

Summary
Background & Aims
Ischemia-reperfusion (I/R) is a major mechanism of liver injury following hepatic surgery or transplantation. Despite numerous reports on the role of oxidative/nitrosative stress and mitochondrial dysfunction in hepatic I/R injury, the proteins that are oxidatively-modified during I/R damage are poorly characterized. This study was aimed at investigating the oxidatively-modified proteins underlying the mechanism for mitochondrial dysfunction in hepatic I/R injury. We also studied the effects of a superoxide dismutase mimetic/peroxynitrite scavenger metalloporphyrin MnTMPyP on oxidatively-modified proteins and their functions.
Methods
The oxidized and/or S-nitrosylated mitochondrial proteins from I/R-injured mouse livers with or without MnTMPyP pretreatment, were labeled with biotin-N-maleimide, purified with streptavidin-agarose and resolved by two-dimensional gel electrophoresis. The identities of the oxidatively-modified proteins were determined using mass-spectrometric analysis. Liver histopathology, serum transaminase levels, nitrosative stress markers and the activities of oxidatively-modified mitochondrial proteins were measured.
Results
Comparative two-dimensional gel analysis revealed markedly increased numbers of oxidized and S-nitrosylated mitochondrial proteins following hepatic I/R injury. Many key mitochondrial enzymes involved in cellular defense, fat metabolism, energy supply, and chaperones were identified as being oxidatively-modified proteins. Pretreatment with MnTMPyP attenuated the I/R-induced increased serum transaminase levels, histological damage, increased iNOS expression, and S-nitrosylation and/or nitration of various key mitochondrial proteins. MnTMPyP pretreatment also restored I/R-induced suppressed activities of mitochondrial aldehyde dehydrogenase, 3-ketoacyl-CoA thiolases, and ATP synthase.
Conclusions
These results suggest that increased nitrosative stress is critically important in promoting S-nitrosylation and nitration of various mitochondrial proteins, leading to mitochondrial dysfunction with decreased energy supply and hepatic injury.
Keywords: Ischemia-reperfusion injury, liver mitochondria dysfunction, redox biology, Cysteine oxidation, S-nitrosylation, metalloporphyrin peroxynitrite scavenger
Introduction
Organ injury, caused by transient ischemia followed by reperfusion (I/R), is a pivotal mechanism of tissue damage in diseases such as stroke, myocardial infarction, organ transplantation and vascular surgeries. Hepatic I/R injury can develop during liver transplantation, surgical removal of hepatic tumors or traumas, circulation shock, and acute exposure to toxic substances, etc. Ischemia (hypoxia) followed by return of oxygenated blood flow (reperfusion) to the liver can eventually lead to hepatic failure with increased mortality and morbidity.1 Reactive oxygen/nitrogen species (ROS/RNS) and pro-inflammatory cytokines play an important role in liver injury depending on the duration of the reperfusion period.1
At earlier stages, increased superoxide and other ROS, derived from the activation of various sources [e.g. xathine oxidoreductases2,3] play a critical role in tissue damage. The activities of these complex enzymes in the mitochondrial respiratory chain are inhibited,4-6 allowing more ROS to leak out of the respiratory chain. The increased superoxide and NO generation during I/R (the latter being mostly derived from increased iNOS induction) favors the formation of the more potent oxidant peroxynitrite (ONOO-),7 causing impaired cellular functions.8
After a longer period of reperfusion, increased amounts of pro-inflammatory cytokines produced from resident macrophage-Kupffer cells and infiltrating leukocytes are critical in inflicting tissue damage.9 Both elevated ROS/RNS and pro-inflammatory cytokines such as TNFα also activate various cell death signaling pathways via c-Jun, N-terminal kinase (JNK)10-12 and p38 kinase,12,13 leading to apoptosis and/or necrosis.10,11,14-18 Consistent with the importance of these pathways, a variety of pharmacological agents such as specific JNK inhibitors11 and various antioxidants (e.g. ascorbate, glutathione, S-adenosylmethionine, vitamine E, etc.) attenuate the severity of I/R-mediated liver injury.3,8,19-21 Accumulation of oxidatively-modified proteins, lipid peroxides, and nucleic acids may lead to mitochondrial dysfunction and tissue damage.8,9,21-25 Despite numerous reports on the role of oxidative stress and mitochondrial dysfunction in hepatic I/R injury,4-9,26-28 the proteins that are oxidatively-modified during I/R injury are poorly characterized. We hypothesized that oxidative modifications of key mitochondrial proteins lead to their inactivation and mitochondrial dysfunction with decreased energy supply, ultimately contributing to cellular damage. To test this hypothesis, we employed a method that uses biotin-N-maleimide (biotin-NM) to detect oxidatively-modified proteins.23,29,30 In this report, we examined the mechanism of inactivation of oxidatively-modified mitochondrial proteins in a murine model of hepatic I/R injury. Furthermore, by using the superoxide dismutase (SOD) mimetic and peroxynitrite decomposition catalyst (termed “peroxynitrite scavenger”) MnTMPyP,20,31,32 we demonstrated that elevated oxidative/nitrosative stress plays a critical role in I/R-mediated mitochondrial dysfunction and hepatic damage.
Materials and Methods
Materials
Biotin-NM, anti-β-ATP synthase antibody, dithiothreitol (DTT), MnTMPyP, and L-sodium ascorbate (Asc) were obtained from Sigma Chemical. Specific antibodies to prohibitin or horse radish peroxidase-conjugated MAb-biotin were purchased from Oncogene Science and Cell Signaling, respectively. Specific antibodies to inducible nitric oxide synthase (iNOS) and 3-nitrotyrosine (3-NT) were purchased from Santa Cruz Biotechnologies and Upstate Biotechnologies, respectively.
Animals, Hepatic I/R Protocol and Histological Analysis
All animal experiments were conducted in accordance with the NIH guidelines and were approved by the Institutional Animal Care and Use Committee. Young male C57BL/6J mice from Jackson Laboratory were anesthetized with a single dose of pentobarbital (65 mg/kg i.p.) and a midline laparotomy incision was performed to expose the liver of anesthesized mice as described.33,34 The hepatic artery and the portal vein were clamped using microaneurysm clamps. This model results in segmental (70%) hepatic ischemia. This method of partial ischemia prevents mesenteric venous congestion by allowing portal decompression throughout the right and caudate lobes of the liver. The liver was kept moist at 37 °C with gauzes soaked with 0.9% saline. Body temperature, monitored with a rectal temperature probe, was maintained at 37 °C using a thermoregulatory heating blanket. Some of the mice were pretreated with MnTMPyP (20 mg/kg i.p.) 1 hour prior to hepatic I/R procedure. Mice injected with the same volume of saline were used as negative controls. Sham surgeries were conducted in a similar manner except that hepatic blood flow was not reduced with a microaneurysm clamp. The duration of warm hepatic ischemia was 1-h, after which the microaneurysm clamps were removed. The duration of reperfusion was 2-h (or 10-h and 24-h shown in Supplementary Figures). After reperfusion, blood was collected and liver tissues were removed, weighed, and snap-frozen in liquid nitrogen. A small frontal piece of the largest lobe of each mouse was fixed in 4 % buffered formalin for histopathological evaluation. After embedding formalin-fixed liver tissues and cutting 5 μm slices, all sections were stained with hematoxylin and eosin (H&E). Histological evaluation was performed in a blinded manner.33,34
Determination of the Levels of Transaminases, Nitrotyrosine, Malondialdehyde, and Enzyme Activities
The activities of alanine aminotransferase (ALT) and aminotransferase (AST) were measured in serum samples using a clinical chemistry analyzer system (PROCHEM-V; Drew Scientific, Oxford, CT).23,33,34 The level of 3-NT was quantified using the HBT Nitrotyrosine ELISA Kit as per manufacturer’s instructions (Cell Sciences, Sharon, MA). Hepatic levels of malondialdehyde (MDA) were measured using a lipid peroxidation assay kit (EMD-Calbiochem, San Diego, CA) as per manufacturer’s instructions. ATP synthase activity was determined using an ATP bioluminescence assay kit (Roche, Mannheim, Germany) following the manufacturer’s instructions. One unit of ATP synthesis activity represents 1 nM ATP produced/min/mg protein at ambient temperature. NADH-ubiquinone oxidoreductase (complex I) activity was determined by measuring OD at 340 nm and subtracting the OD values in the presence of rotenone, an inhibitor of NADH-ubiquinone oxidoreductase.35 One unit represents a reduction of 1 nmol NADH/min/mg protein.
