via phosphorylation through the release of glutamate and NMDA receptors activate NF-kB (see Fig. 1), the neurons were pretreated with MK-801 (1μM) to ensure that the NMDA receptors were blocked.
FIGURE 2.
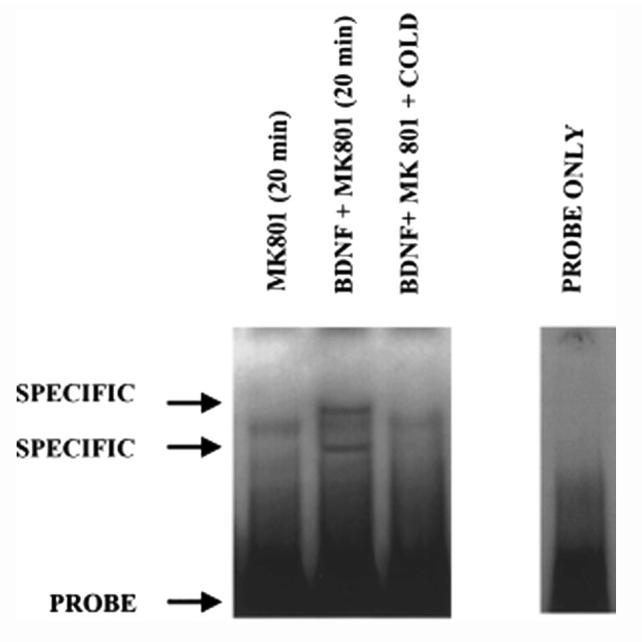
Brain-derived neurotrophic factor activates NF-kB in nuclear extracts prepared from rat cerebellar neurons. Two specific DNA-protein binding complexes were induced in neurons by BDNF (100 ng/mL), a concentration that protects about 30% of the neurons against glutamate excitotoxicity (Marini et al., 1998). The NMDA receptor antagonist, MK-801, was added to the neurons to block NMDA receptors, thereby ensuring that the activation of NF-kB was mediated solely through the activation of TrkB receptors.
NF-kB TARGET DNA BLOCKS NMDA-MEDIATED NEUROPROTECTION
To test the hypothesis that NF-kB was critical for the neuroprotective effect of NMDA in the cultured neurons, we used a small target or “decoy” oligonucleotide described by Mattson et al.43 This target contained the NF-kB-like sequence of the rat BDNF gene that we used in our EMSA-based experiments. The cultured neurons were incubated for twenty-four hours with either the rat BDNF gene-derived NF-kB double-stranded oligonucleotide target or a scramble double-stranded DNA containing the same bases but in a random sequence. Following the addition of either the target or scramble DNA, a maximum neuroprotective concentration of NMDA (100μM) was then added for six hours followed by the addition of an excitotoxic concentration of glutamate (100μM) for twenty-four hours. Neuronal viability was determined using the fluorescein diacetate staining method.13 Pretreatment of the cultured neurons with NMDA (100μM) protected all of the vulnerable neurons against the excitotoxic actions of glutamate acting on NMDA receptors and was comparable to the survival observed in untreated neurons (see Figure 3). In sharp contrast, only about 30% of the neurons survived twenty-four hours after the addition of an excitotoxic concentration of glutamate (100μM). Neuronal loss observed in the presence of the NF-kB target DNA followed by the addition of an identical excitotoxic concentration of glutamate was significantly different compared with neuronal cultures pretreated with either NMDA (100μM) or in the presence of the scramble DNA. These results strongly suggest that the specific factor required for NMDA-mediated neuroprotection is activated NF-kB.
FIGURE 3.
