

Abstract
Dithiolethiones are a well-known class of cancer chemopreventive agents, whose key mechanism of action involves activation of Nrf2 signaling and induction of Phase 2 enzymes. In the past, attention has been focused mainly on 4-methyl-5-pyrazinyl-3H-1,2-dithiole-3-thione (oltipraz), which showed ability as a wide-spectrum inhibitor of chemical carcinogenesis in preclinical models. However, clinical trials of oltipraz have shown questionable efficacy and, at the high doses employed in such studies, significant side effects were observed. Dithiolethiones that are markedly more effective and potent than oltipraz in both induction of Phase 2 enzymes and inhibition of chemical carcinogenesis in preclinical studies have been identified, and these compounds have shown pronounced organ specificity in vivo. Further investigation of these compounds may lead to development of effective and safe agents for cancer prevention in humans.
Keywords: dithiolethiones, chemoprevention, oltipraz, Phase 2 enzymes, cancer prevention
Introduction
3H-1,2-Dithiole-3-thiones (dithiolethiones) are a class of organosulfur compounds of the general structure shown in Fig. 1. The first synthesis of a compound of this class appears to have been by the Italian chemist G. A. Barbaglia who, in 1884, isolated and purified a substance of formula C5H6S3 from the reaction between isovaleraldehyde and sulfur (1). He was unable to determine its structure, but from its melting point and the method of synthesis, it would appear likely that this substance was 4,5-dimethyl-3H-1,2-dithiole-3-thione (Compound 6, Fig. 1). The parent compound of this class, 3H-1,2-dithiole-3-thione (D3T, Compound 1, Fig. 1) was first synthesised in 1948 (2). Many more derivatives were prepared in the late 1940s and 1950s, when considerable interest developed, particularly in France, in the potential industrial and pharmaceutical uses of such compounds (3). Very few achieved commercial success, but two, 5-(4-methoxyphenyl)-1,2-dithiole-3H-3-thione (ADT, Compound 14, Fig. 1) and 4-methyl-5-pyrazinyl-1,2-dithiole-3H-3-thione (oltipraz, Compound 15, Fig. 1), moved on to efficacy trials in humans with regard to therapeutic use. ADT found use as a saliva stimulant (4) and oltipraz was employed in the treatment of schistosomiasis (5, 6).
Figure 1.
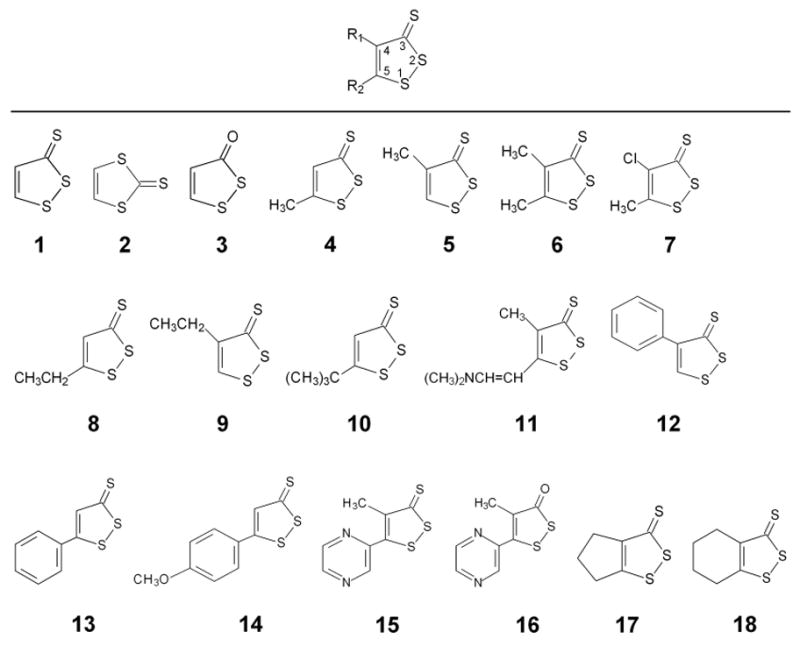
Names and chemical structures of dithiolethiones discussed in this review. Compound 1, 3H-1,2-dithiole-3-thione; Compound 2, 1,3-dithiole-2-thione; Compound 3, 3H-1,2-dithiole-3-one; Compound 4, 5-methyl-3H-1,2-dithiole-3-thione; Compound 5, 4-methyl-3H-1,2-dithiole-3-thione; Compound 6, 4,5-dimethyl-3H-1,2-dithiole-3-thione; Compound 7, 4-chloro-5-methyl-3H-1,2-dithiole-3-thione; Compound 8, 5-ethyl-3H-1,2-dithiole-3-thione; Compound 9, 4-ethyl-3H-1,2-dithiole-3-thione; Compound 10, 5-tert-butyl-3H-1,2-dithiole-3-thione; Compound 11, 5-[2-dimethylamino)vinyl]-4-methyl-3H-1,2-dithiole-3-thione; Compound 12, 4-phenyl-3H-1,2-dithiole-3-thione; Compound 13, 5-phenyl-3H-1,2-dithiole-3-thione; Compound 14, 5-(4-methoxyphenyl)- 3H-1,2-dithiole-3-thione); Compound 15, 4-methyl-5-pyrazinyl-3H-1,2-dithiole-3-thione; Compound 16, 4-methyl-5-pyrazinyl-3H-1,2-dithiole-3-one; Compound 17, 5,6-dihydrocyclopenta[c]-1,2-dithiole-3(4H)-thione; Compound 18, 4,5,6,7-tetrahydrobenzo[c]-1,2-dithiole-3-thione.
In the 1980s, interest in this class of compound was greatly stimulated by the work of Bueding’s group at Johns Hopkins University, who reported that oltipraz and ADT, when fed to mice, significantly elevated hepatic levels of glutathione (GSH) and glutathione S-transferase (GST) and protected against the toxic effects of acetaminophen and carbon tetrachloride (5). Enthusiasm about these and other dithiolethiones was further increased when subsequent studies by Bueding and others showed that the dithiolethiones elevated GST and GSH as well as other cytoprotective proteins in multiple tissues of both rats and mice in vivo, and that oltipraz prevented a number of carcinogens from causing DNA damage and cancer in animals (7–9). Numerous studies of dithiolethiones have since been carried out. Both oltipraz and ADT have undergone several chemopreventive trials in humans; many new analogs have been synthesized and evaluated; and new chemopreventive mechanisms of dithiolethiones have been identified. While no dithiolethione has yet been approved for cancer prevention in humans, the wealth of knowledge accumulated on these compounds, particularly oltipraz, offers both guidance and lessons for further research and development of this interesting family of compounds for cancer prevention. In this review we discuss the preclinical evidence that propelled both oltipraz and ADT to cancer prevention trials in humans, the outcome of these trials, and other dithiolethiones that show promising cancer chemopreventive activity.
Oltipraz
Chemopreventive efficacy and safety in animal models
Oltipraz is by far the most extensively studied cancer chemopreventive agent among dithiolethiones. It has been shown to inhibit carcinogenesis in a variety of organs in rodents, including bladder, blood, colon, kidney, liver, lung, pancreas, stomach and trachea, induced by a large number of carcinogens (Table 1). It has also been reported that oltipraz inhibits cancer development in the mammary gland and skin of rodents (10), and to protect against DNA adduct formation in the aorta of smoke-exposed rats (11). Moreover, oltipraz is effective against many different types of carcinogen (Table 1), some of which are common human carcinogens, such as aflatoxin B1 (AFB1), benzo[a]pyrene (B[a]P) and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Oltipraz is orally active and is effective at dietary levels as low as 200 ppm, which, in the rat, would give a dose of approximately 10 mg/kg/day. In some animal experiments, oltipraz showed efficacy only when given in the initiation phase (during carcinogen exposure). For example, administration of oltipraz to rats after AFB1 exposure had no effect on liver carcinogenesis (12). In other experiments, however, oltipraz showed efficacy when given either in the initiation phase or post-initiation phase, as demonstrated by the inhibition of rat colon carcinogenesis induced by azoxymethane (AOM) (13). Oral administration of oltipraz also significantly inhibited SVR murine angiosarcoma xenograft growth in nude mice (14), suggesting that oltipraz could also inhibit the growth of advanced cancer.
Table 1.