Identification of Oxidatively-Modified Proteins Using Mass Spectrometry
Mitochondrial fractions were prepared from pooled mice livers (n≥5 per group) treated differently using differential centrifugation followed by two separate washing steps, as described.22,23,29 Relative purities of the mitochondrial fractions were determined by immunoblot analysis using a specific antibody against mitochondrial α-ATP synthase or cytosolic peroxiredoxin-II. Little contamination with cytosolic proteins in our mitochondrial fractions and vice versa was observed (Supplementary Fig. 1). Labeling of oxidatively-modified proteins with biotin-NM was performed concurrently for three different groups including the sham-operated mice (Supplementary Figure 2).23,29,30 Purified biotin-NM labeled oxidized and S-nitrosylated proteins bound to the streptavidin-agarose beads were washed twice to remove non-specifically bound proteins before being resolved using 2-dimensional polyacrylamide gel electrophoresis (2-DE), silver-stained, scanned, and analyzed. In-gel digestion of protein gel spots, nanoflow reversed-phase liquid chromatography (nanoRPLC)—tandem mass spectrometry and bioinformatic analyses were performed as described.23,29,30
Data Processing and Statistical Analysis
All data in this report represent results from at least three separate experiments, unless stated otherwise. Statistical analyses were performed using the Student’s t test, with p<0.05 being considered statistically significant. Other materials and methods not described here were performed as previously described.23,29,30,33,34
Results
Serum Transaminase Levels and Hepatic Histopathology
To assess hepatocellular damage following I/R in mouse liver, serum transaminase ALT/AST activities and hepatic histopathology were evaluated. After 1-h of warm ischemia followed by reperfusion for 2-h, serum ALT and AST activities were increased ∼33-fold and 8-fold, respectively, compared to sham-operated controls (Fig. 1A,B). Pretreatment with the peroxynitrite scavenger MnTMPyP significantly reduced the levels of serum ALT and AST following I/R injury. Serum ALT and AST levels were markedly elevated after reperfusion for 10-h and 24-h compared to sham controls, although much higher ALT and AST levels were detected in mice reperfused for 10-h than 24-h (Supplementary Fig. 3). However, MnTMPyP significantly suppressed the elevated ALT and AST levels in I/R-injured mice. Sham-operated mice showed normal hepatic histology (Fig. 1C, top). In contrast, increased signs of blood cell infiltration with sinusoidal congestion and some necrotic hepatocytes were observed in mouse livers reperfused for 2-h (Fig. 1C, middle ). These initial signs of I/R-induced tissue injury were markedly attenuated in mice pretreated with MnTMPyP (Fig. 1C, bottom). Liver histology data also showed marked necrosis after reperfusion for 10-h (Supplementary Figs. 4 and 5) or 24-h (Supplementary Figs. 6 and 7). MnTMPyP significantly attenuated the histopathological damage under these conditions (Supplementary Figs 4-7).
Figure 1.

The levels of serum transaminases and histological evaluation following I/R injury in the absence and presence of MnTMPyP. (A and B) Serum levels of ALT (A), and AST (B) following I/R injury (2-h reperfusion) without or with MnTMPyP are shown. (C) Typical H&E stained liver slides for different groups are also presented. Magnification: left panel, 100x; right panel, 400x. *significantly different from the sham controls at p<0.001; **significantly different from the I/R injured mice at p<0.005; ***significantly different from the sham controls at p<0.0001; ****significantly different from the I/R subjected samples at p< 0.0005.
Increased Amounts of Nitrite, iNOS, and Nitrotyrosine in I/R-Injured Mouse Livers
By using knockout mice,36,37 it was demonstrated that local iNOS contributes to hepatic I/R injury, while endothelial NOS protects against I/R injury. Furthermore, induction of iNOS produces large quantities of NO for extended periods, affecting liver physiology.38,39 To demonstrate elevated nitrosative stress in I/R injured mouse liver (2-h reperfusion), the levels of nitrite, iNOS, and nitrotyrosine were measured. Nitrite levels were significantly elevated from 2.07±0.70 μM to 7.28±1.45 μM after I/R injury (Fig. 2A). Pretreatment with MnTMPyP reduced the nitrite level to 3.50±0.76 μM. I/R injury increased hepatic iNOS expression (Fig. 2B, top), a source for increased nitrosative stress. Increased iNOS expression was attenuated by MnTMPyP pretreatment. In contrast, the amounts of prohibitin were similar in all samples (Fig. 2B, bottom). I/R also increased hepatic 3-NT levels [footprint of peroxynitrite generation and/or nitrative stress].8 Pretreatment with MnTMPyP significantly reduced hepatic 3-NT levels (Fig. 2C).
Figure 2.

The hepatic levels of nitrite, iNOS, and nitrotyrosine following of I/R injury in the absence and presence of MnTMPyP. The hepatic levels of nitrite (A), iNOS (B), and nitrotyrosine (C) without or with MnTMPyP pretreatment are presented. *significantly different from the sham control samples at p< 0.05; **significantly different from the I/R subjected samples (2-h reperfusion) at p<0.05; ***significantly different from the sham control samples at p< 0.005.
Identification of Oxidatively-Modified Mitochondrial Proteins in I/R Injury
Increased ROS/RNS, which can be effectively prevented using various anti-oxidants, plays an important role in hepatic I/R injury.7,8,18-21 Due to elevated levels of nitrite, iNOS, and nitrotyrosine, we hypothesized that many mitochondrial proteins are oxidized and/or S-nitrosylated in the livers of I/R injured mice. To test this hypothesis and selectively identify oxidized proteins versus S-nitrosylated (and/or S-glutathionylated) proteins, N-ethylmaleimide-modified proteins were treated with DTT and Asc, respectively, to specifically reduce oxidized (disulfides and sulfenic acids of Cys residues) versus S-nitrosylated (and/or S-glutathionylated) proteins (see Supplementary Fig. 2 for overview).23 DTT- or Asc-treated proteins were subsequently labeled with biotin-NM. Figure 3 (left panel) depicts a pattern of Coomassie blue staining of mitochondrial proteins, showing that similar levels of proteins were analyzed for all samples. Only a small number of biotin-NM labeled, oxidatively-modified proteins were detected in sham control animals (right panel, lane 1). The levels of biotin-NM labeled oxidized proteins (identified after DTT treatment: lane 2) or S-nitrosylated proteins (identified after Asc treatment: lane 4) were markedly elevated in I/R injured mice (2-h reperfusion) compared to their sham controls, when their relative levels were determined by immunoblot analysis by using MAb-biotin-HRP as a detection probe. Pretreatment with MnTMPyP, however, markedly decreased the intensity of S-nitrosylated proteins (right panel, lane 5). In contrast, MnTMPyP did not decrease the intensity of oxidized (disulfides and/or sulfenic acids) proteins (lane 3).
Figure 3.

Alteration of the levels of oxidatively-modified proteins in mouse livers following I/R injury. (Left panel) Equal amounts of mitochondrial proteins (20 μg/well) were isolated from sham-control or I/R injured mouse livers (2-h reperfusion) with and without MnTMPyP and analyzed by 12% SDS-PAGE subjected to Coomassie blue staining. (Right panel) Biotin-NM labeled oxidized- or S-nitrosylated-mitochondrial proteins (20 μg/well) were separated on 12 % SDS-PAGE, transferred to a PVDF-Immobilon membrane, and subjected to immunoblot analysis using HRP-conjugated monoclonal antibody against biotin (MAb-biotin-HRP). This figure represents a typical result from two separate experiments.
