

Abstract
We have studied the Sphingosine 1-phosphate (S1P) receptor system to better understand why certain molecular targets within a closely related family are much more tractable when identifying compelling chemical leads. Five medically important G protein-coupled receptors for S1P regulate heart rate, coronary artery caliber, endothelial barrier integrity, and lymphocyte trafficking. Selective S1P receptor agonist probes would be of great utility to study receptor subtype-specific function. Through systematic screening of the same libraries, we identified novel selective agonists chemotypes for each of the S1P1 and S1P3 receptors. uHTS for S1P1 was more effective than for S1P3, with many selective, low nanomolar hits of proven mechanism emerging for. Receptor structure modeling and ligand docking reveal differences between the receptor binding pockets, which are the basis for sub-type selectivity. Novel selective agonists interact primarily in the hydrophobic pocket of the receptor in the absence of head-group interactions. Chemistry-space and shape-based analysis of the screening libraries in combination with the binding models explain the observed differential hit rates and enhanced efficiency for lead discovery for S1P1 vs. S1P3 in this closely related receptor family.
Sphingosine 1-phosphate (1, supporting material Figure S1) mediates a wide variety of physiological responses, including heart rate1, 2, coronary artery caliber, endothelial integrity, and lymphocyte recirculation1,3,4,5 through high affinity interactions with five members of the endothelial differentiation gene (EDG) family of plasma membrane-localized GPCRs – the sphingosine lipid receptors S1P1-5.6,7,8 Inhibition of lymphocyte recirculation by nonselective S1P receptor agonist FTY720 (2, Figure S1) produces clinical immunosuppression in multiple sclerosis. This prodrug, once phosphorylated by sphingosine kinase 2, acts as a potent agonist at S1P1, S1P3, S1P4, and S1P5 9. S1P1 is associated with immuno-suppression while other subtypes contribute to dose-limiting mechanism-based bradycardia and broncho-constriction. Understanding the contribution of individual receptors has been limited by the unavailability of selective agonists or antagonists for the 5 receptor subtypes. Selective probes of receptor function synergize with genetic models an allow validation of new therapeutic targets that are chemically tractable.10 S1P receptor subtype selective agonists and antagonists will be of broad utility in understanding cell functions in vitro, and vascular physiology in vivo. Success of the chemical approach for S1P1 suggests that developing selective tools for the resolution of function across this broad lipid receptor family is now possible.11, 12, 13. SEW2871 (3, Figure S1) is an in-vivo active heterocyclic selective S1P1 agonist probe originally identified by high throughput screening.1 SEW2871 unlike FTY720 recapitulates the action of S1P and demonstrates the essential role of the S1P1 receptor in lymphocyte trafficking.4 The development of pharmacophore models of receptor subtype-selective agonist and antagonist interactions would aid rational design and lead optimization, which is still severely limited by the lack of selective agonists for S1P2, S1P3, S1P4 and S1P5 sub-types. The S1P receptors belong to the largest subfamily A of GPCRs for which high resolution x-ray structures are available.14, 15 Homology models of the S1P1 and S1P4 receptors based on a theoretical rhodopsin model and mutagenesis studies have identified three key polar head-group interactions of S1P with its receptors.16, 17 The S1P1 and S1P3 receptors are the most closely related members of this family with about 50% identical and >70% similar amino acids.18 Key residues that constitute the hydrophobic binding pocket of the S1P1 and the S1P3 receptors have been identified.19
Here we report novel S1P1 and S1P3 agonist series identified by high throughout screening. Despite the high homology between these sphingolipid receptors, the confirmed hit rate was much higher for S1P1 than S1P3. Mapping of the hit series on to diversity space demonstrates that S1P1 ligands are better represented in the screening collections. Homology modeling of the S1P1 and S1P3 receptors and induced fit docking studies of the receptor binding interaction of the selective S1P1 and S1P3 agonist series reveal, in part, the basis for S1P1 versus S1P3 agonist specificity. SAR analysis and comparison of the binding modes of the new agonist series and the natural S1P ligand indicate their primary interaction with the hydrophobic binding pocket.
Results and Discussion
S1P1 and S1P3 uHTS and confirmation
In order to identify novel and specific entry points for agonists of the S1P1 and S1P3 receptors we developed CRE (S1P1) and NFAT (S1P3 – coupled via the promiscuous G-protein gα16) promoter β-lactamase reporter cell lines and used these to construct robust HTS assays.20 We examined the structural diversity of the Molecular Libraries Small Molecule Repository (MLSMR) with the Maybridge HitFinder (MBHF) collection using a chemistry space- and fingerprint-based method; the libraries are sufficiently diverse from one-another (see methods and supporting material Figure S2) and we therefore screened both libraries in an effort to identify unique lead series.
The MBHF and MLSMR libraries were screened in 384 and 1536 well formats, respectively. With the S1P1 cell line, two closely related screens were conducted, one for agonists, the second designed to identify potentiators of S1P1 agonists. Thus we used the data from both assays to identify compounds that reproducibly act as S1P1 agonists. Active wells in the primary screening assay were confirmed and counter-screened against the parental cell-line to eliminate compounds activating the reporter nonspecifically. The S1P3 actives were counter-screened against the 5HT1A/Gα16/NFAT-bla CHO-K1 cell line expressing the 5HT1A serotonin receptor subtype, because it is highly unlikely that a true S1P3 agonist compound would also have a high affinity for this receptor. Any S1P1 or S1P3 active compounds also active in the respective counterscreening assay were considered nonspecific and of no further interest. The majority of false positive compounds fall into one of two classes. First, blue fluorescent and green fluorescence quenching compounds modify the 410 nm excitation to 460 nm emission (blue) to 410 nm excitation/590 nm emission intensity (green) ratio of the CCF4 FRET dye such that they appear to be agonists. The second class of compounds activates the promoter for the reporter assay nonspecifically. In either case, activity in the counterscreening assays are used to exclude such hits. Screening statistics and confirmation numbers are summarized in the supporting material (Table S1). Dose response curves were measured using solid compound of confirmed structure for selected, higher potency compounds.
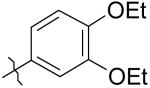
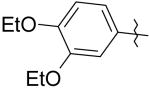
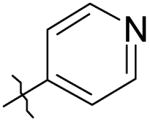
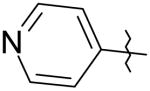
Initial structure activity results were independently hierarchically clustered using Leadscope keys.21 Chemical series and individual compounds with activity in the parental cell lines and reactive or chemically undesired motifs were removed. The statistical characterization of the most active structural series as identified by the initial dose-response confirmation assay for the two receptors are shown in the supporting information (Table S2). A series of 3,5-diaryl-oxadiazole (represented by structure 5, Table S2) is the largest and highest scoring analog series of S1P1 agonists with selectivity against S1P3. Although of lower potency compared to S1P1, a subset of the 3,5-diaryl-oxadiazoles also forms one of the most active clusters of S1P3 agonists; this is because of the overall much lower potency of identified S1P3 vs. S1P1 agonists.
To extend the SAR of the S1P1 agonists we built similarity clusters around the most active compounds (S1P1 EC50 < 1 μM) and removed redundancy by hierarchical clustering using Leadscope keys. This method also brings back compounds that were inactive in the primary screen.
Potencies (confirmed from fresh samples, vide infra), source and diversity (using a 2-dimensional BCUTS chemistry space) of identified S1P1 agonists with an EC50 of less than 1 μM are illustrated in Figure 1. Structures of various scaffolds are exemplified including the most privileged cluster of 3,5-diaryl-oxadiazoles; among them the most active and selective 3-(4-pyridyl)-5-(3,4-diethoxyphenyl)-1,2,4-oxadiazole (6) with an EC50 of 4 nM. SAR of the oxadiazole series in context of the receptor binding model is discussed below (Table 1). Representative S1P1 agonists of the confirmed series are also shown in the supporting material Table S3; additional actives identified from the Maybridge library are shown in Table S4.
Figure 1.

Representative S1P1 agonists with EC50 < 1 μM in a 2-dimensional chemistry space. EC50 values are shown for S1P1 / S1P3 (blue MLSMR, red MBHF, green SEW2871; size is scaled by S1P1 pEC50).
Table 1.
SAR of selected 3,5-diaryloxadiazoles in nM.