Protection against chemical carcinogenesis by oltipraz in rodents
| Carcinogen | Species (sex/strain) | Tumor target organ | Reference |
|---|---|---|---|
| BBN | Mouse (♂, BDF and C57BL/6) | Bladder | (27, 81) |
|
| |||
| PhIP | Rat (♂, F344) | Blood/Lymphoma | (82) |
|
| |||
| AOM | Rat (♂, F344 and Sprague-Dawley) | Colon | (13, 83–86) |
|
| |||
| AFB1 | Rat (♂, F344) | Kidney | (9) |
|
| |||
| AFB1 | Rat (♂, F344) | Liver | (26, 87–90) |
|
| |||
| B[a]P, DEN, UM | Mouse (Mice (♀, ICR/Ha) | Lung | (8) |
| B[a]P | Mouse (Mice (♀, A/J) | Lung | (91) |
| MNU, DEN | Hamster (♂, Syrian) | Lung | (10) |
| BOP | Hamster (Mice (♀, Syrian) | Lung | (92) |
|
| |||
| BOP | Hamster (Mice (♀, Syrian) | Pancreas | (93) |
|
| |||
| DEN | Hamster (♂, Syrian) | Trachea | (94) |
|
| |||
| MNNG | Rat (♂, Wistar) | Glandular stomach | (95) |
| B[a]P | Mice (♀, ICR) | Forestomach | (96) |
AFB1, aflatoxin B1; AOM, azoxymethane; BBN, N-butyl-N-(4-hydroxybutyl) nitrosamine; B[a]P, benzo[a]pyrene; BOP, N-nitrosobis(2-oxopropyl) amine; DEN, diethylnitrosamine; MNNG, N-methyl-N′-nitro-N-nitrosoguanidine; MNU, N-methyl-N-nitrosourea; PhIP, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine; UM, uracil mustard;
Chronic toxicity studies in rats, in which oltipraz was administered daily by gavage for 13 weeks at 5 and 50 mg/kg/day and at 10, 30 and 50 mg/kg/day for 52 weeks, revealed liver enlargement at the intermediate and highest dose-levels, which was associated with diffuse or centrilobular hepatocytic hypertrophy (15). Slight decreases in erythrocyte count, hematocrit and hemoglobin levels were also recorded, which were associated with an increase in reticulocyte count. Similar effects were seen in dogs given oltipraz at 20 and 100 mg/kg/day for 13 weeks and at 5, 15 and 60 mg/kg/day for 52 weeks (15). The reason for the hematological changes, whether reflecting hemolysis or hemorrhage, was not established, but the possible role of the disulfide group of oltipraz needs to be considered, since both aliphatic and aromatic disulfides are hemolytic agents in animals (16). Liver enlargement has also been observed in other animal studies on oltipraz (17, 18).
Chemopreventive activity and safety in humans
Several phase I and phase II cancer chemopreventive trials of oltipraz have been carried out. When human volunteers (6/group) were given a single oral dose of oltipraz at 125, 250, 500, or 1,000 mg/m2, no significant side effects were detected, but the activities of glutamate cysteine synthetase (GCS), GST and NAD(P)H:quinone oxidoreductase 1 (NQO1) in colonic mucosa and NQO1 in peripheral mononuclear cells were significantly increased (19). The induction of gene expression appeared to reach a peak between 2 and 4 days after initiation of treatment and was maximal at a dose of 250 mg/m2. Higher doses were not more effective. However, in a follow-up study in which groups of 7 volunteers were given oral oltipraz twice weekly for 12 weeks at either 125 or 250 mg/m2, no significant modulation of GST and NQO1 in either colon tissue or blood was recorded (20). Moreover, while the low dose was well tolerated by all volunteers, two in the high dose group required dose reductions due to significant fatigue. In a randomized, placebo-controlled study in China in which oltipraz was administered to volunteers at either 125 mg (approximately 71 mg/m2) daily or 500 mg (approximately 286 mg/m2) weekly for 8 weeks (76–80 subjects/arm), a syndrome involving numbness, tingling and pain in the extremities was associated with oltipraz intake (18.4 and 14.1% of the daily 125 and weekly 500 mg arms, respectively, compared with 2.5% in the placebo arm (21). These side effects were not new, however, as similar problems were observed more than ten years earlier in humans taking oltipraz for schistosomiasis treatment (22). Interestingly, in a randomized, placebo-controlled, double-blind phase IIa trial in China, where participants exposed to high levels of dietary AFB1 took oltipraz orally at 125 mg daily or 500 mg weekly for one month (80/arm), similar side effects were not noted (23), suggesting that these side effects occur only after prolonged exposure to this compound. More importantly, the latter study showed that oltipraz at the high dose regimen inhibited Phase 1 activation (reduced formation of urinary aflatoxin M1) and at the low dose regimen increased Phase 2 conjugation (increased formation of urinary aflatoxin-mercapturic acid) of aflatoxin, indicating that oltipraz caused potential protective alterations of aflatoxin metabolism in vivo. In contrast, another randomized, double-blind, placebo-controlled trial in smokers, who were enrolled into one of the three arms: 200 or 400 mg/week oral oltipraz or placebo (12–23 subjects/arm) for 12 weeks, found 15% of those receiving oltipraz experienced grade 2/3 toxicity, predominantly gastrointestinal, while no significant difference was observed among the trial groups with regard to hydrocarbon-DNA adduct levels in lung epithelial cells, blood, oral lining cells, or bladder lining cells (24). The usefulness of oltipraz in cancer chemoprevention in humans must therefore be called into question, since significant side effects of oltipraz occur in humans at dose levels which show no or questionable chemopreventive efficacy.
Mechanism of action
Investigation of the cancer chemopreventive activity of oltipraz was launched initially on the basis of its ability to induce cytoprotective enzymes and to elevate GSH, as mentioned above. Subsequent studies have focused almost exclusively on the extent of modulation of cytoprotection by oltipraz, the underlying mechanism and the extent to which modulation of cytoprotection accounts for the cancer chemopreventive activity of oltipraz.
Oltipraz has been shown to induce Phase 2 enzymes important for detoxification of carcinogens and oxidants, such as NQO1, multiple subunits of GST, GCS, epoxide hydrolase and UDP-glucuronosyltransferase (UGT) (25, 26). A major mechanism by which oltipraz induces Phase 2 enzymes is the activation of Nrf2 transcription factor. Nrf2 is normally bound by its repressor Keap1 and targeted for degradation by the ubiquitin-proteosome system. Inducers of Phase 2 enzymes, including oltipraz (27, 28), disrupt the Nrf2-Keap1 complex and cause Nrf2 to translocate to the nucleus where it dimerizes with its partners such as Maf, binds to the antioxidant response element (ARE, core sequence: 5′TGACnnnGC3′) in the promoter region of Phase 2 genes and stimulates gene transcription (Fig. 2). Knockout of Nrf2 rendered Phase 2 enzymes nonresponsive to oltipraz and caused loss of its cancer chemopreventive efficacy in animal models of both bladder cancer and stomach cancer (27–29). Activation of Nrf2 signaling is therefore critical for inhibition of cancer development. However, it is not clear to what extent the cancer preventive role of Nrf2 depends on induction of Phase 2 enzymes or which specific Phase 2 enzyme is of paramount importance, since Nrf2 also regulates many non-Phase 2 genes (30), and the response of these genes to oltipraz under Nrf2 knockout is probably blocked as well. Modification of critical cysteine residues of Keap1 by inducers and stressors, which frees Nrf2 from Keap1, has been recognized as a key mechanism of Nrf2 activation (31). Although direct reaction of oltipraz with Keap1 cysteine thiols has not yet been demonstrated, dithiolethiones are reactive with thiols (32). Moreover, both oltipraz and D3T have been shown to generate reactive oxygen species (ROS) in cells (33, 34), which may further contribute to Nrf2 activation, as ROS may oxidize the cysteine thiols of Keap1 and/or activate other proteins involved in Nrf2 activation such as protein kinase C (35).
Figure 2.
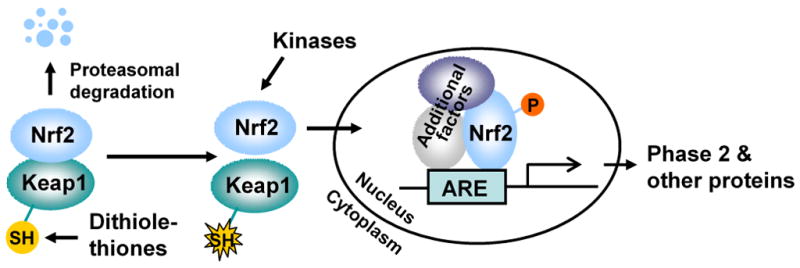
The key cancer chemopreventive mechanism of dithiolthiones. Binding of Keap1 to Nrf2 targets Nrf2 for proteasomal degradation. Disruption of the association of Keap1 with Nrf2 by dithiolethiones through interaction with critical Keap1 cysteine residues causes increased nuclear accumulation and ARE binding of Nrf2, leading to increased transcription of Phase 2 genes and other genes and increased cellular protection.