Summary of Protein Sequencing Analyses of Oxidized and/or S-Nitrosylated Mitochondrial Proteins
Because of the elevated levels of oxidized and/or S-nitrosylated mitochondrial proteins in I/R injured mouse livers, we used mass spectrometry to identify each biotin-NM labeled oxidatively-modified protein. Once again, DTT and Asc were used, respectively, to selectively identify oxidized versus S-nitrosylated (and/or S-glutathionylated) proteins (Fig. 4).23 Consistent with the results shown in Figure 3, only a few biotin-NM labeled oxidized and S-nitrosylated proteins were detected in the sham control mouse livers (Fig. 4A and 4D, respectively). The number and abundance of oxidized (Fig. 4B) and S-nitrosylated proteins (Fig. 4E) found in mouse livers reperfused for 2-h, however, were markedly increased compared to sham controls. Our results showed that MnTMPyP did not decrease the levels of oxidized proteins (Fig. 4C), however, it drastically decreased the levels of S-nitrosylated proteins when mouse livers were reperfused for 2-h (Fig. 4F). More oxidized and S-nitrosylated proteins (Supplementary Figs. 8 and 9, respectively) were observed after reperfusion for 10-h and 24-h without MnTMPyP. Under these conditions, we observed a time-dependent effect of MnTMPyP, which also seems to have affected the oxidized proteins (ie, DTT-treated samples in Supplementary Fig. 8) although a greater effect of MnTMTyP seems to be observed with the S-nitroylated proteins (i.e. Asc-treated samples in Supplementary Fig. 9). However, it is unclear why MnTMPyP reversed the oxidized proteins after reperfusion for 10-h but not for 2-h.
Figure 4.
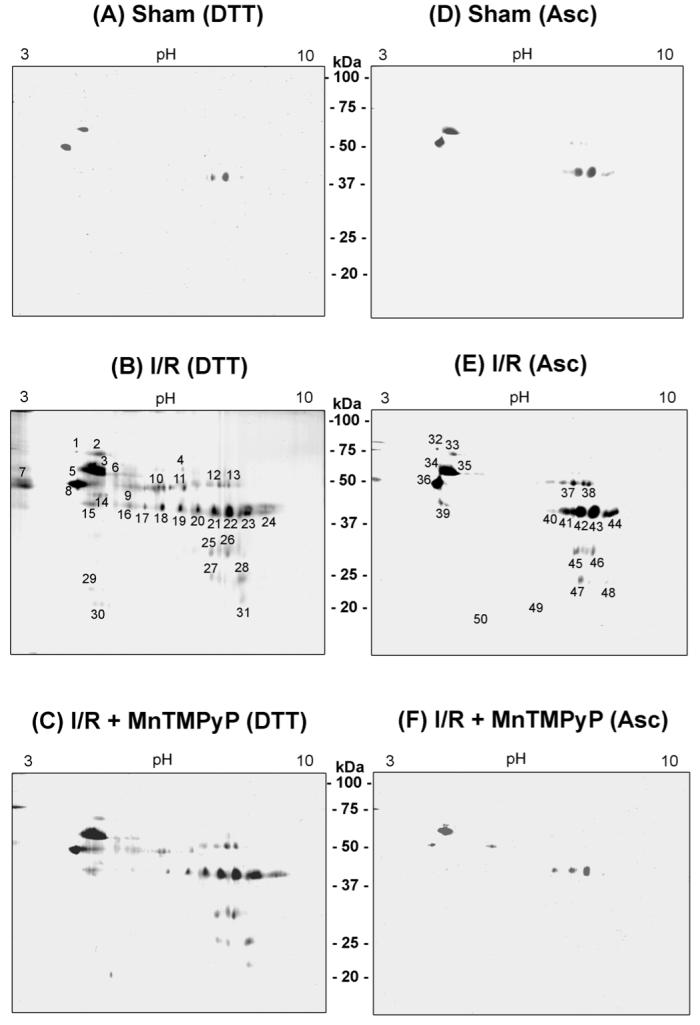
Comparison of oxidized and S-nitrosylated mitochondrial proteins by 2-DE in hepatic I/R injury without or with MnTMPyP. Oxidized mitochondrial proteins (10 mg/sample) from sham-control (A) and I/R injured mice (2-h reperfusion) without (B) or with MnTMPyP (C) (I/R + MnTMPyP) were processed concurrently processed, labeled with biotin-NM in the presence of DTT, and then purified with streptavidin-agarose. S-nitrosylated mitochondrial proteins (10 mg/sample) from sham-control (D) and I/R injured mice (2-h reperfusion) without (E) or with MnTMPyP (F) were labeled with biotin-NM in the presence of Asc and then purified with streptavidin-agarose. Purified biotin-NM labeled oxidized (spots 1-31) and S-nitrosylated (spots 32-50) proteins (0.25 mg/sample) were resolved by 2-DE, and silver stained. Individual protein spots with differential intensities were excised out of this particular gel (pH range 3-10) and subjected to MS analysis following in-gel trypsin digestion. This figure represents a typical result from two separate experiments.
Thirty-one oxidized and nineteen S-nitrosylated proteins acquired from mouse liver mitochondria after I/R injury (2-h reperfusion) that showed an increased staining intensity (Fig. 4B and 4E) were subjected to MS analysis. The protein sequence data acquired for each spot are provided in Table 1 and summarized in Fig. 5. Protein sequence analysis revealed that many mitochondrial proteins are identified as being oxidized as well as S-nitrosylated, suggesting that these two modifications occur concurrently after I/R injury. These proteins include: oxidative phosphorylation [NADH-ubiquinone oxidoreductase (complex I), ubiquinol-cytochrom-c-reductase (complex III), and α- and β-ATP synthase (complex V)]; anti-oxidative defense system [aldehyde dehydrogenase (ALDH2), methylmalonate-semialdehyde dehydrogenase (ALDH6), 4-trimethylaminobutyraldehyde dehydrogenase (ALDH9), glutathione transferase, glutathione peroxidase (GPx), etc]; fatty acid β-oxidation and intermediary metabolism (acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-hydroxyacy-CoA dehydrogenase, 3-ketoacyl-CoA thiolase, acetyl-CoA acetyltransferase, hydroxymethylglutaryl-CoA synthase, hydroxymethylglutaryl-CoA lyase, isovaleryl-CoA dehydrogenase, carbamoyl-phosphate synthase, etc); TCA cycle and energy supply (pyruvate dehydrogenase, dihydrolipoamide succinyltransferase, glutamate dehydrogenase, pyruvate carboxylase, etc); molecular chaperon proteins [heat shock protein (Hsp) 60, Hsp 70.2, Hsp 70.3, Hsp 70.8, glucose-regulated protein (GRP) 75, GRP78, etc]; mitochondrial electron transport and ion channeling proteins [α- or β-subunit of electron transfer flavoprotein (α- or β-ETF) and voltage-dependent anion channel protein1 (VDAC1)].
Table 1.