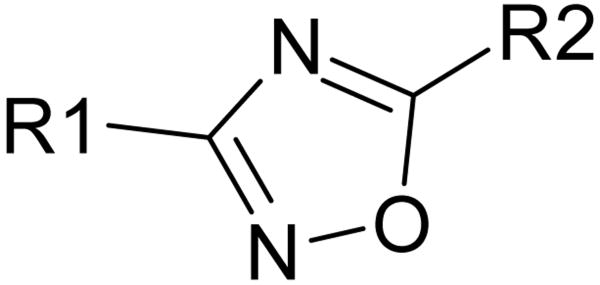 | ||||
|---|---|---|---|---|
| R1 | R2 | EC50 S1P1 |
EC50 S1P3 |
|
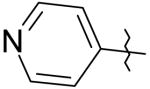
|
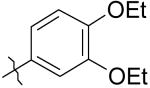
|
6 | 4.4 | 550 |
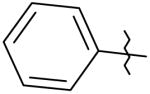
|
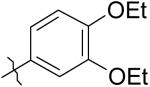
|
5 | 10 | 7,700 |
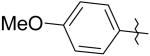
|
|
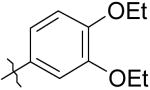
7 | 11 | >50K |
|
|
8 | 17 | >50K |
|
|
9 | 18 | 6,200 |
|
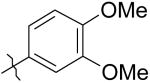
|
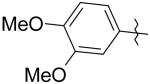
29 | 135 | >50K |
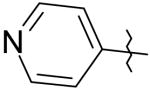
|
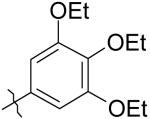
|
30 | >50K | >50K |
Potency, diversity and source of S1P3 agonists are illustrated in Figure 2. Interestingly the prevalence of confirmed hits for S1P3 identified from the Hitfinder library compared to the MLSMR library was the opposite of that seen for S1P1. Because of the generally higher diversity of this collection compared to the MLSMR library only singletons and no structural series were identified; their activities and confirmatory calcium flux assay results are provided in the supporting material (Table S5). The examples include novel nanomolar agonists with selectivity against S1P1 and confirmed mechanism of action. To our knowledge there are currently no selective S1P3 agonists. The most active oxadiazole S1P1 agonist identified from the MLSMR library also is among the most – although much less – active S1P3 agonists. From the MLSMR library we identified dicyclohexylamide 20 as a nanomolar selective S1P3 agonist (S1P3 EC50 = 0.35 μM, S1P1 EC50 >10 μM and inactive against S1P2, S1P4, S1P5 and LPA1 (Table S6). Its 5-methyl derivative, also a S1P3 screening hit, (not shown) is significantly less active (∼11 μM).
Figure 2.

S1P3 agonists with EC50 < 1 μM in a 2-dimensional chemistry space. EC50 values are shown for S1P3 / S1P1 (blue MLSMR, red MBHF, size is scaled by S1P3 pEC50).
Secondary Assay Confirmation and sub-type selectivity of Lead Series
Agonist mediated internalization and lysosomal degradation of the S1P1 receptor by AFD-R (4 supporting material, Figure S1), the native ligand S1P (1), and SEW2871 (3), occurs with a 3 log concentration differential (AFD-R≫S1P>SEW2871).22 Like other S1P agonists, stimulation with the novel agonist 6 (Figure 1, Table 1) causes S1P1-eGFP internalization, protein phosphorylation and polyubiquitination. Thus, 6 is a compound with full agonist potential, clearly differentiated in biological potential from the oft-utilized S1P1 probe of moderate potency, SEW2871. Agonist-stimulated receptor phosphorylation was studied using GFP immunoprecipitates in S1P1-GFP stable cells that were metabolically labeled with 32P prior to agonist incubation. The same immunoprecipitation protocol has been successfully used to report receptor degradation pathways.22 Accordingly, the EC50 for S1P mediated receptor phosphorylation (at 30 min of incubation) was determined to be 5 nM (not shown). Figure 3 shows that incubation of agonists for 30 min resulted in 32P incorporation into the immunoprecipitated receptor, with AFD-R being more efficacious (eliciting the greatest response) relative to S1P or the selective S1P1 agonist 6. S1P3 activation by the agonist 20 (Figures 2, 6) and a subsequent follow-up analog 28 (Figure 6) induced calcium flux to the same extent as S1P (Figure 3 panel C). Compounds 6 and 20 were also tested against S1P2, S1P4, S1P5 and LPA1 and show exquisite selectivity: oxadiazole 6 with an EC50 for S1P1 of 5nM and EC50 for S1P3 of 0.5 μM is inactivity against the other S1P receptors and LPA1; dicyclohexylamidie 20 with an EC50 for S1P3 of 350 nM is inactive for all other S1P receptors and LPA1. Thus we have identified biologically active compounds, specific for either receptor, from uHTS.
Figure 3.
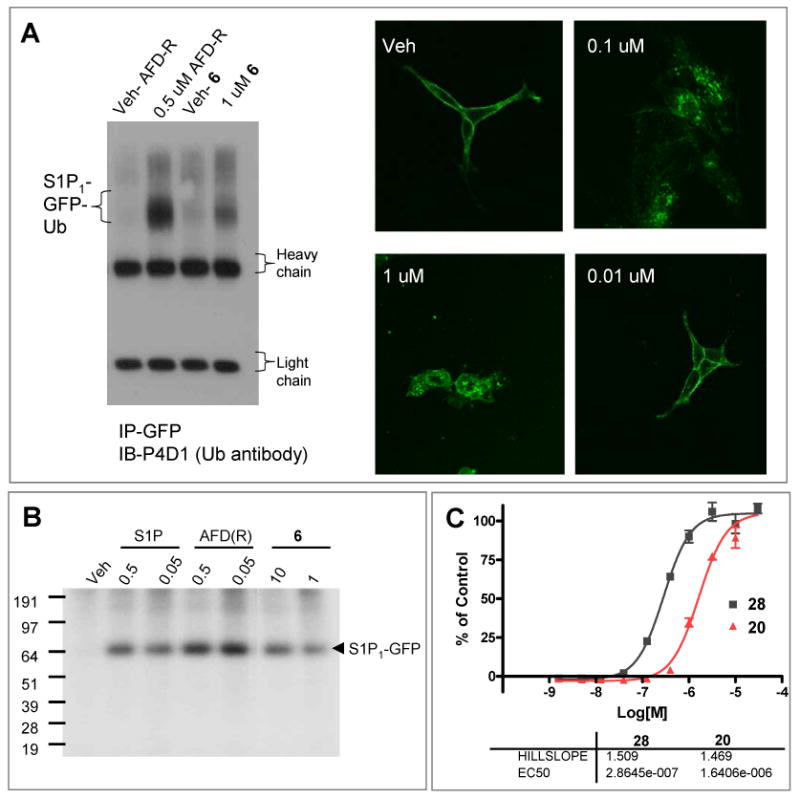
Biological activity of S1P1 and S1P3 agonists. A. S1P1-GFP internalization and ubiquitination by 6. B. Agonist induced 32P incorporation into immunoprecipitated S1P1-GFP. C. S1P3 agonist dose dependent activation of Calcium flux in S1P3/ga16 CHO cells by 20 and 28.
Figure 6.

Structure and activity of S1P3 selective dicyclohexylamides 20 and 28.
S1P1 and S1P3 Receptor Binding Models
The striking difference between the number and potency of confirmed agonists identified in the S1P1 and S1P3 campaigns led us to evaluate the receptor binding models. Among the sphingosine lipid receptors, S1P1 and S1P3 are the most closely related by sequence. Their agonist binding pockets, which consists of an upper polar region16, 17 and a lower hydrophobic pocket18, 19 are even more similar. Yet there is a remarkable difference in small molecule agonist recognition, as demonstrated by the different confirmed hit rates for the high throughput screening campaigns. The development of a structural hypothesis underlying these differences has been limited by the lack of experimental structures of any of the receptors and homology modeling can be a valuable approach to develop structural insight in these receptors.16-19 Our first step therefore was to develop two comparable models of the S1P1 and S1P3 receptors. The initial homology model of S1P1 was built based on a high-resolution crystal structure of chain A of Bovine Rhodopsin (PDB 1u19, 2.2 Å resolution)15 with retinal in the unactivated (cis) conformation, which was refined involving repositioning the second extracellular loop connecting TM4 and TM5, local and global minimizations and side chain optimization of the hydrophobic part of the pocket using induced fit docking (IFD)11 runs with S1P and FTY720-P as known ligands as described in the methods section. The final S1P1 model was then used as a template to build the corresponding S1P3 model. Although the receptor side chain positions may vary depending on the specific receptor-ligand complex our goal was to minimize modeling bias and develop structures that are exactly aligned and in which the binding pockets can therefore be directly compared. We also generated models based on a more recent structure of lumirhodopsin (PDB 2hpy, 2.8 Å resolution)23 – a photoactivated all-trans retinal intermediate of the activated receptor (see methods section). For all docking studies we used the optimized structures based on the higher resolution ‘dark-state’ rhodopsin, modeling a prospective snapshot of ligand binding prior to receptor activation.