Additional mechanisms may be involved in the regulation of certain Phase 2 enzymes by oltipraz. Human NQO1 promoter is known to contain an AP-1 binding site, i.e., TPA response element (TRE, core sequence 5′TGACTCA3′) (36). In HT29 human colon adenocarcinoma cells, oltipraz significantly elevated the mRNA levels of AP-1 factors Jun and Fos as well as that of redox factor-1 (Ref-1) which enhances AP-1 binding, and the removal of the AP-1 site in the NQO1 promoter in a reporter construct resulted in 65% loss of the induction by oltipraz (37). Rat UGT 1A6 carries a xenobiotic response element (XRE, core sequence 5′TGCGTG3′), which was shown to mediate at least partly the induction of this protein by oltipraz in primary rat hepatocytes (38). XRE is recognized by a heterodimeric complex of aryl hydrocarbon receptor (AHR) and AHR nuclear translocator (ARNT). Treatment of human hepatoma HepG2 cells with oltipraz led to increased binding of AHR and ARNT to the UGT1A6 XRE (38).
Oltipraz has also been shown to protect against certain carcinogens by inhibiting cytochrome P450s (CYPs or Phase 1 enzymes). Metabolic activation of AFB1 by Phase 1 enzymes is required for its carcinogenic activity (32), and oltipraz was shown to inhibit AFB1 activation by acting as both a competitive and irreversible inhibitor of both CYP1A2 and CYP3A4 expressed in bacteria (39) and in primary human hepatocytes (40) and of CYP2B in primary rat hepatocytes (41). Rat experiments showed that oltipraz promoted the degradation of CYP2B1 and inhibited CYP1A2-mediated metabolism of AFB1 in vivo (42, 43). Moreover, a single 125-mg dose in humans appeared to reduce CYP1A2 activity by approximately 75%, as measured by the clearance of caffeine by demethylation, and daily administration of 125 mg oltipraz for 8 days appeared to result in further inhibition of CYP1A2 (44). However, the impact of oltipraz on CYPs is apparently not always inhibitory. Oltipraz was shown to significantly induce CYP1A2 and CYP3A2 in rat liver in vivo (25, 45), CYP1A in both rat kidney and lung in vivo (42), and CYP1A1 in rat hepatoma H4IIE cells and human colon carcinoma Caco-2 cells in culture (46, 47). Interestingly, the pyrazinyl structure is probably principally, if not entirely, responsible for the inductive effect of oltipraz on CYPs, since the unsubstituted compound, D3T, is largely devoid of such effect (26, 48).
In addition to modulating Phase 1 and Phase 2 enzymes, one study also reported antiangiogenic activity of oltipraz, showing inhibition of microvessel formation in both in vitro, ex vivo, and in vivo assays, which was accompanied by strong inhibition of angiosarcoma xenograft in nude mice (14).
Pharmacokinetics and metabolism
14C-labelled oltipraz was administered in a single oral dose to Rhesus monkeys (20 mg/kg), Sprague-Dawley rats (50 mg/kg) and CD1 mice (100–250 mg/kg) (49). Only the results in monkeys and rats are discussed below, because the results in mice were compromised due to infection with Schistosoma mansoni. Radioactivity levels in plasma reached a maximum at 3 h in both the rat and monkey, while plasma elimination half-lives were 2.5 h in the rat and 3.5–7 h in the monkeys. 57% and 52% of the administered dose was eliminated in the urine in four days by the monkeys and rats, respectively, and the fecal elimination was 23% and 39% for the monkey and rat. In the blood, unchanged oltipraz accounted for <1% of the radioactivity in the monkeys and 4–10% in the rats. The amount of unchanged oltipraz eliminated in the urine in 3–4 days was <0.5–1%, whereas the amount of unchanged oltipraz eliminated in the feces during the same period was substantially more: 5% and 13% of the dose in the rat and monkey. Data on plasma levels of oltipraz in animals receiving this substance at dose-levels shown to prevent cancer are not presently available, although such information would be of great value in interpreting the results of trials involving oltipraz in humans, since it would permit comparison with the pharmacokinetic profile of oltipraz in humans (50). Plasma concentrations of unchanged oltipraz in humans reached a maximum at 2.2 h after a single oral dose (125–1000 mg/m2); maximal plasma concentrations (158–671 ng/ml or 0.7–3.0 μM) were proportional to the dose; plasma elimination half-lives ranged from 9.3–22.7 h, and were inversely correlated with dose. These plasma concentrations of oltipraz, however, were significantly lower than those required to significantly induce Phase 2 enzymes in cultured cells (10–100 μM) (51–54). Another study showed that urinary concentrations of unchanged oltipraz in humans were similar to those in animals (55). Oltipraz is extensively metabolized – a total of 13 metabolites were detected in the plasma and urine specimens of animals and humans dosed with this substance (55). 4-Methyl-5-(2-pyrazinyl)-3H-1,2-dithiole-3-one (oltipraz ketone, Compound 16, Fig. 1), which was formed in only trace amounts but did not appear to be further metabolized (50, 55), was the only metabolite to retain the dithiole structure. In the human colon cancer HT29 cell line in vitro, the activity of oltipraz ketone as a Phase 2 enzyme inducer was shown to be similar to oltipraz itself (53), but its inductive activity was lower than the parent compound when administered to rats (26). An intermediate metabolite of oltipraz, 7-methyl-6,8-bis(methylthio)H-pyrrolo[1,2-a]pyrazine, also induced Phase 2 enzymes and activated Nrf2 in cultured cells with similar efficacy to oltipraz, although the oxidation products of this, in which one or both of the sulfide groups were converted to a sulfoxide, were without inductive effect (54). There is also evidence that the pyrrolopyrazine thione precursor of this substance is an inducer, possibly via a mechanism involving the formation of ROS through reaction with cytochrome c within mitochondria (56, 57).
ADT
Information on ADT is rather limited in comparison with oltipraz. Oral administration of this substance led to induction of Phase 2 enzymes such as GST and NQO1 in multiple tissues in rats and mice, protection against carbon tetrachloride, acetaminophen and hexachloro-1,3-butadiene toxicities in rodents (5, 7, 58), inhibition of tumor multiplicity in rats challenged with 7,12-dimethylbenz[a]anthracene (59), and decreases in both the incidence and multiplicity of colon tumors in animals dosed with azoxymethane (60). In cultured human Jurkat T cells, ADT pretreatment protected cells against oxidative stress-induced cytotoxicity, which was associated with induction of catalase and glutathione reductase and elevation of GSH levels (61). Moreover, ADT is reported to be a free radical scavenger and to inhibit lipid peroxidation both in vitro and in vivo (62, 63). However, a summary of trials with ADT under the NCI Chemoprevention Drug Development Program in the U.S. reported no effect of this substance against several animal models of lung, colon, mammary gland and bladder cancer (10). In a randomized phase IIb trial of ADT in smokers with bronchial dysplasia, ADT at 25 mg orally thrice daily for 6 months led to a moderate but statistically significant decrease in the progression of pre-existing dysplastic lesions, and the treatment was associated with only minor gastrointestinal symptoms (64).
Autoradiographic studies on mice dosed orally with low doses of 14C-ADT (~5 mg/kg) showed the presence of the isotope in the intestine, liver, gall bladder, kidney and urinary bladder. High concentrations persisted in most tissues for 12 h, but none was seen in any organ after 24 h, indicating rapid excretion (65). In contrast, a large dose (1 g/kg) of ADT was excreted slowly by rats and mice. No unchanged ADT was found in the urine of rats, but appreciable amounts of ADT were found in the urine of mice for up to 5 days. In both rats and mice, the major metabolite was desmethyl ADT [5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione], the urinary excretion of which peaked at 2 days in rats and 4 days in mice (66).
Other dithiolethiones
More recently, many analogs of oltipraz and ADT have been synthesized and evaluated in preclinical models. There were, to our knowledge, no clear features of structure-activity relationship that were used to select these compounds. Rather, compounds were selected on the basis of giving a broad range of representation of structural types. Indeed, studies of these compounds, as described below, did not reveal obvious indicators for high activity. None of these compounds has yet entered clinical trials, but a number of them have shown activities that are superior to those of oltipraz and ADT in animal tests. Induction of Phase 2 enzymes remains fundamental to their chemopreventive activity, but their activities have been shown to be associated with significant tissue specificity.