Summary of nanoRP LC-MS/MS peptide sequence analyses for oxidized and S-nitrosylated proteins in ischemia/reperfusion injury mouse liver mitochondria.
| Spot No. | Protein Identified | Accession Number |
No. of Peptides Identified |
|---|---|---|---|
| 1 | 78 kDa glucose-regulated protein (GRP 78) | P200029 | 20 |
| 2 | 75 kDa glucose-regulated protein (GRP 75) | P38647 | 14 |
| 2 | Heat shock cognate 71 kDa protein (Hsp 70.8) | P63017 | 5 |
| 2 | Heat shock-related 70 kDa protein 2 (Hsp 70.2) | P17156 | 4 |
| 2 | Heat shock 70 kDa protein 1A (Hsp 70.3) | Q61696 | 2 |
| 3 | 60 kDa heat shock protein, mitochondrial (Hsp 60) | P63038 | 6 |
| 4 | Not identified | ||
| 5 | 60 kDa heat shock protein, mitochondrial (Hsp 60) | P63038 | 33 |
| 5 | ATP synthase beta chain, mitochondrial (β-ATP synthase) | P56480 | 2 |
| 6 | Protein disulfide isomerase A3 precursor | Q3TIL2 | 16 |
| 6 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 4 |
| 6 | 60 kDa heat shock protein, mitochondrial (Hsp 60) | P63038 | 2 |
| 7 | ATP synthase beta chain, mitochondrial (β-ATP synthase) | P56480 | 6 |
| 7 | Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P47738 | 2 |
| 8 | ATP synthase beta chain, mitochondrial (β-ATP synthase) | P56480 | 21 |
| 8 | Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P47738 | 2 |
| 9 | Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P47738 | 16 |
| 9 | Dihydrolipoamide succinyltransferase | Q9D2G2 | 6 |
| 9 | 60 kDa heat shock protein, mitochondrial (Hsp 60) | P63038 | 2 |
| 9 | Carbamoyl-phosphate synthase | Q8C196 | 2 |
| 9 | Dihydrolipoamide branched chain transacylase | P53395 | 2 |
| 9 | Pyruvate dehydrogenase protein X component (PDH-X) | Q8BKZ9 | 2 |
| 10 | Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P47738 | 13 |
| 10 | 4-Trimethylaminobutyraldehyde dehydrogenase (ALDH9) | Q3TG52 | 3 |
| 11 | Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P47738 | 16 |
| 11 | 4-Trimethylaminobutyraldehyde dehydrogenase (ALDH9) | Q3TG52 | 2 |
| 11 | Hydroxymethylglutaryl-CoA synthase, mitochondrial | P54869 | 2 |
| 12 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 5 |
| 12 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q3TDA2 | 5 |
| 13 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q3TDA2 | 6 |
| 13 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 4 |
| 14 | Ubiquinol-cytochrome-c reductase complex core protein I | Q9CZ13 | 9 |
| 15 | Not identified | ||
| 16 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 10 |
| 16 | Isovaleryl-CoA dehydrogenase, mitochondrial | Q9JHI5 | 2 |
| 17 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 12 |
| 17 | 2-Oxoisovalerate dehydrogenase alpha subunit, mitochondrial | P50136 | 2 |
| 18 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 15 |
| 18 | 2-Oxoisovalerate dehydrogenase alpha subunit, mitochondrial | P50136 | 3 |
| 18 | Isovaleryl-CoA dehydrogenase, mitochondrial | Q9JHI5 | 2 |
| 19 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 12 |
| 19 | Isovaleryl-CoA dehydrogenase, mitochondrial | Q9JHI5 | 3 |
| 20 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 16 |
| 20 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 3 |
| 20 | Isovaleryl-CoA dehydrogenase, mitochondrial | Q9JHI5 | 3 |
| 20 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 2 |
| 21 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 17 |
| 21 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 5 |
| 21 | 3-Ketoacyl-CoA thiolase A, peroxisomal | Q921H8 | 3 |
| 21 | Acyl-CoA dehydrogenase, medium-chain, mitochondrial | P45952 | 3 |
| 22 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 24 |
| 22 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 7 |
| 22 | 3-Ketoacyl-CoA thiolase A, peroxisomal | Q921H8 | 4 |
| 22 | Acyl-CoA dehydrogenase, medium-chain, mitochondrial | P45952 | 4 |
| 22 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 4 |
| 23 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 18 |
| 23 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 5 |
| 23 | 3-Ketoacyl-CoA thiolase A, peroxisomal | Q921H8 | 4 |
| 24 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 21 |
| 24 | 3-Ketoacyl-CoA thiolase A, peroxisomal | Q921H8 | 7 |
| 24 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 3 |
| 25 | Electron transfer flavoprotein α-subunit (α-ETF) | Q99LC5 | 4 |
| 26 | Electron transfer flavoprotein α-subunit (α-ETF) | Q99LC5 | 4 |
| 26 | Hydroxymethylglutaryl-CoA lyase, mitochondrial | P38060 | 2 |
| 27 | Electron transfer flavoprotein, β-subunit (β-ETF) | Q810V3 | 6 |
| 27 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 5 |
| 28 | Electron transfer flavoprotein, β-subunit (β-ETF) | Q810V3 | 6 |
| 28 | 3-Hydroxyacyl-CoA dehydrogenase type II | O08756 | 3 |
| 28 | Enoyl-CoA hydratase, mitochondrial | Q8BH95 | 3 |
| 28 | Hydroxyacyl-CoA dehydrogenase type II | Q99N15 | 2 |
| 28 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 2 |
| 29 | NADH-ubiquinone oxidoreductase 24 kDa subunit, mitochondrial | Q9D6J6 | 2 |
| 30 | ATP synthase D chain, mitochondrial | Q9DCX2 | 8 |
| 31 | Glutathione S-transferase P 1 | P19157 | 4 |
| 31 | Glutathione peroxidase 1 (GPx) | P11352 | 3 |
| 31 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 2 |
| 31 | Acyl-protein thioesterase 1 | P97823 | 2 |
| 32 | 78 kDa glucose-regulated protein (GRP 78) | P200029 | 14 |
| 33 | Stress-70 protein, mitochondrial (GRP 75) | P38647 | 22 |
| 33 | Heat shock cognate 71 kDa protein (HSP 70.8) | P63017 | 10 |
| 33 | 78 kDa glucose-regulated protein (GRP 78) | P200029 | 6 |
| 33 | 60 kDa heat shock protein, mitochondrial (Hsp 60) | P63038 | 5 |
| 33 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 5 |
| 33 | Heat shock-related 70 kDa protein 2 (Hsp 70.2) | P17156 | 5 |
| 33 | Pyruvate carboxylase | Q3T9S7 | 2 |
| 33 | Heat shock 70 kDa protein 1A (Hsp 70.3) | Q61696 | 2 |
| 34 | 60 kDa heat shock protein, mitochondrial (Hsp 60) | P63038 | 54 |
| 34 | ATP synthase beta chain, mitochondrial (β-ATP synthase) | P56480 | 4 |
| 34 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q3TDA2 | 2 |
| 35 | 60 kDa heat shock protein, mitochondria (Hsp 60) | P63038 | 15 |
| 35 | Protein disulfide isomerase A3 precursor | Q3TIL2 | 12 |
| 35 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q3TDA2 | 2 |
| 35 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 2 |
| 36 | ATP synthase beta chain, mitochondrial (β-ATP synthase) | P56480 | 40 |
| 36 | Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P47738 | 9 |
| 36 | 60 kDa heat shock protein, mitochondrial (Hsp 60) | P63038 | 2 |
| 36 | 4-Trimethylaminobutyraldehyde dehydrogenase (ALDH9) | Q3TG52 | 2 |
| 36 | Protein disulfide isomerase A3 precursor | Q3TIL2 | 2 |
| 37 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 19 |
| 37 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q3TDA2 | 10 |
| 37 | Glutamate dehydrogenase 1, mitochondrial (GDH) | P26443 | 6 |
| 37 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 5 |
| 37 | ATP synthase beta chain, mitochondrial (β-ATP synthase) | P56480 | 2 |
| 38 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 19 |
| 38 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q3TDA2 | 9 |
| 38 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 2 |
| 39 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 5 |
| 39 | Succinyl-CoA ligase beta-chain, mitochondrial | Q9Z2I9 | 3 |
| 39 | ATP synthase beta chain, mitochondrial (β-ATP synthase) | P56480 | 2 |
| 40 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 15 |
| 40 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 5 |