Our models suggest one of the major differences of the binding pockets is determined by Leu276 in S1P1 vs. Phe263 in S1P3 as illustrated by the surface representation of both receptors with the S1P and FTY720-P agonists docked into the pocket (Figure 4). The Phe263 residue results in a contraction of the pocket between the lower hydrophobic pocket and the upper polar section by 1.5 to 1.8Å compared to the corresponding S1P1 Leu276 residue; this also results in a difference in binding pocket volume between 50 and 100Å3 (see methods section). Additional illustrations of the S1P1 and S1P3 receptor binding sites with docked S1P and FTY720-P and illustrations of their detailed binding interactions in the receptor models are provided in the supporting material (Figures S4 and S5). The difference in spatial constraint between the polar and hydrophobic sections of the binding pockets of S1P1 and S1P3 may explain the lower frequency and potency of S1P3 vs. S1P1 agonists (vide infra); presumably the S1P1 pocket may accommodate more rigid and perhaps larger ligands compared to S1P3.
Figure 4.
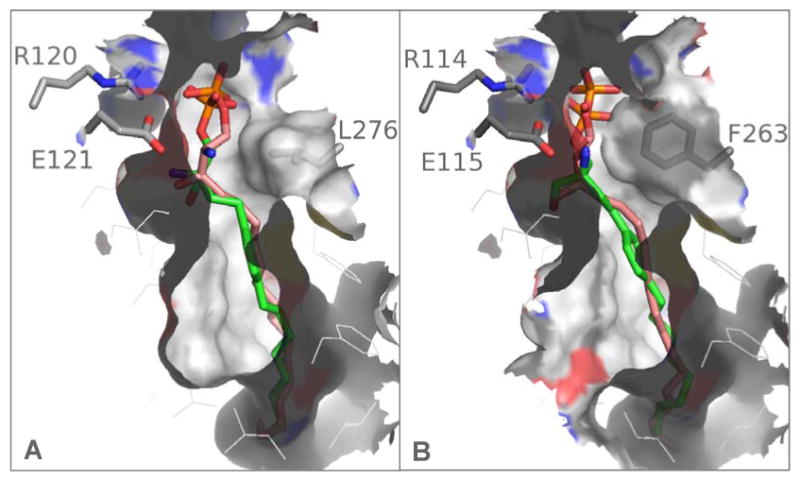
Comparison of the binding pocket of the S1P1 (A) and S1P3 (B) receptors with S1P and FTY720-P docked into each receptor. The main difference in the binding pocket derives from the S1P1 Leu276 and S1P3 Phe263 side chains; in S1P3 the binding pocket is narrowed by 1.5 to 2 Å compared to the same region in S1P1.
Compared to an available model of S1P124 the 7 transmembrane domains are overall well aligned with our models, but with some of the active site residues in a different orientation. The largest variation is in the position of the second extracellular loop which would interfere with S1P binding. Recently a study comparing models of all five S1P receptors in the context of known modulators including S1P, FTY720-P and SEW2871 was published.25 The described key residues involved in recognition of S1P and FTY720-P indicate that our models are largely in agreement although this study does not refer to the influence of Leu276 vs. Phe263. The main difference between S1P1 and S1P3 was described as the length of the binding pocket and illustrated in the docked conformations of S1P and FTY720-P; our models are different in that respect as a result of the modeling process. Our models seem in best agreement with the studies by Parent et al18 who also identify the Leu276 and Phe363 as a key determinant of the S1P1/3 selectivity. Structure models based on templates of only remote (< 20 %) homology such as ours and other reported S1P receptor structures clearly can be useful to suggest pharmacological trends, however their absolute accuracy remains unknown and they are therefore most effectively applied in the context of experimental data.
Binding hypothesis of selective agonists for S1P1 and S1P3
Examples of the 3,5-diaryl-oxadiazole series of selective S1P1 agonists are given in Table 1 to illustrate the influence of hydrophobic space and positioning of the two aryl moieties.
The four most active compounds were docked, aligned and minimized in the receptor site as described in the methods section (Figure 5). An overlay of the four structures in the receptor is shown in the supporting material (Figure S6).
Figure 5.
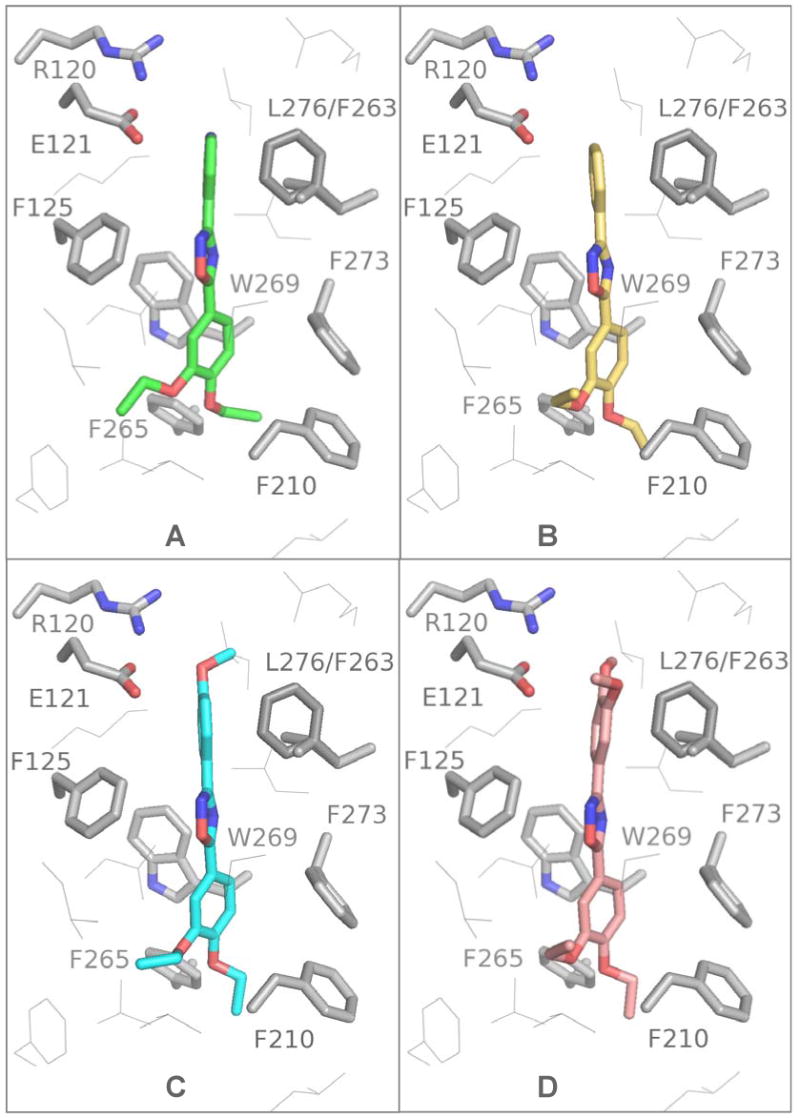
Oxadiazoles docked into the S1P1 receptor: 6 green (A), 5 yellow (B), 7 cyan (C), 8 copper (D). The S1P3 Phe263 residue interferes with these ligand poses explaining the observed selectivity.