Induction of Phase 2 enzymes: structure-activity relationships and identification of exceptional inducers
D3T (Compound 1, Fig. 1) was shown to be a potent inducer of Phase 2 enzymes and a powerful inhibitor of AFB1-induced hepatic toxicity and formation of hepatic preneoplastic lesions, while its isomer, 1,3-dithiolane-2-thione (Compound 2, Fig. 1) was largely ineffective (51, 67), indicating that the 1,2-dithiole structure is essential for chemopreventive activity. In this connection, it is also worth noting that 1,2-dithiole-3-one (Compound 3, Fig. 1) and oltipraz ketone (Compound 16, Fig. 1), both of which retain the 1,2-dithiole structure while the thione group is changed to ketone, were similar to the unchanged compounds in bioactivity in cultured cells (51, 53). The latter finding also led to speculation that these ketone derivatives might be more effective chemopreventive agents than the corresponding dithiolethiones, because oltipraz ketone was shown not to be further metabolized in vivo (55). However, both induction of hepatic Phase 2 enzymes and inhibition of AFB1-induced hepatic DNA damage in rats by either 1,2-dithiole-3-one or oltipraz ketone were weaker than the corresponding dithiolethiones (26).
The inductive activity of many dithiolethiones has been evaluated in cultured cells in vitro. D3T was a more effective inducer of NQO1 than oltipraz in murine hepatoma Hepa1c1c7 cells (68). In a study of 25 dithiolethiones by Kensler and others in Hepa1c1c7 cells (51), 5,6-dihydrocyclopenta[c]-1,2-dithiole-3(4H)-thione (CPDT, Compound 17, Fig. 1) was the most potent inducer of NQO1. This compound was 6, 72 and 88 times more potent than D3T, ADT and oltipraz, respectively, as measured by the concentration of the inducer required to double NQO1 activity in treated cells. It is of note that NQO1 induction in Hepa1c1c7 cells has been a widely used model system for detection of inducers of Phase 2 enzymes (69). Minor structural modifications of CPDT, however, resulted in a marked decrease in inducer activity, as 4,5,6,7-tetrahydrobenzo[c]-1,2-dithiole-3-thione and 4,5-dimethyl-3H-1,2-dithiole-3-thione (Compounds 18 and 6, Fig. 1) were 20- and 320-fold weaker than CPDT, respectively. It is also of note that while dithiolethiones induce Phase 2 enzymes in a wide variety of human cell lines in vitro, the response of such cell was not consistent. Indeed, some lines showed no response at all (70, 71). In NBT-II bladder cancer cells, CPDT and D3T showed similar activity in inducing both GST and NQO1, while both ADT and oltipraz were significantly weaker inducers (72).
In vivo, most attention has been focused on the effects of dithiolethiones on Phase 2 enzymes in the liver. Kensler et al. (26) showed that D3T when fed to rats in the diet at 0.075% for 1 week increased the activity of hepatic Phase 2 enzymes to a much greater degree than 4- and 5-phenyl-3H-1,2-dithiole-3-thione, ADT and oltipraz. In a subsequent study, among seventeen dithiolethiones evaluated in rats for induction of GST and NQO1 in vivo, the magnitude of induction of hepatic activities of GST and NQO1, measured 48 h after a single dose of 1 mmol dithiolethione/kg, by the following compounds was as follows: 4.4 and 20.8 fold (5-[2-dimethylamino]vinyl-4-methyl-, Compound 11, Fig. 1), 2.9 and 10.8 fold (4-methyl-, Compound 5, Fig. 1), 3.6 and 9.5 fold (5-methyl-, Compound 4, Fig. 1), 3.0 and 10.2 fold (D3T), 2.4 and 7.3 fold (CPDT), 1.9 and 2.7 fold (oltipraz), respectively (73). Thus, D3T is a more effective inducer than CPDT, which contradicts the results in Hepa1c1c7 cells or NBT-II cells mentioned above, suggesting that in vitro bioassays do not reliably predict in vivo activity of dithiolethiones. In the latter study, Compound 11 showed exceptional inducer activity and was the most effective inducer among all the compounds tested, and oltipraz was one of the weakest inducers. In another study, D3T and CPDT and 8 other dithiolethiones (Compound 11 was not included) were each administered to rats by oral intubation at 125 μmol/kg once daily for 5 days and tissue activities of GST and NQO1 were measured on the sixth day. Hepatic GST and NQO1 induction levels ranged from 1.2 to 2.0 and 1.6 to 3.5 fold, respectively (72). While most compounds showed similar inductive activity for GST, the order of inductive activity for NQO1 ranked as follows: 4-chloro-5-methyl-3H-1,2-dithiole-3-thione > 5-phenyl-3H-1,2-dithiole-3-thione > 4-phenyl-3H-1,2-dithiole-3-thione > 4,5,6,7-tetrahydrobenzo[c]-1,2-dithiole-3-thione > D3T > 5-methyl-3H-1,2-dithiole-3-thione > CPDT > 4,5-dimethyl-3H-1,2-dithiole-3-thione > oltipraz > ADT (see Fig. 1 for chemical structures). Thus, in this study, both D3T and CPDT demonstrated only modest inductive activities in the liver. While there is a degree of variation for some of the compounds among the results of the above-mentioned studies, possibly reflecting differences in dosing regimen, it is clear that the two most extensively studied dithiolethiones, oltipraz and ADT, are among the weakest inducers of Phase 2 enzymes in the liver in vivo.
Tissue specificity in Phase 2 enzyme induction
While a great deal of data are available on the effects of dithiolethiones on hepatic Phase 2 enzymes, there is much less information on effects in other tissues. In a dose escalation study with D3T, the activities of GST and NQO1 were measured in 13 tissues, including spleen, liver, kidneys, heart, lungs, urinary bladder, glandular stomach, forestomach, duodenum, jejunum, ileum, cecum and colon/rectum, after rats were dosed orally at 0.98–125 μmol/kg once daily for 5 days (74). At the highest dose, D3T caused significant increases in NQO1 and/or GST in all the tissues, with the greatest effects being seen in the kidneys, urinary bladder, forestomach, duodenum and jejunum. With decreasing dose levels of D3T, the degree of induction and the number of tissues showing significant enzyme induction diminished. However, even at the lowest dose level (0.98 μmol/kg), significant enzyme induction was still detected in the glandular stomach, forestomach and duodenum, showing that these tissues are particularly sensitive to D3T. In a comparative study involving D3T and 9 other dithiolethiones (Compounds 4, 6, 7, 12–15, 17, 18), each of which was administered to rats orally at 125 μmol/kg once daily for 5 days (72), high activity of D3T in many tissues with regard to induction of GST and NQO1 was detected, but different dithiolethiones showed different tissue specificities. CPDT was particularly active in the kidney and urinary bladder, 4-phenyl-3H-1,2-dithiole-3-thione in the lungs, 5-methyl-3H-1,2-dithiole-3-thione in the intestines, and 4-chloro-5-methyl-3H-1,2-dithiole-3-thione in the liver. In contrast, enzyme induction by ADT and oltipraz was either absent or statistically insignificant in most tissues.
Anticarcinogenic activity
The anticarcinogenic activities of dithiolethiones other than ADT and oltipraz have been evaluated mainly in AFB1-induced liver carcinogenesis models and have been performed mainly by Kensler and coworkers. Treatment of rats with D3T or one of 7 substituted analogs in the diet at 0.075% for one week induced many Phase 2 enzymes in the liver, and such treatment inhibited hepatic AFB1-DNA adduct formation when the rats were subsequently exposed to AFB1 (26). However, of all the compounds tested, including ADT and oltipraz, D3T was the most significant inducer of Phase 2 enzymes and the most effective inhibitor of AFB1-induced DNA damage. Moreover, this study revealed that, unlike its analogs, D3T had no inductive effects on CYP enzymes. A subsequent study showed that dietary supplementation with D3T at 0.001% inhibited AFB1-induced putative preneoplastic lesions (volume of liver occupied by GGT or GST-P foci) by 80%, and higher D3T doses were more efficacious (48). This study also confirmed the previous observation that D3T up-regulates Phase 2 enzymes without inductive effects on CYP enzymes. However, this property of D3T should be viewed with caution, as a later study showed marked induction of hepatic CYP2C12 in rats gavaged with D3T three times once every other day at 13.4 or 40.2 mg/kg (75). D3T was also the most effective inducer of hepatic GST and NQO1 and inhibitor of AFB1-induced hepatic injury in rats among the 9 dithiolethiones evaluated (40.2 mg/kg, three times a week, by gavage), whereas oltipraz was among the weakest agents (73). It is also important to note that the above study also showed a strong positive correlation between in vivo induction of hepatic Phase 2 enzymes and inhibition of AFB1-induced acute hepatotoxicity or formation of GSTP-P foci, highlighting the importance of induction of Phase 2 enzymes in dithiolethione-induced inhibition of carcinogenesis. D3T was again the most effective agent, while oltipraz was among the least effective. D3T was also the most potent inhibitor of AFB1- induced liver preneoplastic lesions in rats among 8 dithiolethiones including oltipraz in yet another rat study, and microarray analysis of the liver of D3T-treated rats showed induction of two genes known to detoxify aflatoxin, namely, GSTA5 and AFB1 aldehyde reductase, among other genes induced by D3T (75). Further analysis of hepatic gene expression profiles in D3T-treated wild type mice and in Nrf2 deficient mice showed up-regulation of 292 genes in wild-type mice 24 h after treatment with D3T by gavage at 67 mg/kg, 79% of which were induced in wild-type, but not in Nrf2-deficient mice (30). These D3T-induced and Nrf2-dependent genes included many for Phase 2 enzymes but also genes for chaperones, protein trafficking molecules, proteasome subunits, signaling molecules and other proteins.