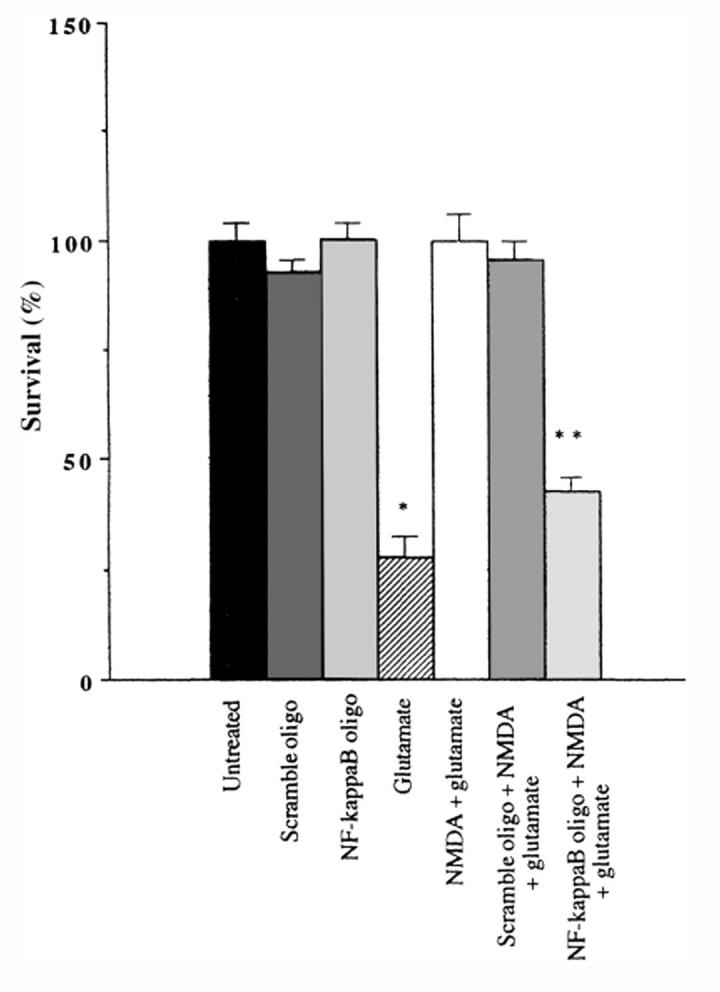
NF-kB DNA oligonucleotide decoy derived from the 5'- flanking region of Exon 3 of the bdnf gene blocks NMDA receptor-mediated neuroprotection. A double-stranded DNA target sequence based upon the 5'- flanking region of exon 3 of the bdnf gene (see Lipsky et al., 2001) was added to granule cell cultures for 24 h (NF-kB oligo). Neurons were also pretreated with a random sequence of the NF-kB oligonucleotide (scramble oligo). On the following day some culture dishes were treated with a maximum neuroprotective concentration of NMDA (100 μM) for six hours followed by the addition of an excitotoxic concentration of glutamate (100 μM) for 24 hours. Neuronal viability was determined by the fluorescein diacetate staining method (Marini and Paul, 1992). Note that pretreatment of the cultured neurons with NMDA for six hours followed by the addition of glutamate protects all of the vulnerable neurons against glutamate. Glutamate alone kills about 70% of the neurons. Pretreatment with the specific NF-kB oligo in the presence of a maximum neuroprotective concentration of NMDA completely blocked NMDA neuroprotection. Data are expressed as mean ± SE, where n = 6; *p < 0.001 compared with untreated neurons; **p < 0.003 compared with neurons treated with NMDA + glutamate.
NMDA NEUROPROTECTION INCREASES BCL-2 GENE EXPRESSION FOLLOWING EXCITOTOXICITY
Glutamate excitotoxicity induces apoptosis in cerebellar granule cells cultured in depolarizing concentrations of potassium chloride.44 Because apoptosis or programmed cell death reduces antiapoptotic genes in the Bcl-2 family, we hypothesized that glutamate-mediated neuronal cell death may involve a reduction in Bcl-2 mRNA, a major antiapoptotic protein in the cultured neurons. Glutamate (100μM) rapidly attenuates mRNA levels of bcl-2. This effect was observed at three and six hours (data not shown), and twenty-four hours following treatment (see Figure 4). We hypothesized that NMDA neuroprotection might be mediated, in part, by blocking the reduction in bcl-2 mRNA. By itself, neuroprotective concentrations of NMDA (100μM) increased bcl-2 (Fig. 4) mRNA levels. Thus, NMDA (100μM) pretreatment reversed the effects of excitotoxicity by increasing bcl-2 mRNA levels 24h after glutamate (100μM) addition (Fig. 4, hatched bars). Pretreatment with MK-801 (1μM) had substantially the same effect.