In our models the diethoxyphenyl moieties reach down into the hydrophobic part of the pocket with the pyridyl or phenyl substituents reaching towards the upper more polar portion of the pocket between Glu121 and Leu276. In this orientation the top aryl moiety interferes with Phe263 of S1P3, which can rationalize the observed selectivity (Figure 5). Oxadiazoles with spatially more demanding substituents 4-methylphenyl and 3,4-dimethylphenyl are less potent against S1P1, but become completely inactive for S1P3. This spatially more constrained section of the S1P3 binding pocket may have a larger effect as the size of the interacting substituent increases. The higher activity of the pyridine-(6) vs. phenyl-substituted compound (5) may relate to the more polar environment of this part of the pocket, although the distance of the pyridine N and Arg120 NH (3.7 Å) seems too large for a direct hydrogen bond and an interaction with Glu121 is not expected at neutral pH unless a water molecule participates. In all ligands the aromatic ring systems are slightly out of plane and not in their optimal geometric (all planar) conformation; in particular the 3-substituent that reaches up towards the more polar region of the binding site. The most active compounds all share the hydrophobic 2,3-diethoxy-phenyl moiety. Introduction of an additional ethoxy substituent results in loss of activity (compare 6 and 30). This is explained given the binding model in Figure 4 which indicates interference of any additional substituent on the phenyl with Trp269 and Phe273 of TM6. A smaller group, 2,3-dimethoxy-phenyl vs. 2,3-diethoxy-phenyl (6 vs. 29) results in decreased activity (30 fold), probably due to less effective interaction with hydrophobic residues required for receptor activation. Reversing the orientation of the oxadiazole (i.e. swapping the 3 and 5 substituents as in 6 vs. 9) results in decreased activity for both receptors. A previous study reports activity of different 5-membered heterocycles against S1P1 S1P3 and S1P5.26 Although their compounds all include a carboxylic acid head group presumably interacting with one or more of the Arg side chains, the authors report a decease in S1P1 activity when the 3,5-1,2,4-oxadiazole is replaced for example against a 2,5-1,3,4-oxadiazole core in otherwise identical structures. In our homology model Cys200 can be oriented to form a hydrogen bond with the oxadiazole oxygen (S-O-distance ∼ 2.2 Å) – a potential explanation for this observed effect (not shown).
From the MLSMR library we identified the nanomolar S1P3 selective agonist 20 (Figure 6). Based on the hypothesis that the dicyclohexylamide would interact in the hydrophobic binding pocket of S1P3 we tested additional commercial analogs, one of which, 28, was found to be even more active (but less selective). Docking of these ligands confirms this possible binding mode (Figure 7). The propyl-isoxazole moiety of the relatively compact 20 warps underneath, slightly behind Phe263. Similarly, the furanyl- moiety of 28 avoids the contraction of the S1P3 binding pocket. The overlaid structures in the receptor are shown in the supporting material (Figure S7). Interestingly, the docked structures in Figure 7 do interfere with Leu276 of S1P1, which may explain their selectivity. A possible orientation of 28, which is only three times selective over S1P1, in the S1P1 receptor is illustrated in figure S9 in the supporting material (see methods section).
Figure 7.
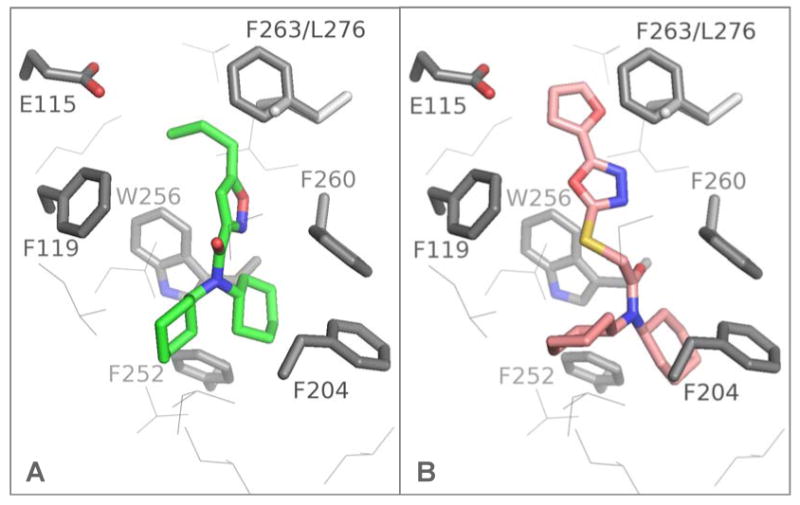
S1P3-selective dicyclohexylamides docked into the S1P3 receptor hydrophobic binding pocket: green 20 (A), copper 28 (B).
When comparing the poses of the selective diaryloxadiazole S1P1 and the above S1P3 agonists, the respective diethoxyphenyl and dicyclohexylamide moieties reaching into the hydrophobic pocket align reasonably with respect to shape. Our structural models and docking poses suggest that the moieties reaching towards the polar region at least in part determine selectivity. In case of S1P1, the relatively rigid diaryl-oxadiazoles occupy space between Glu121 and Leu276 with the plane of the upper 3-aryl moiety aligned between the TM3 and TM6 domains. The potent and selective S1P3 agonists are less spatially demanding in that region and more flexible to adopt a conformation in which this ‘upper’ ligand moiety can adapt to the spatial constraint of the pocket imposed by Phe263. In case of 28 the orientation of the plane of the furanyloxadiazole in S1P3 is almost perpendicular to the one of the 3-aryl plane of the oxadizoles in S1P1. In the pose of 28 in S1P1 these aryl moieties align parallel.
As described above, computer-generated models of the S1P1 and S1P3 receptor ligand binding pockets differ most strikingly with Leucine 276 of S1P1 replaced with Phenyalanine 263 of S1P318. Docking of the S1P3 specific compound 20, into the S1P1 model was at least partially blocked by Leucine 276 and generates structures with low docking scores. In order to evaluate the validity of the S1P1 modeling we generated the S1P1 L276F mutation and measured ERK phosphorylation in response to S1P and the S1P3 specific compound 20. Indeed, 20, while inactive at 30uM on wild type S1P1 elicits a striking pERK response (EC50 1.4uM) in the Leu276Phe mutant (Figure S12). Thus the L276F mutation results in a gain of function with the S1P3 selective compound 20.
Ligand shape confers receptor specificity. Our results suggest the more linear and rigid aromatic diaryloxadiazole ligands fit the S1P1 pocket while the more compact flexible diyclohexylamide derivatives prefer S1P3. Mutations of residues lining the ligand binding pocket to smaller side chains frequently lead to loss of receptor activation by S1P.19 The S1P1 and by extension the highly related S1P3 receptors are only activated by ligands of sufficient volume to interact in the hydrophobic pocket of the receptors. Our models and docking studies suggest that in contrast to any of the known S1P1 agonists, both of our selective S1P receptor agonist series have no obvious interaction with any of the Glu (E121) or Arg (R120; R292) residues that form their well established polar binding sites and, in this respect, constitute ligands with a novel binding mode. This hypothesis is now experimentally confirmed by mutation studies and further analogs of 6 (manuscript in preparation). Our S1P receptor agonists interact in the lower hydrophobic binding pocket while their selectivity can be rationalized by the receptor specific ligand conformations matching the spatial requirements towards the upper section of the binding pocket.
Conclusion and potential for discovery of selective agonists
In contrast to S1P1 fewer and less potent agonists were identified for S1P3. It can be qualitatively expected that a binding pocket that is spatially constrained in one area may have fewer potential ligands, which – on average – may interact with lower potency, because conformational flexibility and ligand size relate to binding energy. However the relative confirmed hit rates are a function of the screening library composition. To quantify this effect we compared the shape of the docked conformations of the most active and selective agonists for S1P1 and S1P3 respectively against the entire MLSMR and MBHF screening library. The results are illustrated in the supporting material (Figure S10) and confirm that there are many more shape similar compounds for the S1P1 diaryloxadizole agonist 6 compared to the dicyclohexylamide 20. This reflects the composition of typical screening libraries relative to the identified selective S1P1 and S1P3 agonists 6 and 20 in their preferred docked conformations. An analysis of the density of the chemistry space that characterizes the MLSMR and MBHF libraries shows that the majority of the identified S1P1 agonists fall into higher occupied regions compared to the S1P3 agonists (supporting material Figure S11). This is a receptor-structure independent perspective also suggesting screening library bias towards S1P1 vs. S1P3.