| 40 | Pyruvate dehydrogenase E1, alpha subunit | P35486 | 4 |
| 40 | 2-Oxoisovalerate dehydrogenase alpha subunit | P50136 | 2 |
| 40 | Acyl-CoA dehydrogenase, medium-chain | P45952 | 2 |
| 41 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 17 |
| 41 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 6 |
| 41 | Acyl-CoA dehydrogenase, medium-chain | P45952 | 4 |
| 41 | Fumaryl acetoacetase | P35505 | 4 |
| 41 | Isovaleryl-CoA dehydrogenase, mitochondrial | Q9JHI5 | 3 |
| 41 | Acetyl-CoA acyltransferase A | Q921H8 | 2 |
| 41 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 2 |
| 42 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 24 |
| 42 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 7 |
| 42 | Acyl-CoA dehydrogenase, medium-chain | P45952 | 6 |
| 42 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 5 |
| 42 | Acetyl-CoA acyltransferase A | Q921H8 | 3 |
| 42 | Hydroxyacyl-CoA dehydrogenase | Q5U5Y5 | 2 |
| 43 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 32 |
| 43 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 9 |
| 43 | Acyl-CoA dehydrogenase, medium-chain | P45952 | 6 |
| 43 | Carbamoyl-phosphate synthase, mitochondrial | Q8C196 | 2 |
| 44 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 24 |
| 44 | Acetyl-CoA acetyltransferase, mitochondrial | Q8QZT1 | 9 |
| 44 | Acetyl-CoA acyltransferase A | Q921H8 | 4 |
| 44 | Aspartate aminotransferase, mitochondrial | P05202 | 2 |
| 44 | Hydroxyacyl-CoA dehydrogenase | Q5U5Y5 | 2 |
| 45 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 6 |
| 45 | Electron transfer flavoprotein α-subunit (α-ETF) | Q99LC5 | 6 |
| 45 | Voltage-dependent anion-selective channel protein 1 (VDAC1) | Q60932 | 3 |
| 45 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 2 |
| 45 | Hydroxymethylglutaryl-CoA lyase, mitochondrial | P38060 | 2 |
| 46 | Electron transfer flavoprotein α-subunit (α-ETF) | Q99LC5 | 7 |
| 46 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 5 |
| 47 | Electron transfer flavoprotein, β-subunit (β-ETF) | Q810V3 | 12 |
| 47 | 3-Hydroxyacyl-CoA dehydrogenase type II | O08756 | 6 |
| 47 | Short chain L-3-hydroxyacyl-CoA dehydrogenase | Q99N15 | 4 |
| 48 | Electron transfer flavoprotein, β-subunit (β-ETF) | Q810V3 | 5 |
| 48 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 4 |
| 48 | Short chain L-3-hydroxyacyl-CoA dehydrogenase | Q99N15 | 3 |
| 48 | Enoyl-CoA hydratase, mitochondrial | Q8BH95 | 3 |
| 48 | Glutathione S-transferase P 1 | P19157 | 3 |
| 48 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 2 |
| 48 | Trifunctional enzyme beta subunit, mitochondrial | Q99JY0 | 2 |
| 49 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 16 |
| 49 | Glutathione peroxidase 1 (GPx) | P11352 | 7 |
| 49 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 4 |
| 49 | Glutathione S-transferase P 1 | P19157 | 3 |
| 50 | ATP synthase D chain, mitochondrial | Q9DCX2 | 11 |
| 50 | Glutathione peroxidase 1 (GPx) | P11352 | 3 |
| 50 | Glutathione S-transferase P 1 | P19157 | 3 |
| 50 | 3-Ketoacyl-CoA thiolase, mitochondrial | Q8BWT1 | 2 |
| 50 | ATP synthase alpha chain, mitochondrial (α-ATP synthase) | P02535 | 2 |
Biotin-NM labeled oxidized- or S-nitrosylated proteins were purified with streptavidinagarose beads, washed twice, resolved by 2-DE, and silver stained. Each protein spot as indicated was picked up with a razor blade and subjected to protein identification by mass spectrometric analysis.23,29,30 Since our original aim was to identify early signs of oxidative modifications, we only identified the oxidatively-modified proteins after 2-h reperfusion.
Figure 5.

Summary of oxidized and S-nitrosylated mitochondrial proteins following hepatic I/R injury in the absence of MnTMPyP. Mitochondrial proteins that are simulataneously oxidized- and S-nitrosylated proteins are summarized with respects to the function of each protein.
Many proteins such as ATP synthase (spots 12,13,37,38), ALDH2 (spots 7-11), ALDH6 (spots 12,13,34-38), acetyl-CoA acetyltransferase (spots 20-24,40-44), acyl-CoA dehydrogenase (spots 21,22,40-43), 3-ketoacyl-CoA thiolase (spots 16-24,39-44), etc. were repeatedly detected from spots with different pI values, suggesting they were also covalently modified by either phosphorylation or hyper-oxidation to sulfinic and/or sulfonic acids. Several proteins were also identified from spots with apparently lower molecular weights of the parent protein (e.g. spots 27-28,45,48-50 for αATP synthase). These spots likely represent fragments of the full-length proteins (e.g. 58 kDa for α-ATP synthase). Increased degradation of oxidized proteins was also observed with β-ATP synthase, 3-ketoacyl-CoA thiolase and other proteins even after 2-h reperfusion.
Reversible inactivation of oxidized and/or S-nitrosylated mitochondrial enzymes in I/R injured mice without and with MnTMPyP
We recently reported that the activities of many mitochondrial proteins are inhibited through S-nitrosylation of Cys residue(s) and nitration of Tyr residue(s).22,23,29 Indeed, several proteins such as ALDH240 and 3-ketoacyl-CoA thiolase41 are known to contain Cys residue(s) within their catalytic sites. Therefore, we evaluated whether the catalytic activities of ALDH2, 3-ketoacyl-CoA thiolase, and ATP synthase were altered in I/R injured mice and if this activity could be restored in the presence of MnTMPyP. Hepatic ALDH2 activity in sham control mice was 3.30±0.13 units compared to 1.52±0.58 and 2.74±0.17 units in 2-h reperfused mice without and with MnTMPyP, respectively. These data suggest that mouse ALDH2 activity was markedly reduced in the absence of MnTMPyP and that its activity recovered significantly with MnTMPyP treatment (Fig. 6A). Significant suppression of ALDH2 activity was also observed after reperfusion for 10-h or 24-h. However, MnTMPyP treatment fully restored the ALDH2 activity (Supplementary Fig. 10A). Since ALDH2 is known to metabolize toxic lipid aldehydes such as malondialdehyde (MDA),42 we also measured the level of MDA, as an indicator of lipid peroxidation, in different groups. The level of MDA, which was low in sham control mice, was increased by 45% in mouse livers reperfused for 2-h (Fig. 6B). The increased MDA level in I/R injured mice, was abolished by pretreatment with MnTMPyP. MDA levels were also elevated in 10-h or 24-h reperfused mice and significantly attenuated by MnTMPyP pretreatment (Supplementary Fig. 10B).
Figure 6.

Inactivation of mitochondrial ALDH2 activity and the level of lipid peroxidation in I/R injured mice without or with MnTMPyP. (A) Catalytic activities of mitochondrial ALDH2 from the indicated liver samples (2-h reperfusion) were determined. (B) Hepatic malondialdehyde (MDA) levels in different mice were measured and presented. *significantly different from the sham controls at p<0.05; **different from the I/R subjected samples at p=0.056; ***significantly different from the sham controls at p< 0.025; ****significantly different from the I/R subjected samples at p< 0.01.
One His (His352) and two Cys residues (Cys92 and Cys382) are essential for the activity of 3-ketoacyl-CoA thiolase.41 These residues are likely targets of oxidative/nitrosative modification, thereby inactivating the protein. Indeed, mitochondrial 3-ketoacyl-CoA thiolase showed an approximately 61% decrease in activity in I/R injured mice compared to sham control mice (5.82±0.01 unit). The suppressed activity of 3-ketoacyl-CoA thiolase was restored to 92.3% of control animals when I/R-injured mice were pretreated with MnTMPyP (data not shown).