FIGURE 4.
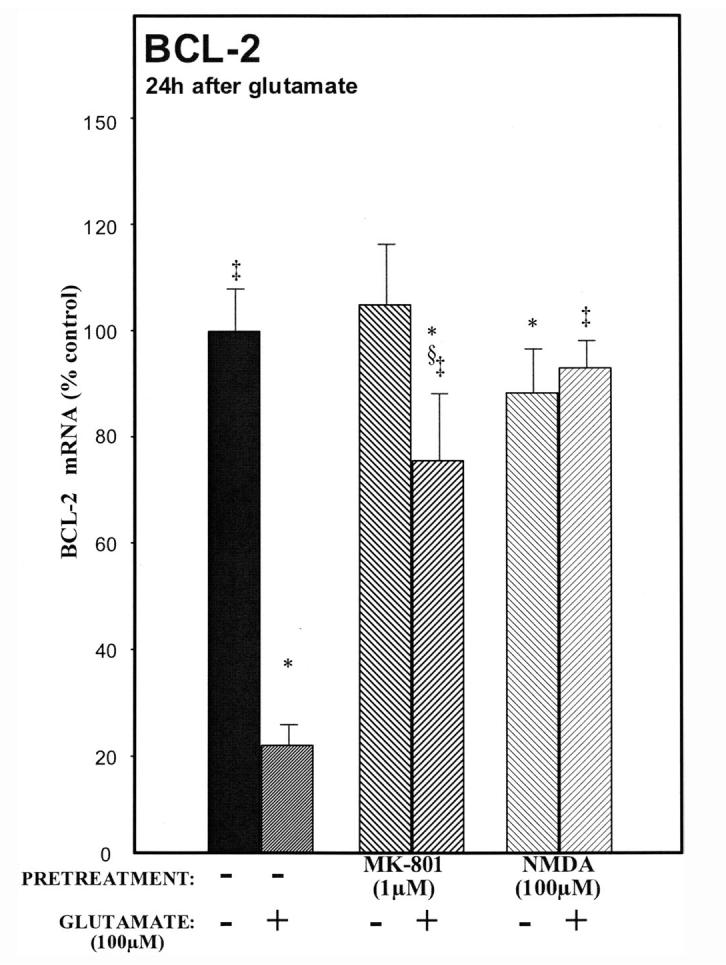
A maximum neuroprotective concentration of NMDA (100 μM) induces Bcl-2 mRNA. A marked decrease in bcl-2 mRNA was observed at twenty-four hours in glutamate-treated neurons (hatched bars). In sharp contrast, preincubation with NMDA (100 μM) followed by the addition of glutamate (100 μM, light hatch) blocked the glutamate-induced decrease in bcl-2 mRNA. This supports the hypothesis that another mechanism exists where NMDA promotes neuronal survival through the anti-apoptotic bcl family of proteins. p < 0.05 versus ‡glutamate, *untreated, or §MK-801.
OVERVIEW OF NMDA RECEPTOR-MEDIATED NEURONAL SURVIVAL
The central hypothesis of the activity-dependent release of BDNF by NMDA receptors from cultured rat cerebellar granule cells is shown in Figure 5. Activation of NMDA receptors results in an influx of calcium through NMDA receptor-associated channels, and BDNF is released, which in turn binds to and activates TrkB receptors. This is the early response in NMDA receptor-mediated neuroprotection against glutamate excitotoxicity. Activation of TrkB receptors along with the influx of calcium through NMDA receptors leads to an activation of the transcription factor nuclear factor kappaB (NF-kB) within 40 minutes that we showed was critical for NMDA neuroprotection (see Fig. 3; Ref. 14). Thus, activation of the transcription factor NF-kB, and possibly other transcription factors such as CREB,42 increase the expression of BDNF mRNA and synthesis of BDNF protein.33 The increase in BDNF mRNA and protein is the later response in NMDA receptor-mediated neuroprotection. In addition, NMDA increases the synthesis of Bcl-2 mRNA, suggesting that the Bcl family of anti-apoptotic proteins play a role in the survival effects of NMDA.