Summary
Selective S1P agonist probes can be important tools to causally-associate biological events with specific receptor-derived signals. We have identified, through systematic high throughput and directed follow-up screening, a number of novel chemotypes of S1P1 and S1P3 selective agonist. Much greater numbers and more potent S1P1 agonists were identified compared to S1P3. We report, to our knowledge, the first S1P3 agonist with selectivity against S1P1. Through receptor modeling and ligand docking studies we have characterized the difference of the receptor pockets and provide insights into the specific interactions that determine selectivity. These studies suggest that the diaryloxadiazole S1P1 and dicyclohexylamide S1P3 selective agonist interact primarily the hydrophobic binding pocket and, in contrast to previously reported agonists, that there are no obvious interactions with the Glu and Arg residues characterizing the polar region of the binding site. Thus, our receptor structure models can aid in rational design and further optimization of selective ligands. The full agonist potential of the best compounds was characterized biologically by showing receptor internalization, protein phosphorylation, polyubiquitinylation and calcium flux for S1P1 and S1P3 respectively. Analysis of the chemistry space characterizing the MLSMR and MBHF screening libraries and shape comparison of these libraries against the docked conformations of the selective agonists rationalizes the observed greater hit rates and on average higher potency of the S1P1 vs. S1P3 agonists.
Methods
Materials
CHO-K1 CRE-bla and CHO-K1 NFAT-bla, and 5HT1A/ga16 CHO K1 NFAT-bla cells lines and pcDNA3.1(+)-hygromycin, lipofectamine 2000, LiveBlazer dye and all media components except serum were purchased from Invitrogen. pcDNA3-S1P1, pcDNA3-S1P2 and pcDNA3-S1P3 were obtained from the Missouri S&T cDNA Resource Center (www.cdna.org). The ga16 gene was subcloned from pcDNAI into pcDNA3.1(+)/hygromycin. Forskolin, probenecid and fatty acid free BSA were purchased from Sigma. Fetal calf serum (FCS), Bovine Growth Serum (BGS) and charcol dextran stripped serum (CDS) was obtained from Hyclone. CA4 dye was purchased from Molecular Probes. BAS3430808 (28) was purchased from Asinex.
Cell Line Generation
CHO K1 S1P1/CRE-bla, CHO K1 S1P2/CRE-bla and CHO-K1 S1P3-ga16/NFAT-bla cell lines were generated by standard methods20: Cells were cultured in growth media (DMEM, 10% heat inactivated bovine growth serum, 0.1 mM NEAA, 1 mM Sodium Pyruvate, 25 mM HEPES, 5 mM L-Glutamine, 2 mg/ml Geneticin, and 1× penicillin-streptomycin. The media for S1P3-ga16/NFAT-bla cell line also includes 200 ug/ml hygromycin.
S1P1 CRE-bla CHO Reporter Assay
Cells were suspended (0.3 million/mL 384 well-format or 1.25 million/mL 1536 well format) in phenol red free Dulbecco's Modified Eagle's Media containing 0.5% charcoal/dextran treated fetal bovine serum, 0.1 mM NEAA, 1 mM Sodium Pyruvate, 25mM HEPES, and 5mM L-Glutamine. The assay began by dispensing 10 uL (4 uL for 1536 well format) of cell suspension to each well of the assay plates. The cells were then allowed to incubate in the plates for 20 hrs at 37 C in 5% CO2. Then test compound or controls were added by pintool, followed by forskolin (2 uM 384 well format, 4 uM 1536 well format) SEW2871 was added to 1 uM to positive control wells. Plates were then incubated at 37 C in 5% CO2 for 4 hrs. After the incubation, GeneBLAzer fluorescent substrate mixture, containing 15 mM probenecid, was added. After 2 hours of incubation at room temperature in dark, plates were read.on an Envision (384 well format), or a ViewLux plate-reader (1536 well format).
CRE-bla Reporter Counterscreen Assays
Cells were suspended (0.3 million/mL 384 well-format or 1.25 million/mL 1536 well format) in phenol red free Dulbecco's Modified Eagle's Media containing 0.5% charcoal/dextran treated fetal bovine serum, 0.1 mM NEAA, 1 mM Sodium Pyruvate, 25mM HEPES, and 5mM L-Glutamine. The assay began by dispensing 10 uL (4 uL for 1536 well format) of cell suspension to each well of the assay plates. The cells were then allowed to incubate in the plates for 20 hrs at 37 C in 5% CO2. Then test compound or controls were added by pintool, followed by forskolin (2 uM 384 well format, 4 uM 1536 well format) SEW2871 was added to 1 uM to positive control wells. Plates were then incubated at 37 C in 5% CO2 for 4 hrs. After the incubation, GeneBLAzer fluorescent substrate mixture, containing 15 mM probenecid, was added. After 2 hours of incubation at room temperature in dark, plates were read.on an Envision (384 well format), or a ViewLux plate-reader (1536 well format).
S1P3-gα16-NFAT-bla Reporter Assay
Cells were suspended to a concentration of 1 million/milliliter in phenol red free Dulbecco's Modified Eagle's Media containing 0.5% charcoal/dextran treated fetal bovine serum, 0.1 mM NEAA, 1 mM Sodium Pyruvate, 25 mM HEPES, and 5 mM L-Glutamine. The assay began by dispensing 5 uL of cell suspension to each test well of a 1536 well plate (10 uL for 384 well format). The plated cells incubated overnight at 37 deg °C in 5% CO2. Test compound or controls were added by pintool. The S1P positive control was also added to the appropriate control wells to a final concentration of 1 micromolar. Plates were then incubated at 37 deg C in 5% CO2 for 4 hrs. After the incubation, GeneBLAzer fluorescent substrate mixture, containing 15 mM probenecid, was added. After 2 hours of incubation at room temperature in dark, plates were read on an Envision (384 well format), or a ViewLux plate-reader (1536 well format).
5HT1A/Gα16-NFAT-bla Reporter Counterscreen Assay
5HT1A/ga16 CHO K1 NFAT-bla cells were suspended to a concentration of 1 million/milliliter in phenol red free Dulbecco's Modified Eagle's Media containing 2% charcoal/dextran treated fetal bovine serum, 0.1 mM NEAA, 1 mM Sodium Pyruvate, 25 mM HEPES, and 5 mM L-Glutamine. The assay began by dispensing 10 uL of cell suspension to each test well of a 384 well plate. The plated cells incubated overnight at 37 deg °C in 5% CO2. Test compound or controls were added by pintool. The 5-carboxamidotryptamine (5-CT) positive control was added to the positive control wells to a final concentration of 125 micromolar. Plates were then incubated at 37 deg C in 5% CO2 for 4 hrs. After the incubation, GeneBLAzer fluorescent substrate mixture, containing 15 mM probenecid, was added. After 2 hours of incubation at room temperature in dark, plates were read on an Envision plate-reader
Dose Response Curve fitting
For each compound, percent activation was plotted against compound concentration and fitted to a four parameter equation describing a sigmoidal concentration-response curve with adjustable baseline using Assay Explorer software by MDL. The reported EC50 values are generated from fitted curves by solving for x-intercept at the 50% activity level of Y-intercept. In cases where the highest concentration tested (45 micromolar) did not result in > 50% inhibition or where no curve fit was achieved, the EC50 was determined manually depending on the observed inhibition at the individual concentrations. Compounds with EC50 values of greater than 10 micromolar were considered inactive, compounds with EC50 equal to less than 10 micromolar are considered active.
PubChem unique Assay Identifiers (AIDs) associated with uHTS assay results
Data associated with the reported results have been deposited into PubChem with the following AIDs: 449 and 1044: S1P1 primary agonist and agonism potentiator assay for MLSMR and MBHF library respectively; 466, 467, 468: S1P1 concentration response data for potentiator, parental cell line, agonist respectively; 373: S1P3 primary agonist assay, 439, 1192 S1P3 concentration-response and purchased analogs.