Mitochondrial dysfunction with decreased activities of the complex enzymes in the respiratory chain has been reported in I/R injury.4-6,43 It is possible that I/R injury results in S-nitrosylation, nitration or other modification of critical Cys or Tyr residues in the active site of ATP synthase and other components of the mitochondrial respiratory chain under elevated levels of nitrite and/or peroxynitrite. In fact, Tomkins et al recently reported that ROS can be generated through thiol modification of mitochondrial complex I.43 Therefore, we measured the activity of ATP synthase (complex V) in mice with or without MnTMPyP pretreatment. The ATP synthase activity was 472.8±7.5 units in sham control mice, and 265.1±41.8 and 444.4±10.2 units, respectively, in 2-h reperfused mice without and with MnTMPyP treatment (Fig. 7A). Inhibition of ATP synthase activity was also observed after reperfusion for 10-h or 24-h, however, this activity was restored following MnTMPyP treatment (Supplementary Fig. 11A). These data indicate that mitochondrial ATP synthase activities are significantly inhibited in I/R injured mouse livers, and this suppression can be restored by MnTMPyP. To further elucidate the mechanism for I/R-mediated inhibition of ATP synthase activity, we determined the levels of 3-NT in immunoprecipitated β-ATP synthase from mitochondria of sham controls and I/R-injured mice with or without MnTMPyP. Immunoblot analysis showed that similar levels of β-ATP synthase exist in all conditions (Fig. 7B, left panel). However, 3-NT was only detected in mouse livers reperfused for 2-h and not in the sham controls (right panel). Furthermore, the 3-NT immunoreactive band was absent in mice pretreated with MnTMPyP. These data support the hypothesis that ATP synthase is inactivated in I/R injured mice through nitration of its Tyr residue(s), including the Tyr residues within the catalytic site,23 and that its inactivation could be prevented with the peroxynitrite scavenger MnTMPyP. Furthermore, the hepatic activity of NADH-ubiquinone oxidoreductase (complex I) was also markedly inhibited in 2-h reperfused mice compared to sham controls (Fig. 7C); an effect that was prevented by MnTMPyP pretreatment (p<0.007). Inhibition of complex I activity was also observed after reperfusion for 10-h or 24-h, while MnTMPyP significantly restored its activity (Supplementary Fig. 11B).
Figure 7.

Inhibition of ATP synthase and complex I activities following hepatic I/R injury and nitration of Tyr residue in β-ATP synthase. (A) Mitochondrial ATP synthase activities from sham control and I/R injured mouse livers (2-h reperfusion) without or with MnTMPyP (I/R + MnTMPyP) are presented. (B) Mitochondrial β-ATP synthase (1 mg proteins) in different groups as indicated were immunoprecipitated with the anti-β-ATP synthase antibody as described,23 separated on 12% SDS-PAGE, and subjected to immunoblot analysis using the anti-β-ATP synthase antibody (left) or the anti-3-NT antibody (right). (C) NADH-ubiquinone oxidoreductase (Complex I) activities in indicated groups were determined and presented. *significantly different from the sham controls at p< 0.05; **significantly different from the I/R subjected samples at p< 0.05; ***significantly different from the sham controls at p< 0.001; ****significantly different from the I/R subjected samples at p< 0.01.
Discussion
Despite the well-established role of ROS/RNS in I/R injury, the protein targets that are oxidatively-modified by elevated ROS/RNS remain elusive. Virtually no systematic approaches to characterize oxidatively-modified proteins (especially for mitochondrial proteins) during hepatic I/R injury have previously been taken. Recent reports show that a few proteins are altered in humans undergoing liver transplantation.25,44,45 In addition, some other proteins were also altered in rabbit hearts following I/R injury.24,46 However, these studies were conducted using whole cell homogenate without subcellular fractionation and functional characterization of those proteins altered during I/R injury.25,44,46 In this study, we specifically aimed at identifying the oxidatively-modified target proteins by focusing on redox modulation of Cys residues and evaluating their functional roles in acute hepatic I/R injury, in the presence or absence of a SOD mimetic/peroxynitrite scavenger. The early time point of reperfusion (2-h) was intentionally chosen to identify early signs of oxidative modifications, which may lead to mitochondrial dysfunction prior to a secondary wave of severe liver damage inflicted by infliltrating immune cells during reperfusion for longer times.1,9,33,34 Because of the involvement of neutrophils in the longer reperfusion periods, neutrophil-derived oxidative modifications and clorination of proteins may take place and provide extra insult to the body system, contributing to more severe hepatotoxicity. In fact, our results in liver tissues reperfused for 10-h or 24-h showed fulminant liver damage (Supplementary Figures 4-7), despite similar degrees of inhibition of the mitochondrial enzymes measured in this study following reperfusion for 2-h versus 10-h or 24-h. These data support our hypothesis that mitochondrial dysfunction with decreased energy supply occurs prior to necrotic liver damage. These results also suggest that other events such as activation of the cell death-related JNK and/or p38 kinase likely occur and accelerate severe tissue damage.10-13
Numerous mitochondrial proteins involved in energy supply and electron transport system, molecular chaperone activity, anti-oxidant defense, the urea cycle, degradation of fatty acids, etc, have been simultaneously oxidized and S-nitrosylated (Fig. 4 and Table 1) during I/R injury, as observed in ethanol-exposed animals,23 often leading to decreased activity/function. These results suggest that oxidative and nitrosative stress can simultaneously affect numerous proteins during the I/R injury process. Thus, it is extremely difficult to clearly separate oxidized proteins from S-nitrosylated or nitrated proteins especially for the liver tissues reperfused for longer periods. Although the activities of all of modified proteins have not been studied, their activities may have been attenuated as a result of oxidative modifications. For instance, ALDH2, ALDH5, ALDH6, and ALDH9 isozymes also have a highly conserved Cys residue within their active sites.40 Although we did not show S-nitrosylation of Cys residue(s) within these ALDH isozymes, it is likely that these enzymes undergo S-nitrosylation, as recently demonstrated.22,23,40 It was also reported that Cys323 is critical for the catalytic activity of glutamate dehydrogenase47 and its activity is significantly inhibited during hepatic I/R injury.21 Based on the inactivation of these critical enzymes (e.g. ALDH2 and ATP synthase) and possibly many others, we believe that oxidative modifications of the proteins listed in Table 1 likely contribute to mitochondrial dysfunction, leading to I/R-related cellular damage at later stage. Furthermore, inactivation of the mitochondrial respiratory chain complex enzymes through oxidative/nitrosative modifications lead to more ROS leakage with reduced efficiency of oxidative phosphorylation and decreased ATP synthesis.4-6,18,43 Based on the positive correlation between the pattern of S-nitrosylated and nitrated proteins and hepatic injury (including ALT/AST levels, histology, nitrite and 3-NT levels, etc) and on the beneficial effects of MnTMPyP pre-treatment against hepatic damage, increased iNOS-derived NO coupled with superoxide formation and consequent generation of peroxynitrite play a critical role in causing I/R-mediated hepatic injury.7-9
Our current results conducted with mouse liver reperfused for 2-h show that many oxidatively-modified mitochondrial proteins exhibited multiple pI values on 2-DE. These multiple pI shifts may represent phosphorylations of Ser/Thr/Tyr residues by JNK and p38 kinase, which are known to be activated during the process of I/R injury.10-13 Alternatively, multiple pI values of these proteins may represent hyper-oxidation of Cys residues to sulfinic and/or sulfonic acids that are not easily reduced by DTT or Asc.23 We also identified smaller fragments of many mitochondrial proteins such as ATP synthase and 3-ketoacyl-CoA thiolase following I/R injury. These smaller protein fragments may increase the susceptibility of oxidized mitochondrial proteins to ubiquitin-dependent and -independent proteases activated under oxidative/nitrosative stress.30,48,49 Alternatively, they may simply represent spontaneous fragmentation of oxidized proteins, as observed with cytosolic proteins in alcohol-exposed mouse livers.23,30 Increased degradation of oxidatively-modified proteins may contribute to the reduced levels of several mitochondrial proteins, as recently reported using rabbit hearts exposed to I/R injury.24,46 Furthermore, we expect that similar covalent modifications (e.g. phosphorylation) and/or fragmentations of mitochondrial proteins occur in 10-h or 24-h reperfused mice, based on the similar 2-DE patterns of oxidatively-modified proteins despite appearance of additional protein spots (Supplementary Figs. 8 and 9) compared to those in 2-h reperfused tissues (Fig. 4).