FIGURE 5.

Overview of the neuroprotective activity of N-methyl-d-aspartate. Activation of NMDA receptors results in the influx of extracellular calcium intracellularly and the immediate release (within two minutes) of brain-derived neurotrophic factor (BDNF). Extracellular BDNF then binds to and activates its cognate receptor, TrkB. Thus, coactivation of NMDA and TrkB receptors occurs within ten minutes (see Marini et al., 1998). The influx of calcium and possibly other second messenger systems leads to the phosphorylation of I-kB, the inhibitor of NF-kB. Once phosphorylated, I-kB is rapidly degraded by the ubiquitin-proteosome-mediated pathway resulting in the release of NF-kB. Activated NF-kB translocates to the nucleus where it binds to the 5'- flanking region of exon 3 of the bdnf gene and likely to the promoter of the bcl-2 gene to activate gene transcription to protect vulnerable neurons against the excitotoxic effects of glutamate acting on NMDA receptors. The question marks indicate gaps in knowledge of the interaction between NMDA and TrkB receptors and the signal transduction pathways that mediate the survival effects of coactivation of NMDA and TrkB receptors.
The notion that pretreatment with a subtoxic concentration of NMDA protects neurons against an excitotoxic concentration of glutamate in physiologic concentrations of glucose and oxygen can be viewed as preconditioning. Classic cerebral ischemic preconditioning involves brief episodes of sublethal ischemia, which provide resistance against damage incurred by a lethal ischemic insult.1-3,45 It has also been shown that NMDA receptors play a major role in ischemic preconditioning,46 whereas cycloheximide treatment blocks the neuroprotective effect mediated by ischemic preconditioning in vivo.47 These findings are reminiscent of our own, where pretreatment of cultured neurons with subtoxic concentrations of NMDA are neuroprotective and NMDA neuroprotection is blocked by cycloheximide.13 Thus, our model reflects physiologic preconditioning rather than ischemic preconditioning. Although the molecular mechanisms underlying ischemic preconditioning are unclear, we show that NMDA receptor activation results in the release of BDNF, which plays a major role in establishing the neuroprotective state in the cultured neurons. In addition, the transcriptional activation of BDNF mRNA by NMDA requires NF-kB, a critical factor in NMDA neuroprotection, which increases exon 3 of the bdnf gene.14 Other molecular mechanisms leading to the synthesis and release of BDNF mRNA and protein are under current investigation, but our results suggest that there is crosstalk between NMDA and TrkB receptors.
Crosstalk between two profoundly different receptors promotes neuronal survival by signaling through common pathways to converge on the activation of NF-kB (see Figs. 1 and 2). In this way, excitatory amino acids communicate with postsynaptic neurons through neurotransmission and by regulating their own survival pathways.
REFERENCES
- 1.Kirino T, Tsujita Y, Tamura A. Induced tolerance to ischemia in gerbil hippocampal neurons. J. Cereb. Blood Flow Metab. 1991;11:299–307. doi: 10.1038/jcbfm.1991.62. [DOI] [PubMed] [Google Scholar]
- 2.Kitagawa K, Matsumoto M, Tagaya M, et al. “Ischemic tolerance” phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Kato H, Nakata N, Kogure K. Protection of rat hippocampus against ischemic neuronal damage by pretreatment with sublethal ischemia. Brain Res. 1992;586:121–124. doi: 10.1016/0006-8993(92)91380-w. [DOI] [PubMed] [Google Scholar]
- 4.Kitagawa K, Matsumoto M, Kuwabara K, et al. “Ischemic tolerance” phenomenon detected in various brain regions. Brain Res. 1991;561:203–211. doi: 10.1016/0006-8993(91)91596-s. [DOI] [PubMed] [Google Scholar]
- 5.Abe H, Nowak TS., Jr. Gene expression and induced ischemic tolerance following brief insults. Acta Neurobiol. Exp. 1996;56:3–8. doi: 10.55782/ane-1996-1096. [DOI] [PubMed] [Google Scholar]