S1P1 Secondary Assays
S1P1-GFP internalization and Ubiquitination western blots were performed as previously described.22
Intact cell S1P1 phosphorylation
Phosphorylation experiments were carried out in monolayers of confluent stable 293-S1P1-GFP cells grown in 6-well plates. Cells were washed twice with serum-free, phosphate-free DMEM, and incubated in the same medium for 2 h prior to labeling with 32P-orthophosphate (80 mCi/ml, 2 h). Following stimulation with the indicated agonists or vehicle control for 30 min at 37°C, the monolayers were washed twice with ice-cold PBS and solubilized in RIPA buffer containing a cocktail of protease inhibitors (Roche) plus 1 mM EDTA, 0.5 M glycerolphosphate, 1 mM phenylmethylsulfonyl fluoride, 1μM Na3VO4 and 1 nM okadaic acid. After centrifugation (16,000 × g for 15 min), the supernatants were first equalized for protein content using the BCA method (Pierce), and subsequently immunoprecipitated with an anti-GFP antibody as reported (ref).22 Following separation of the immunoprecipitated receptor by SDS-PAGE (4-12% gels), the gels were autoradiographed for 24 h at -80 °C, and the extent of ligand-stimulated S1P1-GFP phosphorylation was compared to that of vehicle alone.
S1P3 Calcium Flux Assay
S1P3-gal16-NFAT-bla cells were plated in 25 uL 0.5% CDS media at 12,500 cells/well in 384 well Corning black, clear bottom tissue culture treated plates. After brief centrifugation, CA4 dye, 25 uL, was added and the cells incubated at 37 °C for 1 hour. The calcium flux experiments were collected at 37 °C on a Molecular Devices Flexstation. Fluorescence intensity (485 nm excitation/ 510 nm emission) was collected every 2.5 seconds for 3 minutes. After a 30 second baseline collection compound or S1P was added in quadruplicate. Calcium flux was evaluated as area under the curve for the first 60 seconds following agonist addition. Values are normalized to 1 uM S1P as 100 % of control. The resulting 10 point dose response curves were fitted to a sigmoidal dose response with variable slope in Graph Pad Prizm.
S1P2 CRE-bla, S1P4 TANGO and S1P5 TANGO Reporter Selectivity Assays
CHO K1 S1P2-CRE-bla cells were dispensed (10 μl of 1 million cells/ mL) to 384-well assay plates and incubated overnight at 37 °C in a humidified incubator. Test compounds, S1P or vehicle only were dispensed to all wells. Plates were then incubated at 37 °C in 5 percent CO2 for 4 hours. After the incubation, 2.2 μl/well of the GeneBLAzer fluorescent substrate mixture (prepared according to the manufacturer's protocol and containing 15 mM probenecid) was added. After 2 hours of incubation at room temperature in the dark, plates were read on the EnVision plate reader (PerkinElmer Lifesciences, Turku, Finland) at an excitation wavelength 405 nm and emission wavelengths of 590 nm and 460 nm. The S1P4 and S1P5 TANGO stable cell lines were obtained from Invitrogen and assayed according Invitrogen's protocol with 1 uM S1P as the positive control
S1P1 and S1P1 L276F Erk Phosphorylation Assay
The L276F mutation was generated using the GeneTailor kit (Invitrogen) with pcDNA3-S1P1 as the DNA template and the following oligonucleotides: S1P1 L276F_F 5′-GCACCGCTCTTCATCCTGTTCCTGCTGGATGTG-3′ and S1P1 L276F_R (5′-CAGGATGAAGAGCGGTGCCCAGCAGGCGAT. The mutation was confirmed by sequencing. CHO-K1 cells were transfected overnight (Fugene, Roche) in 70% confluent 10 cm dishes with wild type S1P1 or L276 mutant S1P1 DNA. Each 10 cm dish harvested and seeded into 3 6-well plates in 10%FCS DMEM supplemented with 0.1% NEAA and sodium pyruvate and incubated overnight. The media was changed to DMEM for 5 hours and each well was treated with S1P or compound for 5 minutes. The wells were washed with ice-cold PBS, harvested by scraping the well in 1 ml lysis buffer (Cell Signaling). The samples were centrifuged, diluted 1:1 with sample diluent and 100 μl added to wells of phospho-ERK1/2 ELISA plate, incubated overnight and washed. Wells were developed as described in the product manual. OD450 values for each transfection were normalized to 100 % of high control (1 μM S1P) and 0% of control (vehicle treated wells).
S1P1 and S1P3 receptor structure models
The initial model for the S1P1 receptor was generated using the Structfast algorithm,27 a recent profile-profile alignment algorithm using convergent island statistics28 to determine local alignment score significance. This algorithm is implemented in the TIP software system29, 30. The high-resolution crystal structure of chain A of Bovine Rhodopsin (PDB 1u19, 2.2 Å resolution)15 with retinal in the unactivated (cis) conformation was used as template aligning residues 41 to 351 of S1P1 to residues 34 to 349 of the rhodopsin template. This initial homology model was then optimized by repositioning the second extracellular loop connecting the TM4 and TM5 domains, because of the absence of the template disulfide bridge connecting this loop with the TM3 domain and because this loop interferes with residues Arg120, Glu121, Arg292 in S1P1, which have been shown to be essential for binding of the natural S1P ligand.16 In the resulting structure the transmembrane helices TM4, TM5 and TM7 and the connecting loop between TM4 and TM5 were independently optimized followed by a global minimization of the structure to 0.05 kcal/mol·Å root mean square gradient using the OPLS (Optimized Potential for Liquid Simulations) force field implemented in Schrodinger Macromodel.31 During the global minimization the α-helix backbone atoms were frozen +/-1 Å to avoid local movements of the helices that was observed otherwise; required rearrangement of the transmembrane helices associated with receptor activation make it plausible that the unactivated receptor is not in a globally minimized conformation. For comparison we generated a model based on a more recent structure of lumirhodopsin (PDB 2hpy, 2.8 Å resolution)23 – a photoactivated all-trans retinal intermediate of the activated receptor. The model differs slightly in the middle of helix three and in the loop connecting TM4 and TM5; the side-chain minimized models also differ in the orientation of the Glu121 and Arg120 residues indicating that local structural movements in these areas may be associated with receptor activation. Previous S1P-receptor homology models were based on lower resolution or modeled rhodopsin structures.18, 19
The final S1P1 docking model was obtained by optimizing the binding pocket side chains by induced fit docking (IFD)11 with S1P and FTY720-P (S-isomer) as ligands and using the default settings (other than indicated below). The IFD protocol is implemented in Schrodinger Maestro and includes a constraint receptor minimization step followed by initial flexible Glide docking of the ligands using a softened potential (reduced Coulomb van der Waals cutoff and scaling van der Waals radii to 0.5) to generate a diverse ensemble of potential poses.
For each pose the nearby receptor structure (using a cut-off of 4 Å) is refined using Prime (prediction of side chain orientation followed minimization in presence of the docked ligand). Each ligand is then re-docked (using Glide) into its corresponding optimized low-energy receptor structure and ranked by GlideScore. Based on the established binding mode two of the three Arg120, Glu121, Arg292 polar side chains know to interact with S1P were constraint as required hydrogen bonds during the IFD runs.16 The resulting receptor-ligand complexes were evaluated based on docking score, emodel energy, and Prime energies, and visually to select the best and most abundant side chain orientations among the IFD results.
The S1P3 receptor model was then build based on the above S1P1 structure template using the Prime comparative modeling module; both receptor structures were capped after their terminal secondary structure residues aligning S1P1 Glu42-Met326 to S1P3 Ser36-Val313 with identical positions of the backbone atoms except a gap of 7 amino acids of S1P3 in the loop connecting TM5 and TM6.