D3T thus shows highly promising chemopreventive activity. A short-term toxicological study in rats, however, has revealed the potential for toxicity at dose-levels similar to or lower than the chemopreventive doses described above. CD rats were dosed orally with D3T for 14 days at 2, 6, 20 and 60 mg/kg/day (76). At the two highest dose levels, the animals became lethargic and showed piloerection. At necropsy, hyperplasia and necrosis of the epithelium of the glandular stomach were observed. At 60 mg/kg/day, anemia was recorded in female rats, whereas male rats showed decreased body weight gain and increased vacuolation of adrenal cortical cells. Moreover, liver enlargement, associated with changes in blood chemistry indicative of altered liver function, was seen in animals at 6 mg/kg or above. In another study, in which female Sprague-Dawley rats were dosed by oral intubation with D3T once daily for 5 days (74), liver weight was dose-dependently and significantly increased at 2.1 mg/kg and above, and the weights of glandular stomach, forestomach, duodenum, kidney, jejunum, ileum, cecum and colon plus rectum were increased at 8.4 mg/kg and above.
Other chemopreventive activity
In addition to inducing Phase 2 enzymes, D3T has been shown to increase the catalytic subunits of the 26S proteasome, including PSMB5, PSMB6 and PSMB7 in a number of mouse tissues following oral administration (77), and to stimulate heat shock protein 70 in cultured cells (78). As mentioned above, analysis of hepatic gene expression profiles in D3T-treated wild type mice and in Nrf2 deficient mice showed up-regulation of a large number of genes in Nrf2-dependent manner, including genes for Phase 2 enzymes, chaperones, protein trafficking molecules, proteasome subunits, signaling molecules and other proteins, showing an extensive cellular response to D3T.
Summary and future perspectives
While the studies of oltipraz and ADT have yielded valuable information on the potential chemopreventive action of dithiolethiones and on the mechanism of such action, recent work suggests that other dithiolethiones may be more promising in preventing cancer. There is a need for more work in several areas in order to identify and develop dithiolethiones that are ultimately useful for cancer chemoprevention in humans.
Structure-activity relationships among dithiolethiones are by no means clear-cut, and no specific structural feature associated with potent chemopreventive activity has been identified. Progress in this area would facilitate the design and development of dithiolethiones of high activity. Activation of Nrf2 and induction of Phase 2 enzymes has been demonstrated as the fundamental chemopreventive mechanism of dithiolethiones. Hence, identification of structural features that convey potent activation of Nrf2 and induction of Phase 2 enzymes is of particular importance. Further determination of the mechanism whereby these compounds activate Nrf2 and Phase 2 enzymes as well as discovery of novel mechanisms would be of considerable value, especially for identification of biomarkers useful for evaluation of in vivo efficacy of dithiolethiones. Metabolic studies on simple dithiolethiones, such as D3T, are required in order to establish if it is the parent compound or metabolites that are responsible for the observed activity. While much information is available on oltipraz in this regard, this compound is not a typical dithiolethione because of the complex metabolic transformations involving the pyrazinyl ring. New information on metabolism of dithiolethiones may also lead to better understanding of the tissue specificity of dithiolethiones. For example, it has been shown that the high activity of isothiocyanates, another group of well-known chemopreventive agents in the bladder of animals, is due to their metabolism and disposition in the body, leading to the delivery of high concentrations of inducers to the bladder via the urine (79, 80). D3T and CPDT are also highly effective inducers of Phase 2 enzymes in the bladder, but whether this reflects a high urinary excretion of the parent compound or bioactive metabolites is not presently known. Toxicities of oltipraz observed in humans contributed to the difficulty for its further development, but better insight into the tissue specificity of the chemopreventive activity of dithiolethiones may lead to development of agents that are effective in specific tissue(s) and safe for use in humans.
In the final analysis, the most important property of dithiolethiones for cancer chemoprevention is the ability to stimulate the Nrf2-Phase 2 enzymes system. Future work should focus on identifying compounds that are particularly effective in this regard, recognizing the need for whole animal studies to identify specific tissues in which these substances are particularly effective. In this way, dithiolethiones may be identified that are able to protect against carcinogenesis in human tissues at very low dose-levels, and efficacy may be achieved while avoiding the side effects seen in previous studies with dithilethiones.
Acknowledgments
NIH Grant CA120533.
References
- 1.Barbaglia GA. Ueber den Sulfovaleraldehyd. Ber Dtsch Chem Ges. 1884;17:2654–5. [Google Scholar]
- 2.Lüttringhaus A, König HB, Böttcher B. Über Trithione II. Konstitution und neue Bildungsweisen Liebigs Ann Chem. 1948;560:201–14. [Google Scholar]
- 3.Lozac’h N, Vialle J. The chemistry of the 1,2-dithiole ring. In: Kharasch H, Meyers CY, editors. The chemistry of organic sulfur compounds . Vol. 2. Pergamon Press; 1966. pp. 257–85. [Google Scholar]
- 4.Epstein JB, Decoteau WE, Wilkinson A. Effect of Sialor in treatment of xerostomia in Sjogren’s syndrome. Oral Surg Oral Med Oral Pathol. 1983;56:495–9. doi: 10.1016/0030-4220(83)90096-8. [DOI] [PubMed] [Google Scholar]
- 5.Ansher SS, Dolan P, Bueding E. Chemoprotective effects of two dithiolthiones and of butylhydroxyanisole against carbon tetrachloride and acetaminophen toxicity. Hepatology. 1983;3:932–5. doi: 10.1002/hep.1840030608. [DOI] [PubMed] [Google Scholar]
- 6.Bella H, Rahim AG, Mustafa MD, Ahmed MA, Wasfi S, Bennett JL. Oltipraz--antischistosomal efficacy in Sudanese infected with Schistosoma mansoni. Am J Trop Med Hyg. 1982;31:775–8. doi: 10.4269/ajtmh.1982.31.775. [DOI] [PubMed] [Google Scholar]
- 7.Ansher SS, Dolan P, Bueding E. Biochemical effects of dithiolthiones. Food Chem Toxicol. 1986;24:405–15. doi: 10.1016/0278-6915(86)90205-x. [DOI] [PubMed] [Google Scholar]
- 8.Wattenberg LW, Bueding E. Inhibitory effects of 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (Oltipraz) on carcinogenesis induced by benzo[a]pyrene, diethylnitrosamine and uracil mustard. Carcinogenesis. 1986;7:1379–81. doi: 10.1093/carcin/7.8.1379. [DOI] [PubMed] [Google Scholar]
- 9.Kensler TW, Egner PA, Trush MA, Bueding E, Groopman JD. Modification of aflatoxin B1 binding to DNA in vivo in rats fed phenolic antioxidants, ethoxyquin and a dithiothione. Carcinogenesis. 1985;6:759–63. doi: 10.1093/carcin/6.5.759. [DOI] [PubMed] [Google Scholar]