- 6.Kawahara N, Croll SD, Wiegand SJ, Klatzo I. Cortical spreading depression induces long-term alterations of BDNF levels in cortex and hippocampus distinct from lesion effects: implications for ischemic tolerance. Neurosci. Res. 1997;29:37–47. doi: 10.1016/s0168-0102(97)00069-2. [DOI] [PubMed] [Google Scholar]
- 7.Obrenovitch TP. Excitotoxicity in neurological disorders: an alternative viewpoint. In: Lo EH, Marwah J, editors. Neuroprotection: Basic and Clinical Aspects. Prominent Press; Scottsdale: 2001. pp. 353–377. [Google Scholar]
- 8.Obrenovitch T, Urenjak J, Wang M. Nitric oxide formation during cortical spreading depression is critical for rapid subsequent recovery of ionic homeostasis. J. Cereb. Blood Flow Metab. 2002;22:680–688. doi: 10.1097/00004647-200206000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Obrenovitch TP, Godukhin OV, Chazot PL. Repetitive spreading depression induces nestin protein expression in the cortex of rats and mice. Is this upregulation initiated by N-methyl-d-aspartate receptors? Neurosci. Lett. 2002;320:161–163. doi: 10.1016/s0304-3940(02)00046-0. [DOI] [PubMed] [Google Scholar]
- 10.Godukhin OV, Obrenovtich TP. Asymmetric propagation of spreading depression along the anteroposterior axis of the cerebral cortex in mice. J. Neurophysiol. 2001;86:2109–2111. doi: 10.1152/jn.2001.86.4.2109. [DOI] [PubMed] [Google Scholar]
- 11.Simon RP, Niiro M, Gwinn R. Prior ischemic stress protects against experimental stroke. Neurosci. Lett. 1993;163:135–137. doi: 10.1016/0304-3940(93)90364-q. [DOI] [PubMed] [Google Scholar]
- 12.Nakata N, Kato H, Kogure K. Ischemic tolerance and extracellular amino acid concentrations in gerbil hippocampus measured by intracerebral microdialysis. Brain Res. Bull. 1994;35:247–251. doi: 10.1016/0361-9230(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 13.Marini AM, Paul SM. N-Methyl-d-aspartate receptor-mediated neuroprotection requires new RNA and protein synthesis. Proc. Natl. Acad. Sci. USA. 1992;89:6555–6559. doi: 10.1073/pnas.89.14.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lipsky RH, Xu K, Zhu D, et al. Nuclear factor kB is a critical determinant in N-methyl-d-aspartate receptor-mediated neuroprotection. J. Neurochem. 2001;78:254–264. doi: 10.1046/j.1471-4159.2001.00386.x. [DOI] [PubMed] [Google Scholar]
- 15.Jonas W, Lin Y, Tortella F. Neuroprotection from glutamate toxicity with ultra-low dose glutamate. Neuroreport. 2001;12:335–339. doi: 10.1097/00001756-200102120-00031. [DOI] [PubMed] [Google Scholar]
- 16.Skaper SD, Facci L, Strijbos PJ. Neuronal protein kinase signaling cascades and excitotoxic cell death. Ann. N.Y. Acad. Sci. 2001;939:11–22. doi: 10.1111/j.1749-6632.2001.tb03606.x. [DOI] [PubMed] [Google Scholar]
- 17.Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J. Neurosci. 1999;19:1657–1662. doi: 10.1523/JNEUROSCI.19-05-01657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nandagopal K, Dawson TM, Dawson VL. Critical role for nitric oxide signaling in cardiac and neuronal ischemic preconditioning and tolerance. J. Pharmacol. Exp. Ther. 2001;297:474–478. [PubMed] [Google Scholar]
- 19.Lindholm D, Dechant G, Heisenberg C-P, Thoenen H. Brain-derived neurotrophic factor is a survival factor for cultured rat cerebellar granule cell neurons and protects them against glutamate-induced neurotoxicity. Eur. J. Neurosci. 1993;5:1455–1464. doi: 10.1111/j.1460-9568.1993.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 20.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu. Rev. Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 21.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 22.Hohn A, Leibrock J, Bailey K, Barde Y-A. Identification and characterization of a novel member of the nerve growth factor/brain-derived neurtrophic factor family. Nature. 1990;344:339–341. doi: 10.1038/344339a0. [DOI] [PubMed] [Google Scholar]