Agonist docking
Docking was performed in Schrodinger Glide32 using the above receptor structures of S1P1 and S1P3. A starting 3D conformation for the ligands was generated using the ChemAxon13 conformer plugin33, 34, ligands (S1P, FTY720-P) were ionized at pH 7.4 using the pKa plugin.35. In case of S1P and FTY720-P (S-isomer) the Arg120 and Glu121 in S1P1 and Arg114 and Glu115 in S1P3 were constraint as hydrogen bond donors and acceptors respectively. Up to 10 best poses per ligand were retained and analyzed by docking score, emodel energy, and visually. The 1,3-diaryl-1,2,4-oxadiazoles 5–8 were first docked flexibly into the S1P1 receptor using Glide XP to better account for the hydrophobic interaction in the bottom part of the pocket. Poses that interacted with the repositioned loop (compare model development above) were removed and the remaining poses evaluated by glide score, emodel energy and visually. The best pose was selected and all structures were aligned to this pose followed by docking optimization of the so prepositioned ligands. Structures were ranked by glide score and emodel energy and the most comparable poses are reported to visualize the binding mode in the S1P1 receptor (figure 5). These poses when scored against the S1P3 receptor grid have Coulomb-vdW energies of 10,000 due to interaction with Phe263 (docking score 0). When docking the oxadiazole ligands flexibly into the S1P3 receptor grid resulted we obtained poses that appear less favorable after evaluating docking energies and visual inspection; for example the diethoxyphenyl moiety points towards the polar region of the pocket and does not interact in the hydrophobic region or the ligands are structurally distorted. A pose of oxadiazole 6 that was obtained is provided in the supporting material (figure S8); the orientation of the ligand is rotated around the axis of the three aromatic rings (to avoid interaction with the Phe263 residue) and the pyridyl aromatic plane appears almost perpendicular to the oxadiazole; although the diethoxyphenyl moiety does not align well with the corresponding S1P1 orientation one of the ethoxy residues still interacts in the hydrophobic region. The S1P3-selective dicyclohexylamide structures 20 and 28 were docked flexibly into the S1P3 receptor using Glide XP. The poses in which the dicycohexylamide moieties align best were selected and – with the cyclohexy substituent in an equatorial position – were minimized using the OLPS force field followed by docking optimization in the receptor grid. The best poses are reported (figure 7). When evaluating these poses in the S1P1 receptor grid they are invalid (by Coulomb-vdW, docking score 0). When docking flexibly into the S1P1 receptor all obtained poses have different orientations and in none of the poses did the dicyclohexylamide moieties of the two ligands align in the hydrophobic part of the binding pocket (in particular the cyclohexyl moieties of 20 would not interact in the hydrophobic pocket). When optimizing the S1P3 pose of 28 in the S1P1 receptor the ligand is rotated so that the aromatic (furyl-oxadiazole) plane orients between the TM6 and TM3 domains, parallel to the corresponding aryl moiety of the S1P1 diaryloxadizole ligands (supporting material figure S9).
All visualizations were prepared in PyMOL.36
Receptor binding site volumes were calculated in MOE37; the receptor site volumes differ by ∼50 to 100 Å3 when measures inside a box around the diaryloxadizole compounds (1780 vs 1727 Å3 to 1,953 vs. 1,864 Å3); the S1P3 pocket is contracted by 1.5 to 1.8 Å compared to S1P1 (as measured point to point between S1P1 Leu276-Glu121 and S1P3 Phe263-Glu115.
Library comparison
The MLSMR (∼60,000 at the time of screening) and Maybridge HitFinder™ (MBHF, 16,000 diverse structures) libraries were compared using a cell-based and a fingerprint-based approach. Extended connectivity fingerprints (ECFP6) and Tanimoto similarities were calculated in Scitegic Pipeline Pilot.38 Neighborhood statistics show that the majority of each library is less similar than 0.4 to the other (Figure S2, supporting material). An optimized 6-dimensional BCUTS39 chemistry space for the combined libraries using the standard 3D hydrogen suppressed descriptors was generated using Diverse Solutions.40 Diversity analysis using 6 bins per dimension (6^6 cells) shows an overlap of 0.68 and 0.52 respectively and a cell-based Tanimoto index of 0.42. To illustrate global library diversity (Figure S3, supporting material) and the diversity of hits for S1P1 and S1P3 (Figures 1, 2) an optimized 2-dimensional chemistry space was used. The chemistry space density / occupancy was calculated as the sum of structures falling into each cell (supporting material Figure S10).
Cluster and similarity analysis of active series
508 compounds with EC50 results for S1P1 were hierarchically clustered using Leadscope21 keys and a cluster height of 0.6. For each cluster Z-scores (defined as the number of standard deviations that the mean of each cluster is away from the mean of the entire data set) were calculated based on pEC50 values of the agonist, potentiator and parental assays. Clusters and singletons with activity in the parental (CHO CRE BLA) cell line or high activity in another GPCR beta-lactamase reporter assay (5HT1A) were removed for likely unspecific effects. Reactive or undesired structural features were also removed. A similar procedure was used to identify the active series of the S1P3 agonist assay. The Z-scores based on the primary assay results (supporting material Table S2) illustrate that the clusters are derived from only the most active compounds confirmed in a dose-response assay. They also indicate selectivity where initial dose response data was not available for both receptors. To follow-up the most interesting series and develop initial SAR, structures with S1P1 EC50 ≤ 1 μM (in the initial HTS dose response assay) were selected as seeds and 80% similar structures were added from the entire library. The resulting compounds were hierarchically clustered with a cutoff threshold of 0.7. Selected examples of the re-tested 3,5-diaryl-1,2,4-oxadiazole series are given in Table 1. The most active representatives are summarized in the supporting material (Table S3). To follow up on S1P3 compounds with an EC50 < 10 μM were selected as seeds and in addition to the 80% similarity expansion, structures with > 60% similarity to the 5-(2,2,-diphenylethyl)-3-4-pyridyl-oxadiazole 16 were added because of the much smaller number of actives and generally lower activities. One series of 5-sulfanyl-cyano-tetrahydroisoquinolines was removed entirely.
Shape-based comparison
A 3D conformer library (10 Mio structures) was generated from the combined MLSMR and HitFinder™ using Omega41 from Open Eye.42 Shape similarities43 of this 3D conformer library were generated against the best docking poses of the S1P3-selective dicyclohexylamide 20 and the S1P1-selective diaryl-oxadiazole 6 using Open Eye ROCS and the largest ShapeTanimoto for each query structure of the library was used to plot the number of shape similar compounds as a function of similarity cut-off (supporting material Figure S11).
Supplementary Material
Figure S1. S1P1 agonists S1P (1), FTY720 (2), SEW2871 (3), and AFD-R (4).
Figure S2. Similarity comparison of the MLSMR and MBHF libraries; for both libraries the number of samples is shown for which the most similar compound of the other library corresponds to given similarity value; using extended connectivity fingerprints with a path length of 6 (ECFP-6) and the Tanimoto metric.
Figure S3. Coverage of chemical space of the MLSMR (blue) and the MBHF (red) libraries; illustrated in an optimized two-dimensional BCUTS space; figures 1 and 2 illustrate the same space.
Figure S4. S1P1 (A) and S1P3 (B) receptors with S1P (copper) and FTY720-P (green) docked into the binding site.
Figure S5. Detailed binding sites of S1P receptors with agonists S1P (copper) docked into S1P1 (A) and S1P3 (C) and FTY720-P (green) docked into S1P1 (B) and S1P3 (D). Docking scores correspond to reported EC50 values and binding affinities with S1P-S1P3 < S1P-S1P1, and FTY720P-S1P1 < FTY720P-S1P3; note that in D, FTY720-P is shifted lower in the S1P3 pocket compared to the poses in A-C due to interaction with Phe263, this removes the interaction of the phosphate ester with Leu279.
Figure S6. Overlay of the docked MLSMR diarloxadiazole agonists in the S1P1 receptor model (showing secondary structure): 6 green, 5 yellow, 7 cyan, 8 copper. The S1P3 Phe263 interferes with these ligand poses explaining the observed selectivity.
Figure S7. S1P3-selective dicyclohexylamides docked into the S1P3 receptor model (showing secondary structure): green SID 20, copper 28. Docking scores of these poses in the corresponding S1P1 receptor model are 0.
Figure S8. Potential pose of oxadizole 6 in the S1P3 receptor (green) in comparison to the pose in S1P1 (white).
Figure S9. Potential pose of dicyclohexylamide 28 in the S1P1 receptor (copper) in comparison to the pose in S1P3 (gray). Pose obtained by optimizing 28 in S1P1 starting from the docked orientation in S1P3.
Figure S10. Number of identified compounds (MLSMR plus MBHF) by shape similarity (blue triangle S1P1 selective diaryloxadiazole 6; red circle, S1P3 selective dicyclohexylamine 20).
Figure S11. Population density (occupancy) of the regions of chemistry space that include identified S1P1 or S1P3 agonists. The optimized 6D BCUTS chemistry space characterizing the MLSMR and MBHF libraries is split into 46,656 cells (6 bins per dimension), 6,438 of which are occupied. Red: Cells with S1P1 agonists, green, cells with S1P3 agonists, blue, cells with both S1P1 and S1P3 agonists, size by pEC50 of the most active agonist in the cell.