- 10.Steele VE, Moon RC, Lubet RA, et al. Preclinical efficacy evaluation of potential chemopreventive agents in animal carcinogenesis models: methods and results from the NCI Chemoprevention Drug Development Program. J Cell Biochem Suppl. 1994;20:32–54. doi: 10.1002/jcb.240560905. [DOI] [PubMed] [Google Scholar]
- 11.Izzotti A, Balansky RM, D’Agostini F, et al. Modulation of biomarkers by chemopreventive agents in smoke-exposed rats. Cancer Res. 2001;61:2472–9. [PubMed] [Google Scholar]
- 12.Maxuitenko YY, MacMillan DL, Kensler TW, Roebuck BD. Evaluation of the post-initiation effects of oltipraz on aflatoxin B1-induced preneoplastic foci in a rat model of hepatic tumorigenesis. Carcinogenesis. 1993;14:2423–5. doi: 10.1093/carcin/14.11.2423. [DOI] [PubMed] [Google Scholar]
- 13.Rao CV, Rivenson A, Katiwalla M, Kelloff GJ, Reddy BS. Chemopreventive effect of oltipraz during different stages of experimental colon carcinogenesis induced by azoxymethane in male F344 rats. Cancer Res. 1993;53:2502–6. [PubMed] [Google Scholar]
- 14.Ruggeri BA, Robinson C, Angeles T, Wilkinson JT, Clapper ML. The chemopreventive agent oltipraz possesses potent antiangiogenic activity in vitro, ex vivo, and in vivo and inhibits tumor xenograft growth. Clin Cancer Res. 2002;8:267–74. [PubMed] [Google Scholar]
- 15.Crowell JA, Page JG, Rodman LE, et al. Chronic toxicity studies of 5-(2-pyrazinyl)-4-methyl-1,2-dithiole-3-thione, a potential chemopreventive agent. Fundam Appl Toxicol. 1997;35:9–21. doi: 10.1006/faat.1996.2256. [DOI] [PubMed] [Google Scholar]
- 16.Munday R. Toxicity of thiols and disulphides: involvement of free-radical species. Free Radic Biol Med. 1989;7:659–73. doi: 10.1016/0891-5849(89)90147-0. [DOI] [PubMed] [Google Scholar]
- 17.Stohs SJ, Lawson TA, Anderson L, Bueding E. Effects of oltipraz, BHA, ADT and cabbage on glutathione metabolism, DNA damage and lipid peroxidation in old mice. Mech Ageing Dev. 1986;37:137–45. doi: 10.1016/0047-6374(86)90071-0. [DOI] [PubMed] [Google Scholar]
- 18.Davies MH, Blacker AM, Schnell RC. Dithiolthione-induced alterations in hepatic glutathione and related enzymes in male mice. Biochem Pharmacol. 1987;36:568–70. doi: 10.1016/0006-2952(87)90370-4. [DOI] [PubMed] [Google Scholar]
- 19.O’Dwyer PJ, Szarka CE, Yao KS, et al. Modulation of gene expression in subjects at risk for colorectal cancer by the chemopreventive dithiolethione oltipraz. J Clin Invest. 1996;98:1210–7. doi: 10.1172/JCI118904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szarka CE, Yao KS, Pfeiffer GR, et al. Chronic dosing of oltipraz in people at increased risk for colorectal cancer. Cancer Detect Prev. 2001;25:352–61. [PubMed] [Google Scholar]
- 21.Jacobson LP, Zhang BC, Zhu YR, et al. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: study design and clinical outcomes. Cancer Epidemiol Biomarkers Prev. 1997;6:257–65. [PubMed] [Google Scholar]
- 22.Archer S. The chemotherapy of schistosomiasis. Annu Rev Pharmacol Toxicol. 1985;25:485–508. doi: 10.1146/annurev.pa.25.040185.002413. [DOI] [PubMed] [Google Scholar]
- 23.Wang JS, Shen X, He X, et al. Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J Natl Cancer Inst. 1999;91:347–54. doi: 10.1093/jnci/91.4.347. [DOI] [PubMed] [Google Scholar]
- 24.Kelley MJ, Glaser EM, Herndon JE, 2nd, et al. Safety and efficacy of weekly oral oltipraz in chronic smokers. Cancer Epidemiol Biomarkers Prev. 2005;14:892–9. doi: 10.1158/1055-9965.EPI-04-0585. [DOI] [PubMed] [Google Scholar]
- 25.Buetler TM, Gallagher EP, Wang C, Stahl DL, Hayes JD, Eaton DL. Induction of phase I and phase II drug-metabolizing enzyme mRNA, protein, and activity by BHA, ethoxyquin, and oltipraz. Toxicol Appl Pharmacol. 1995;135:45–57. doi: 10.1006/taap.1995.1207. [DOI] [PubMed] [Google Scholar]
- 26.Kensler TW, Egner PA, Dolan PM, Groopman JD, Roebuck BD. Mechanism of protection against aflatoxin tumorigenicity in rats fed 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (oltipraz) and related 1,2-dithiol-3-thiones and 1,2-dithiol-3-ones. Cancer Res. 1987;47:4271–7. [PubMed] [Google Scholar]
- 27.Iida K, Itoh K, Kumagai Y, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64:6424–31. doi: 10.1158/0008-5472.CAN-04-1906. [DOI] [PubMed] [Google Scholar]
- 28.Ramos-Gomez M, Kwak MK, Dolan PM, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M, Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis. 2003;24:461–7. doi: 10.1093/carcin/24.3.461. [DOI] [PubMed] [Google Scholar]
- 30.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–45. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 31.Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol. 2005;18:1779–91. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- 32.Kensler TW, Groopman JD, Sutter TR, Curphey TJ, Roebuck BD. Development of cancer chemopreventive agents: oltipraz as a paradigm. Chem Res Toxicol. 1999;12:113–26. doi: 10.1021/tx980185b. [DOI] [PubMed] [Google Scholar]
- 33.Velayutham M, Villamena FA, Fishbein JC, Zweier JL. Cancer chemopreventive oltipraz generates superoxide anion radical. Arch Biochem Biophys. 2005;435:83–8. doi: 10.1016/j.abb.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 34.Jia Z, Zhu H, Trush MA, Misra HP, Li Y. Generation of superoxide from reaction of 3H-1,2-dithiole-3-thione with thiols: implications for dithiolethione chemoprotection. Mol Cell Biochem. 2008;307:185–91. doi: 10.1007/s11010-007-9598-z. [DOI] [PubMed] [Google Scholar]
- 35.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–80. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaiswal AK. Human NAD(P)H:quinone oxidoreductase (NQO1) gene structure and induction by dioxin. Biochemistry. 1991;30:10647–53. doi: 10.1021/bi00108a007. [DOI] [PubMed] [Google Scholar]
- 37.Yao KS, O’Dwyer PJ. Role of the AP-1 element and redox factor-1 (Ref-1) in mediating transcriptional induction of DT-diaphorase gene expression by oltipraz: a target for chemoprevention. Biochem Pharmacol. 2003;66:15–23. doi: 10.1016/s0006-2952(03)00163-1. [DOI] [PubMed] [Google Scholar]
- 38.Auyeung DJ, Kessler FK, Ritter JK. Mechanism of rat UDP-glucuronosyltransferase 1A6 induction by oltipraz: evidence for a contribution of the aryl hydrocarbon receptor pathway. Mol Pharmacol. 2003;63:119–27. doi: 10.1124/mol.63.1.119. [DOI] [PubMed] [Google Scholar]
- 39.Langouët S, Furge LL, Kerriguy N, Nakamura K, Guillouzo A, Guengerich FP. Inhibition of human cytochrome P450 enzymes by 1,2-dithiole-3-thione, oltipraz and its derivatives, and sulforaphane. Chem Res Toxicol. 2000;13:245–52. doi: 10.1021/tx990189w. [DOI] [PubMed] [Google Scholar]
- 40.Langouët S, Coles B, Morel F, et al. Inhibition of CYP1A2 and CYP3A4 by oltipraz results in reduction of aflatoxin B1 metabolism in human hepatocytes in primary culture. Cancer Res. 1995;55:5574–9. [PubMed] [Google Scholar]
- 41.Langouët S, Mahéo K, Berthou F, et al. Effects of administration of the chemoprotective agent oltipraz on CYP1A and CYP2B in rat liver and rat hepatocytes in culture. Carcinogenesis. 1997;18:1343–9. doi: 10.1093/carcin/18.7.1343. [DOI] [PubMed] [Google Scholar]