Oxidative modification of mitochondrial enzymes involved in the oxidation of fatty acids was also detected in this study. The inhibition of the mitochondrial fat oxidation pathway, as evidenced by inactivation of 3-ketoacyl-CoA thiolase, may explain the underlying mechanism for the low success rate in liver transplantation when steatotic livers are used as donor organs.11,50 These results not only represent new information, but also imply possible limited energy supply derived from the β-oxidation pathway of fatty acids during I/R. In addition to blocking the fat oxidation pathway, inactivation of ATP synthase (Fig. 7) may explain the decreased level of ATP and thus favoring necrosis in I/R injury.15,17,19 Furthermore, inactivation of complex I of the respiratory chain and other enzymes such as ALDH isoenzymes, glutathione peroxidase and others listed in Table 1 may cause a marked imbalance between increased ROS/RNS during I/R and cellular antioxidant defense systems. Therefore, our current data supports the pivotal role of ROS/RNS in mitochondrial dysfunction and cell damage during hepatic I/R injury, and provides a possible underlying molecular mechanism(s).
In conclusion, numerous oxidatively-modified mitochondrial proteins from the livers of mice exposed to acute I/R injury have been identified. Inactivation of some of these oxidized/S-nitrosylated and nitrated proteins and others not tested herein, likely contribute to mitochondrial dysfunction and ultimate hepatic damage following acute hepatic ischemia-reperfusion.
Supplementary Material
Acknowledgements
We thank Dr. Norman Salem Jr. for the support. This research was supported by the Intramural Research Program of National Institute on Alcohol Abuse and Alcoholism and National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400.
Abbreviations
- I/R
ischemia-reperfusion
- ALDH2
mitochondrial aldehyde dehydrogenase 2
- ROS/RNS
reactive oxygen/nitrogen species
- MnTMPyP
metalloporphyrin peroxynitrite scavenger/SOD mimetics
- NO
nitric oxide
- iNOS
inducible nitric oxide synthase
- Asc
sodium ascorbate
- biotin-NM
biotin-N-maleimide
- DTT
dithiothreitol
- 3-NT
3-nitrotyrosine
- 2-DE
two-dimensional polyacrylamide gel electrophoresis
- α-ATP synthase
ATP synthase α-subunit
- β-ATP synthase
ATP synthase β-subunit
- HSP
heat shock protein
- GRP
glucose-regulated protein
- MDA
malondialdehyde
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- Complex I
NADH—ubiquinone oxidoreductase
- SOD
superoxide dismutase
- H&E
hematoxylin and eosin
- GPx
glutathione peroxidase
Footnotes
All authors do not have any conflict of interest to disclose.
E-mail addresses of other co-authors are: Kwan-Hoon Moon: kwanhoonm@mail.nih.gov Brian L. Hood: hoodb@upmc.edu Partha Mukhopadhyay: mpartha@mail.nih.gov Rajesh Mohanraj: mohanrajr@mail.nih.gov Mohamed A. Abdelmegeed: abdelmegeedm@mail.nih.gov Yong-Il Kwon: kbgy@hallym.or.kr Thomas P. Conrads: conradstp@upmc.edu Timothy D. Veenstra: veenstra@ncifcrf.gov.
References
- 1.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 2.Engerson TD, McKelvey TG, Rhyne DB, Boggio EB, Snyder SJ, Jones HP. Conversion of xanthine dehydrogenase to oxidase in ischemic rat tissues. J Clin Invest. 1987;79:1564–1570. doi: 10.1172/JCI112990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. doi: 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeong C, Lee SM. The beneficial effect of ATP-MgCl(2) on hepatic ischemia/reperfusion-induced mitochondrial dysfunction. Eur J Pharmacol. 2000;403:243–250. doi: 10.1016/s0014-2999(00)00485-4. [DOI] [PubMed] [Google Scholar]
- 5.Kaibori M, Inoue T, Tu W, Oda M, Kwon AH, Kamiyama Y, Okumura T. FK506, but not cyclosporin A, prevents mitochondrial dysfunction during hypoxia in rat hepatocytes. Life Sci. 2001;69:17–26. doi: 10.1016/s0024-3205(01)01098-0. [DOI] [PubMed] [Google Scholar]
- 6.Caraceni P, Bianchi C, Domenicali M, Pertosa A Maria, Maiolini E, Castelli G Parenti, Nardo B, et al. Impairment of mitochondrial oxidative phosphorylation in rat fatty liver exposed to preservation-reperfusion injury. J Hepatol. 2004;41:82–88. doi: 10.1016/j.jhep.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 7.Ma TT, Ischiropoulos H, Brass CA. Endotoxin-stimulated nitric oxide production increases injury and reduces rat liver chemiluminescence during reperfusion. Gastroenterology. 1995;108:463–469. doi: 10.1016/0016-5085(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 8.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaeschke H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083–1088. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 10.Bradham CA, Stachlewitz RF, Gao W, Qian T, Jayadev S, Jenkins G, Hannun Y, et al. Reperfusion after liver transplantation in rats differentially activates the mitogen-activated protein kinases. Hepatology. 1997;25:1128–1135. doi: 10.1002/hep.510250514. [DOI] [PubMed] [Google Scholar]
- 11.Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, Brenner DA. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42:850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 12.Massip-Salcedo M, Casillas-Ramirez A, Franco-Gou R, Bartrons R, Mosbah I Ben, Serafin A, Rosello-Catafau J, et al. Heat shock proteins and mitogen-activated protein kinases in steatotic livers undergoing ischemia-reperfusion: some answers. Am J Pathol. 2006;168:1474–1485. doi: 10.2353/ajpath.2006.050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi M, Takeyoshi I, Yoshinari D, Matsumoto K, Morishita Y. P38 mitogen-activated protein kinase inhibition attenuates ischemia-reperfusion injury of the rat liver. Surgery. 2002;131:344–349. doi: 10.1067/msy.2002.121097. [DOI] [PubMed] [Google Scholar]
- 14.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 15.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281:21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 16.Levrand S, Vannay-Bouchiche C, Pesse B, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radic Biol Med. 2006;41:886–895. doi: 10.1016/j.freeradbiomed.2006.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 18.Gero D, Szabo C. Role of the peroxynitrite-poly (ADP-ribose) polymerase pathway in the pathogenesis of liver injury. Curr Pharm Des. 2006;12:2903–2910. doi: 10.2174/138161206777947579. [DOI] [PubMed] [Google Scholar]
- 19.Jeon BR, Lee SM. S-adenosylmethionine protects post-ischemic mitochondrial injury in rat liver. J Hepatol. 2001;34:395–401. doi: 10.1016/s0168-8278(00)00045-3. [DOI] [PubMed] [Google Scholar]
- 20.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 21.Lee WY, Lee JS, Lee SM. Protective effects of combined ischemic preconditioning and ascorbic acid on mitochondrial injury in hepatic ischemia/reperfusion. J Surg Res. 2007;142:45–52. doi: 10.1016/j.jss.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 22.Moon KH, Kim BJ, Song BJ. Inhibition of mitochondrial aldehyde dehydrogenase by nitric oxide-mediated S-nitrosylation. FEBS Lett. 2005;579:6115–6120. doi: 10.1016/j.febslet.2005.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 24.Kim N, Lee Y, Kim H, Joo H, Youm JB, Park WS, Warda M, et al. Potential biomarkers for ischemic heart damage identified in mitochondrial proteins by comparative proteomics. Proteomics. 2006;6:1237–1249. doi: 10.1002/pmic.200500291. [DOI] [PubMed] [Google Scholar]