- 23.Maisonpierre PC, Belluscio L, Squinto S, et al. NT-3, BDNF, and NGF in the developing rat nervous system: parellel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 24.Jones KR, Reichardt LF. Molecular cloning of a human gene that is a member of the nerve growth factor family. Proc. Natl. Acad. Sci. USA. 1990;87:8060–8064. doi: 10.1073/pnas.87.20.8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenthal A, Goeddel DV, Nguyen T, et al. Primary structure and biological activity of a novel human neurotrophic factor. Neuron. 1990;4:767–773. doi: 10.1016/0896-6273(90)90203-r. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan DR, Hempstead BL, Martin-Zanca D, et al. The trk proto-oncogene product: a signal tranducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Tebar A, Dechant G, Barde YA. Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron. 1990;4:487–492. doi: 10.1016/0896-6273(90)90107-q. [DOI] [PubMed] [Google Scholar]
- 29.Lamballe F, Klein R, Barbacid M. TrkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell. 1990;66:967–979. doi: 10.1016/0092-8674(91)90442-2. [DOI] [PubMed] [Google Scholar]
- 30.Klein R, Nanduri V, Jing SA, et al. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zirriebel OU, Ohga Y, Carter B, et al. Characterization of TrkB receptor-mediated signaling pathways in rat cerebellar granule neurons: involvement of protein kinase C in neuronal survival. J. Neurochem. 1995;65:2241–2250. doi: 10.1046/j.1471-4159.1995.65052241.x. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J. Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 33.Marini AM, Rabin SJ, Lipsky RH, Mocchetti I. Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J. Biol. Chem. 1998;273:29394–29399. doi: 10.1074/jbc.273.45.29394. [DOI] [PubMed] [Google Scholar]
- 34.Acheson AJ, Conover C, Fandl JP, et al. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 35.Baeuerle PA, Henkel T. Function and activation of NF-kB in the immune system. Annu. Rev. Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 36.Beg A, Baldwin A. The I-kB proteins: multifunctional regulators of Rel/NF-kB transcription factors. Genes Dev. 1993;7:2064–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 37.Brown K, Gerstberger S, Carlson L, et al. Control of I-kB-α proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 38.Guerrini L, Blasi F, Denis-Donini S. Synaptic activation of NF-kB by glutamate in cerebellar granule neurons in vitro. Proc. Natl. Acad. Sci. USA. 1995;92:9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaltschmidt C, Kaltschmidt B, Baeuerle AP. Stimulation of inotropic glutamate receptors activates transcription factor NF-kB in primary neurons. Proc. Natl. Acad. Sci. USA. 1995;92:9618–9622. doi: 10.1073/pnas.92.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Timmusk T, Palm K, Metsis M, et al. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- 41.Metsis M, Timmusk T, Arenas E, Persson H. Differential use of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc. Natl. Acad. Sci. USA. 1993;90:8802–8806. doi: 10.1073/pnas.90.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao X, Finkbeiner S, Arnold DB, et al. Calcium influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1997;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 43.Mattson MP, Goodman Y, Luo H, et al. Activation of NF-kB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J. Neurosci. Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 44.Banaudha K, Marini AM. AMPA prevents glutamate-induced neurotoxicity and apoptosis in cultured cerebellar granule cell neurons. Neurotox. Res. 2000;2:51–61. doi: 10.1007/BF03033327. [DOI] [PubMed] [Google Scholar]
- 45.Kato H, Liu Y, Araki T, Kogure K. Temporal profile of the effects of pretreatment with brief cerebral ischemia on the neuronal damage following secondary ischemic insult in the gerbil: cumulative damage and protective effects. Brain Res. 1991;553:238–242. doi: 10.1016/0006-8993(91)90831-f. [DOI] [PubMed] [Google Scholar]
- 46.Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci. Lett. 1992;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- 47.Barone FC, White RF, Spera PA, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke. 1998;29:1937–1950. doi: 10.1161/01.str.29.9.1937. [DOI] [PubMed] [Google Scholar]