- 42.Le Ferrec E, Ilyin G, Mahéo K, et al. Differential effects of oltipraz on CYP1A and CYP2B in rat lung. Carcinogenesis. 2001;22:49–55. doi: 10.1093/carcin/22.1.49. [DOI] [PubMed] [Google Scholar]
- 43.Scholl P, Musser SM, Kensler TW, Groopman JD. Inhibition of aflatoxin Ml excretion in rat urine during dietary intervention with oltipraz. Carcinogenesis. 1996;17:1385–8. doi: 10.1093/carcin/17.6.1385. [DOI] [PubMed] [Google Scholar]
- 44.Sofowora GG, Choo EF, Mayo G, Shyr Y, Wilkinson GR. In vivo inhibition of human CYP1A2 activity by oltipraz. Cancer Chemother Pharmacol. 2001;47:505–10. doi: 10.1007/s002800000245. [DOI] [PubMed] [Google Scholar]
- 45.Buetler TM, Bammler TK, Hayes JD, Eaton DL. Oltipraz-mediated changes in aflatoxin B1 biotransformation in rat liver: implications for human chemointervention. Cancer Res. 1996;56:2306–13. [PubMed] [Google Scholar]
- 46.Le Ferrec E, Lagadic-Gossmann D, Rauch C, et al. Transcriptional induction of CYP1A1 by oltipraz in human Caco-2 cells is aryl hydrocarbon receptor- and calcium-dependent. J Biol Chem. 2002;277:24780–7. doi: 10.1074/jbc.M111319200. [DOI] [PubMed] [Google Scholar]
- 47.Miao W, Hu L, Kandouz M, Batist G. Oltipraz is a bifunctional inducer activating both phase I and phase II drug-metabolizing enzymes via the xenobiotic responsive element. Mol Pharmacol. 2003;64:346–54. doi: 10.1124/mol.64.2.346. [DOI] [PubMed] [Google Scholar]
- 48.Kensler TW, Groopman JD, Eaton DL, Curphey TJ, Roebuck BD. Potent inhibition of aflatoxin-induced hepatic tumorigenesis by the monofunctional enzyme inducer 1,2-dithiole-3-thione. Carcinogenesis. 1992;13:95–100. doi: 10.1093/carcin/13.1.95. [DOI] [PubMed] [Google Scholar]
- 49.Heusse D, Marlard M, Bredenbac J, et al. Disposition of 14C-oltipraz in animals. Pharmacokinetics in mice, rats and monkeys. Comparison of the biotransformation in the infected mouse and in the schistosomes. Arzneimittelforschung. 1985;35:1431–6. [PubMed] [Google Scholar]
- 50.O’Dwyer PJ, Szarka C, Brennan JM, Laub PB, Gallo JM. Pharmacokinetics of the chemopreventive agent oltipraz and of its metabolite M3 in human subjects after a single oral dose. Clin Cancer Res. 2000;6:4692–6. [PubMed] [Google Scholar]
- 51.Egner PA, Kensler TW, Prestera T, et al. Regulation of phase 2 enzyme induction by oltipraz and other dithiolethiones. Carcinogenesis. 1994;15:177–81. doi: 10.1093/carcin/15.2.177. [DOI] [PubMed] [Google Scholar]
- 52.De Long MJ, Dolan P, Santamaria AB, Bueding E. 1,2-Dithiol-3-thione analogs: effects on NAD(P)H:quinone reductase and glutathione levels in murine hepatoma cells. Carcinogenesis. 1986;7:977–80. doi: 10.1093/carcin/7.6.977. [DOI] [PubMed] [Google Scholar]
- 53.O’Dwyer PJ, Clayton M, Halbherr T, Myers CB, Yao K. Cellular kinetics of induction by oltipraz and its keto derivative of detoxication enzymes in human colon adenocarcinoma cells. Clin Cancer Res. 1997;3:783–91. [PubMed] [Google Scholar]
- 54.Ko MS, Lee SJ, Kim JW, Lim JW, Kim SG. Differential effects of the oxidized metabolites of oltipraz on the activation of CCAAT/enhancer binding protein-beta and NF-E2-related factor-2 for GSTA2 gene induction. Drug Metab Dispos. 2006;34:1353–60. doi: 10.1124/dmd.106.009514. [DOI] [PubMed] [Google Scholar]
- 55.Bieder A, Decouvelaere B, Gaillard C, et al. Comparison of the metabolism of oltipraz in the mouse, rat and monkey and in man. Distribution of the metabolites in each species. Arzneimittelforschung. 1983;33:1289–97. [PubMed] [Google Scholar]
- 56.Velayutham M, Muthukumaran RB, Sostaric JZ, McCraken J, Fishbein JC, Zweier JL. Interactions of the major metabolite of the cancer chemopreventive drug oltipraz with cytochrome c: a novel pathway for cancer chemoprevention. Free Radic Biol Med. 2007;43:1076–85. doi: 10.1016/j.freeradbiomed.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Velayutham M, Villamena FA, Navamal M, Fishbein JC, Zweier JL. Glutathione-mediated formation of oxygen free radicals by the major metabolite of oltipraz. Chem Res Toxicol. 2005;18:970–5. doi: 10.1021/tx049687h. [DOI] [PubMed] [Google Scholar]
- 58.Bouthillier L, Charbonneau M, Brodeur J. Assessment of the role of glutathione conjugation in the protection afforded by anethol dithiolthione against hexachloro-1,3-butadiene-induced nephrotoxicity. Toxicol Appl Pharmacol. 1996;139:177–85. doi: 10.1006/taap.1996.0156. [DOI] [PubMed] [Google Scholar]
- 59.Lubet RA, Steele VE, Eto I, Juliana MM, Kelloff GJ, Grubbs CJ. Chemopreventive efficacy of anethole trithione, N-acetyl-L-cysteine, miconazole and phenethylisothiocyanate in the DMBA-induced rat mammary cancer model. Int J Cancer. 1997;72:95–101. doi: 10.1002/(sici)1097-0215(19970703)72:1<95::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 60.Reddy BS, Rao CV, Rivenson A, Kelloff G. Chemoprevention of colon carcinogenesis by organosulfur compounds. Cancer Res. 1993;53:3493–8. [PubMed] [Google Scholar]
- 61.Khanna S, Sen CK, Roy S, Christen MO, Packer L. Protective effects of anethole dithiolethione against oxidative stress-induced cytotoxicity in human Jurkat T cells. Biochem Pharmacol. 1998;56:61–9. doi: 10.1016/s0006-2952(98)00113-0. [DOI] [PubMed] [Google Scholar]
- 62.Christen MO, Fackir L, Jore D. Use of linoleic acid radiolysis for assay of antioxidant action of anethole dithiolethione. Methods Enzymol. 1995;252:324–31. doi: 10.1016/0076-6879(95)52035-x. [DOI] [PubMed] [Google Scholar]
- 63.Mansuy D, Sassi A, Dansette PM, Plat M. A new potent inhibitor of lipid peroxidation in vitro and in vivo, the hepatoprotective drug anisyldithiolthione. Biochem Biophys Res Commun. 1986;135:1015–21. doi: 10.1016/0006-291x(86)91029-6. [DOI] [PubMed] [Google Scholar]
- 64.Lam S, MacAulay C, le Riche JC, et al. A randomized phase IIb trial of anethole dithiolethione in smokers with bronchial dysplasia. J Natl Cancer Inst. 2002;94:1001–9. doi: 10.1093/jnci/94.13.1001. [DOI] [PubMed] [Google Scholar]
- 65.Isacsson G, Singer P. Studies on the whole-body distribution of 14C-trithioparamethoxyphenylpropene in mice. Scand J Dent Res. 1985;93:249–52. doi: 10.1111/j.1600-0722.1985.tb01953.x. [DOI] [PubMed] [Google Scholar]
- 66.Masoud AN, Bueding E. Identification and quantitation of a metabolite of anethol dithiolthione in rat and mouse urine using high-performance liquid chromatography. J Chromatogr. 1983;276:111–9. doi: 10.1016/s0378-4347(00)85071-9. [DOI] [PubMed] [Google Scholar]
- 67.Maxuitenko YY, Curphey TJ, Kensler TW, Roebuck BD. Protection against aflatoxin B1-induced hepatic toxicity as short-term screen of cancer chemopreventive dithiolethiones. Fundam Appl Toxicol. 1996;32:250–9. doi: 10.1006/faat.1996.0128. [DOI] [PubMed] [Google Scholar]
- 68.Prochaska HJ, Santamaria AB. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: a screening assay for anticarcinogenic enzyme inducers. Anal Biochem. 1988;169:328–36. doi: 10.1016/0003-2697(88)90292-8. [DOI] [PubMed] [Google Scholar]