- 25.Vascotto C, Cesaratto L, D’Ambrosio C, Scaloni A, Avellini C, Paron I, Baccarani U, et al. Proteomic analysis of liver tissues subjected to early ischemia/reperfusion injury during human orthotopic liver transplantation. Proteomics. 2006;6:3455–3465. doi: 10.1002/pmic.200500770. [DOI] [PubMed] [Google Scholar]
- 26.Jeon BR, Yeom DH, Lee SM. Protective effect of allopurinol on hepatic energy metabolism in ischemic and reperfused rat liver. Shock. 2001;15:112–117. doi: 10.1097/00024382-200115020-00006. [DOI] [PubMed] [Google Scholar]
- 27.Grattagliano I, Vendemiale G, Lauterburg BH. Reperfusion injury of the liver: role of mitochondria and protection by glutathione ester. J Surg Res. 1999;86:2–8. doi: 10.1006/jsre.1999.5620. [DOI] [PubMed] [Google Scholar]
- 28.Okatani Y, Wakatsuki A, Enzan H, Miyahara Y. Edaravone protects against ischemia/reperfusion-induced oxidative damage to mitochondria in rat liver. Eur J Pharmacol. 2003;465:163–170. doi: 10.1016/s0014-2999(03)01463-8. [DOI] [PubMed] [Google Scholar]
- 29.Suh SK, Hood BL, Kim BJ, Conrads TP, Veenstra TD, Song BJ. Identification of oxidized mitochondrial proteins in alcohol-exposed human hepatoma cells and mouse liver. Proteomics. 2004;4:3401–3412. doi: 10.1002/pmic.200400971. [DOI] [PubMed] [Google Scholar]
- 30.Kim BJ, Hood BL, Aragon RA, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics. 2006;6:1250–1260. doi: 10.1002/pmic.200500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfeiffer S, Schrammel A, Koesling D, Schmidt K, Mayer B. Molecular actions of a Mn(III)Porphyrin superoxide dismutase mimetic and peroxynitrite scavenger: reaction with nitric oxide and direct inhibition of NO synthase and soluble guanylyl cyclase. Mol Pharmacol. 1998;53:795–800. doi: 10.1124/mol.53.4.795. [DOI] [PubMed] [Google Scholar]
- 32.Ferrer-Sueta G, Batinic-Haberle I, Spasojevic I, Fridovich I, Radi R. Catalytic scavenging of peroxynitrite by isomeric Mn(III) N-methylpyridylporphyrins in the presence of reductants. Chem Res Toxicol. 1999;12:442–449. doi: 10.1021/tx980245d. [DOI] [PubMed] [Google Scholar]
- 33.Batkai S, Osei-Hyiaman D, Pan H, El-Assal O, Rajesh M, Mukhopadhyay P, Hong F, et al. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. Faseb J. 2007;21:1788–1800. doi: 10.1096/fj.06-7451com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajesh M, Pan H, Mukhopadhyay P, Batkai S, Osei-Hyiaman D, Hasko G, Liaudet L, et al. Pivotal Advance: Cannabinoid-2 receptor agonist HU-308 protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress, inflammatory response, and apoptosis. J Leukoc Biol. 2007 doi: 10.1189/jlb.0307180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardoso SM, Pereira C, Oliveira R. Mitochondrial function is differentially affected upon oxidative stress. Free Radic Biol Med. 1999;26:3–13. doi: 10.1016/s0891-5849(98)00205-6. [DOI] [PubMed] [Google Scholar]
- 36.Lee VG, Johnson ML, Baust J, Laubach VE, Watkins SC, Billiar TR. The roles of iNOS in liver ischemia-reperfusion injury. Shock. 2001;16:355–360. doi: 10.1097/00024382-200116050-00006. [DOI] [PubMed] [Google Scholar]
- 37.Hines IN, Kawachi S, Harada H, Pavlick KP, Hoffman JM, Bharwani S, Wolf RE, et al. Role of nitric oxide in liver ischemia and reperfusion injury. Mol Cell Biochem. 2002;234-235:229–237. [PubMed] [Google Scholar]
- 38.Billiar TR. Nitric oxide. Novel biology with clinical relevance. Ann Surg. 1995;221:339–349. doi: 10.1097/00000658-199504000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harbrecht BG, Billiar TR. The role of nitric oxide in Kupffer cell-hepatocyte interactions. Shock. 1995;3:79–87. [PubMed] [Google Scholar]
- 40.Moon KH, Abdelmegeed MA, Song BJ. Inactivation of cytosolic aldehyde dehydrogenase via S-nitrosylation in ethanol-exposed rat liver. FEBS Lett. 2007;581:3967–3972. doi: 10.1016/j.febslet.2007.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng J, Li D. Expression and purification of his-tagged rat mitochondrial 3-ketoacyl-CoA thiolase wild type and His355 mutant proteins. Prot Expr Purif. 2004;35:320–326. doi: 10.1016/j.pep.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 42.Hartley DP, Ruth JA, Petersen DR. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Arch Biochem Biophys. 1995;316:197–205. doi: 10.1006/abbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- 43.Tompkins AJ, Burwell LS, Digerness SB, Zaragoza C, Holman WL, Brookes PS. Mitochondrial dysfunction in cardiac ischemia-reperfusion injury: ROS from complex I, without inhibition. Biochim Biophys Acta. 2006;1762:223–231. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Avellini C, Baccarani U, Trevisan G, Cesaratto L, Vascotto C, D’Aurizio F, Pandolfi M, Adani GL, Tell G. Redox proteomics and immunohistology to study molecular events during ischemia-reperfusion in human liver. Transplant Proc. 2007;39:1755–1760. doi: 10.1016/j.transproceed.2007.05.082. [DOI] [PubMed] [Google Scholar]
- 45.Emadali A, Muscatelli-Groux B, Delom F, Jenna S, Boismenu D, Sacks DB, Metrakos PP, Chevet E. Proteomic analysis of ischemia-reperfusion injury upon human liver transplantation reveals the protective role of IQGAP1. Mol Cell Proteomics. 2006;5:1300–1313. doi: 10.1074/mcp.M500393-MCP200. [DOI] [PubMed] [Google Scholar]
- 46.White MY, Tchen AS, McCarron HC, Hambly BD, Jeremy RW, Cordwell SJ. Proteomics of ischemia and reperfusion injuries in rabbit myocardium with and without intervention by an oxygen-free radical scavenger. Proteomics. 2006;6:6221–6233. doi: 10.1002/pmic.200600219. [DOI] [PubMed] [Google Scholar]
- 47.Yang SJ, Cho EH, Choi MM, Lee HJ, Huh JW, Choi SY, Cho SW. Critical role of the cysteine 323 residue in the catalytic activity of human glutamate dehydrogenase isozymes. Mol Cells. 2005;19:97–103. [PubMed] [Google Scholar]
- 48.Widmer R, Kaiser B, Engels M, Jung T, Grune T. Hyperammonemia causes protein oxidation and enhanced proteasomal activity in response to mitochondria-mediated oxidative stress in rat primary astrocytes. Arch Biochem Biophys. 2007;464:1–11. doi: 10.1016/j.abb.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 49.Venugopal SK, Chen J, Zhang Y, Clemens D, Follenzi A, Zern MA.Role of MAP kinase phosphatase-1 in sustained activation of C-JUN N-terminal kinase (JNK) during ethanol-induced apoptosis in hepatocyte-like VL-17A cells J Biol Chem 2007Sep 11:[Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 50.Imber CJ, Peter SD, Handa A, Friend PJ. Hepatic steatosis and its relationship to transplantation. Liver Transpl. 2002;8:415–423. doi: 10.1053/jlts.2002.32275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.