- 69.Fahey JW, Dinkova-Kostova AT, Stephenson KK, Talalay P. The “Prochaska” microtiter plate bioassay for inducers of NQO1. Methods Enzymol. 2004;382:243–58. doi: 10.1016/S0076-6879(04)82014-7. [DOI] [PubMed] [Google Scholar]
- 70.Doherty GP, Leith MK, Wang X, Curphey TJ, Begleiter A. Induction of DT-diaphorase by 1,2-dithiole-3-thiones in human tumour and normal cells and effect on anti-tumour activity of bioreductive agents. Br J Cancer. 1998;77:1241–52. doi: 10.1038/bjc.1998.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Begleiter A, Leith MK, Doherty GP, Digbya TJ, Pan S. Factors influencing the induction of DT-diaphorase activity by 1,2-dithiole-3-thione in human tumor cell lines. Biochem Pharmacol. 2001;61:955–64. doi: 10.1016/s0006-2952(01)00537-8. [DOI] [PubMed] [Google Scholar]
- 72.Munday R, Zhang Y, Munday CM, Li J. Structure-activity relationships in the induction of Phase II enzymes by derivatives of 3H-1,2-dithiole-3-thione in rats. Chem Biol Interact. 2006;160:115–22. doi: 10.1016/j.cbi.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 73.Maxuitenko YY, Libby AH, Joyner HH, et al. Identification of dithiolethiones with better chemopreventive properties than oltipraz. Carcinogenesis. 1998;19:1609–15. doi: 10.1093/carcin/19.9.1609. [DOI] [PubMed] [Google Scholar]
- 74.Munday R, Munday CM. Induction of phase II enzymes by 3H-1,2-dithiole-3-thione: dose-response study in rats. Carcinogenesis. 2004;25:1721–5. doi: 10.1093/carcin/bgh162. [DOI] [PubMed] [Google Scholar]
- 75.Roebuck BD, Curphey TJ, Li Y, et al. Evaluation of the cancer chemopreventive potency of dithiolethione analogs of oltipraz. Carcinogenesis. 2003;24:1919–28. doi: 10.1093/carcin/bgg173. [DOI] [PubMed] [Google Scholar]
- 76.Brown AP, Morrissey RL, Tolhurst TA, Crowell JA, Levine BS. Oral toxicity of 1,2-dithiole-3-thione, a potential cancer chemopreventive agent, in the rat. Int J Toxicol. 2000;19:375–81. [Google Scholar]
- 77.Kwak MK, Huang B, Chang H, Kim JA, Kensler TW. Tissue specific increase of the catalytic subunits of the 26S proteasome by indirect antioxidant dithiolethione in mice: enhanced activity for degradation of abnormal protein. Life Sci. 2007;80:2411–20. doi: 10.1016/j.lfs.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 78.Andringa G, Jongenelen CA, Halfhide L, Drukarch B. The thiol antioxidant 1,2-dithiole-3-thione stimulates the expression of heat shock protein 70 in dopaminergic PC12 cells. Neurosci Lett. 2007;416:76–81. doi: 10.1016/j.neulet.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 79.Munday R, Mhawech-Fauceglia P, Munday CM, et al. Inhibition of urinary bladder carcinogenesis by broccoli sprouts. Cancer Res. 2008 Mar 1;68(5):1593–600. doi: 10.1158/0008-5472.CAN-07-5009. [DOI] [PubMed] [Google Scholar]
- 80.Tang L, Zhang Y. Isothiocyanates in the chemoprevention of bladder cancer. Curr Drug Metab. 2004;5:193–201. doi: 10.2174/1389200043489027. [DOI] [PubMed] [Google Scholar]
- 81.Moon RC, Kelloff GJ, Detrisac CJ, Steele VE, Thomas CF, Sigman CC. Chemoprevention of OH-BBN-induced bladder cancer in mice by oltipraz, alone and in combination with 4-HPR and DFMO. Anticancer Res. 1994;14:5–11. [PubMed] [Google Scholar]
- 82.Rao CV, Rivenson A, Zang E, Steele V, Kelloff G, Reddy BS. Inhibition of 2-Amino-1-methyl-6-phenylimidazo[4,5]pyridine-induced lymphoma formation by oltipraz. Cancer Res. 1996;56:3395–8. [PubMed] [Google Scholar]
- 83.Rao CV, Tokomo K, Kelloff G, Reddy BS. Inhibition by dietary oltipraz of experimental intestinal carcinogenesis induced by azoxymethane in male F344 rats. Carcinogenesis. 1991;12:1051–5. doi: 10.1093/carcin/12.6.1051. [DOI] [PubMed] [Google Scholar]
- 84.Begleiter A, Sivananthan K, Curphey TJ, Bird RP. Induction of NAD(P)H quinone: oxidoreductase1 inhibits carcinogen-induced aberrant crypt foci in colons of Sprague-Dawley rats. Cancer Epidemiol Biomarkers Prev. 2003;12:566–72. [PubMed] [Google Scholar]
- 85.Wargovich MJ, Chen CD, Jimenez A, et al. Aberrant crypts as a biomarker for colon cancer: evaluation of potential chemopreventive agents in the rat. Cancer Epidemiol Biomarkers Prev. 1996;5:355–60. [PubMed] [Google Scholar]
- 86.Liu T, Mokuolu AO, Rao CV, Reddy BS, Holt PR. Regional chemoprevention of carcinogen-induced tumors in rat colon. Gastroenterology. 1995;109:1167–72. doi: 10.1016/0016-5085(95)90575-8. [DOI] [PubMed] [Google Scholar]
- 87.Roebuck BD, Liu YL, Rogers AE, Groopman JD, Kensler TW. Protection against aflatoxin B1-induced hepatocarcinogenesis in F344 rats by 5-(2-pyrazinyl)-4-methyl-1,2-dithiole-3-thione (oltipraz): predictive role for short-term molecular dosimetry. Cancer Res. 1991;51:5501–6. [PubMed] [Google Scholar]
- 88.Bolton MG, Muñoz A, Jacobson LP, et al. Transient intervention with oltipraz protects against aflatoxin-induced hepatic tumorigenesis. Cancer Res. 1993;53:3499–504. [PubMed] [Google Scholar]
- 89.Primiano T, Egner PA, Sutter TR, Kelloff GJ, Roebuck BD, Kensler TW. Intermittent dosing with oltipraz: relationship between chemoprevention of aflatoxin-induced tumorigenesis and induction of glutathione S-transferases. Cancer Res. 1995;55:4319–24. [PubMed] [Google Scholar]
- 90.Kensler TW, Davidson NE, Egner PA, et al. Mechanisms of chemoprotection against aflatoxin-induced hepatocarcinogenesis by oltipraz. In: Nygaard OF, Upton AC, editors. Anticarcinogenesis and radiation protection. Vol. 2. New York: Plenum Press; 1991. pp. 315–22. [Google Scholar]
- 91.Sharma S, Gao P, Steele VE. The chemopreventive efficacy of inhaled oltipraz particulates in the B[a]P-induced A/J mouse lung adenoma model. Carcinogenesis. 2006;27:1721–7. doi: 10.1093/carcin/bgl052. [DOI] [PubMed] [Google Scholar]
- 92.Son HY, Nishikawa A, Furukawa F, et al. Organ-dependent modifying effects of oltipraz on N-nitrosobis(2-oxopropyl)amine (BOP)-initiation of tumorigenesis in hamsters. Cancer Lett. 2000;153:211–8. doi: 10.1016/s0304-3835(00)00373-6. [DOI] [PubMed] [Google Scholar]
- 93.Clapper ML, Wood M, Leahy K, Lang D, Miknyoczki S, Ruggeri BA. Chemopreventive activity of Oltipraz against N-nitrosobis(2-oxopropyl)amine (BOP)-induced ductal pancreatic carcinoma development and effects on survival of Syrian golden hamsters. Carcinogenesis. 1995;16:2159–65. doi: 10.1093/carcin/16.9.2159. [DOI] [PubMed] [Google Scholar]
- 94.Moon RC, Rao KVN, Detrisac CJ, Kelloff GJ, Steele VE, Doody LA. Chemoprevention of respiratory tract neoplasia in the hamster by oltipraz, alone and in combination. Int J Oncol. 1994;4:661–7. doi: 10.3892/ijo.4.3.661. [DOI] [PubMed] [Google Scholar]
- 95.Nishikawa A, Tanakamura Z, Furukawa F, et al. Chemopreventive activity of oltipraz against induction of glandular stomach carcinogenesis in rats by N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis. 1998;19:365–8. doi: 10.1093/carcin/19.2.365. [DOI] [PubMed] [Google Scholar]
- 96.Fahey JW, Haristoy X, Dolan PM, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci U S A. 2002;99:7610–5. doi: 10.1073/pnas.112203099. [DOI] [PMC free article] [PubMed] [Google Scholar]