Figure S12. ERK phosphorylation induced by S1P (■) or 20(▲). Results are an average of two independent experiments and are normalized to vehicle (0 percent of control) and 1 uM S1P (100 percent of control).
Table S1. Summary of Screening Campaigns.
Table S2. Most active S1P1 and S1P3 agonist series based on initial data (MLSMR library; # - number of cluster members).
Table S3. Reconfirmed S1P1 agonists representing including the clusters shown in table 2 above.
Table S4. Additional selected S1P1 actives identified from the 16K MBHF library
Table S5. Selected S1P3 actives identified from the 16K MBHF library.
Table S6. S1P receptor subtype selectivity data for S1P1 oxadiazole agonist 6 and S1P3 dicyclohexylamide agonist 20.
Acknowledgments
This work was supported by the National Institutes of Health Molecular Library Screening Center Network grant U54 MH074404-01 and by grants from NIAID to HR. We thank Pierre Baillargeon for compound management and Kashif Hoda for data management with PubChem.
References
- 1.Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279(14):13839–48. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 2.Forrest M, Sun SY, Hajdu R, Bergstrom J, Card D, Doherty G, Hale J, Keohane C, Meyers C, Milligan J, Mills S, Nomura N, Rosen H, Rosenbach M, Shei GJ, Singer II, Tian M, West S, White V, Xie J, Proia RL, Mandala S. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309(2):758–68. doi: 10.1124/jpet.103.062828. [DOI] [PubMed] [Google Scholar]
- 3.Wei SH, Rosen H, Matheu MP, Sanna MG, Wang SK, Jo E, Wong CH, Parker I, Cahalan MD. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary T cells to lymphatic sinuses. Nat Immunol. 2005;6(12):1228–35. doi: 10.1038/ni1269. [DOI] [PubMed] [Google Scholar]
- 4.Jo E, Sanna MG, Gonzalez-Cabrera PJ, Thangada S, Tigyi G, Osborne DA, Hla T, Parrill AL, Rosen H. S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol. 2005;12(6):703–15. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 5.Alfonso C, McHeyzer-Williams MG, Rosen H. CD69 down-modulation and inhibition of thymic egress by short- and long-term selective chemical agonism of sphingosine 1-phosphate receptors. Eur J Immunol. 2006;36(1):149–59. doi: 10.1002/eji.200535127. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92(5):913–22. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 7.Hla T. Signaling and biological actions of sphingosine 1-phosphate. Pharmacol Res. 2003;47(5):401–7. doi: 10.1016/s1043-6618(03)00046-x. [DOI] [PubMed] [Google Scholar]
- 8.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–9. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 9.Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277(24):21453–7. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 10.Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, Derian CK, Andrade-Gordon P, Rosen H, Ruf W. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008 doi: 10.1038/nature06663. [DOI] [PubMed] [Google Scholar]
- 11.Rosen H. Chemical approaches to the lysophospholipid receptors. Prostaglandins Other Lipid Mediat. 2005;77(14):179–84. doi: 10.1016/j.prostaglandins.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Rosen H, Liao J. Sphingosine 1-phosphate pathway therapeutics: a lipid ligand-receptor paradigm. Curr Opin Chem Biol. 2003;7(4):461–8. doi: 10.1016/s1367-5931(03)00085-1. [DOI] [PubMed] [Google Scholar]
- 13.Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, Cahalan MD, Wong CH, Rosen H. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2(8):434–41. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- 14.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 15.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342(2):571–83. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 16.Parrill AL, Wang D, Bautista DL, Van Brocklyn JR, Lorincz Z, Fischer DJ, Baker DL, Liliom K, Spiegel S, Tigyi G. Identification of Edg1 receptor residues that recognize sphingosine 1-phosphate. J Biol Chem. 2000;275(50):39379–84. doi: 10.1074/jbc.M007680200. [DOI] [PubMed] [Google Scholar]
- 17.Inagaki Y, Pham TT, Fujiwara Y, Kohno T, Osborne DA, Igarashi Y, Tigyi G, Parrill AL. Sphingosine 1-phosphate analogue recognition and selectivity at S1P4 within the endothelial differentiation gene family of receptors. Biochem J. 2005;389(Pt 1):187–95. doi: 10.1042/BJ20050046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng Q, Clemas JA, Chrebet G, Fischer P, Hale JJ, Li Z, Mills SG, Bergstrom J, Mandala S, Mosley R, Parent SA. Identification of Leu276 of the S1P1 receptor and Phe263 of the S1P3 receptor in interaction with receptor specific agonists by molecular modeling, site-directed mutagenesis, and affinity studies. Mol Pharmacol. 2007;71(3):724–35. doi: 10.1124/mol.106.029223. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara Y, Osborne DA, Walker MD, Wang DA, Bautista DA, Liliom K, Van Brocklyn JR, Parrill AL, Tigyi G. Identification of the hydrophobic ligand binding pocket of the S1P1 receptor. J Biol Chem. 2007;282(4):2374–85. doi: 10.1074/jbc.M609648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunapuli P, Ransom R, Murphy KL, Pettibone D, Kerby J, Grimwood S, Zuck P, Hodder P, Lacson R, Hoffman I, Inglese J, Strulovici B. Development of an intact cell reporter gene beta-lactamase assay for G protein-coupled receptors for high-throughput screening. Anal Biochem. 2003;314(1):16–29. doi: 10.1016/s0003-2697(02)00587-0. [DOI] [PubMed] [Google Scholar]
- 21.Leadscope Enterprise. Leadscope Inc.; Columbus: 2007. [Google Scholar]
- 22.Gonzalez-Cabrera PJ, Hla T, Rosen H. Mapping pathways downstream of sphingosine 1-phosphate subtype 1 by differential chemical perturbation and proteomics. J Biol Chem. 2007;282(10):7254–64. doi: 10.1074/jbc.M610581200. [DOI] [PubMed] [Google Scholar]
- 23.Nakamichi H, Okada T. Local peptide movement in the photoreaction intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103(34):12729–34. doi: 10.1073/pnas.0601765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Devries ME, Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput Biol. 2006;2(2):e13. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pham TC, Fells JI, Sr, Osborne DA, North EJ, Naor MM, Parrill AL. Molecular recognition in the sphingosine 1-phosphate receptor family. J Mol Graph Model. 2007 doi: 10.1016/j.jmgm.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vachal P, Toth LM, Hale JJ, Yan L, Mills SG, Chrebet GL, Koehane CA, Hajdu R, Milligan JA, Rosenbach MJ, Mandala S. Highly selective and potent agonists of sphingosine-1-phosphate 1 (S1P1) receptor. Bioorg Med Chem Lett. 2006;16(14):3684–7. doi: 10.1016/j.bmcl.2006.04.064. [DOI] [PubMed] [Google Scholar]
- 27.Debe DA, Danzer JF, Goddard WA, Poleksic A. STRUCTFAST: protein sequence remote homology detection and alignment using novel dynamic programming and profile-profile scoring. Proteins. 2006;64(4):960–7. doi: 10.1002/prot.21049. [DOI] [PubMed] [Google Scholar]
- 28.Poleksic A, Danzer JF, Hambly K, Debe DA. Convergent Island Statistics: a fast method for determining local alignment score significance. Bioinformatics. 2005;21(12):2827–31. doi: 10.1093/bioinformatics/bti433. [DOI] [PubMed] [Google Scholar]
- 29.Hambly K, Danzer J, Muskal S, Debe DA. Interrogating the druggable genome with structural informatics. Mol Divers. 2006;10(3):273–81. doi: 10.1007/s11030-006-9035-3. [DOI] [PubMed] [Google Scholar]
- 30.TIP. Eidogen-Sertanty Inc; San Diego: 2007. [Google Scholar]
- 31.Macromodel. Schrodinger LLC; Portland: 2007. [Google Scholar]
- 32.Glide. Schrödinger, LLC; Portland, OR: 2007. [Google Scholar]
- 33.Imre G, Kalászi A, Jákli I, Farkas Ö. Advanced Automatic Generation of 3D Molecular Structures. 1st European Chemistry Congress; Budapest, Hungary. 2006. [Google Scholar]
- 34.ChemAxon: Budapest, 2006.
- 35.Szegezdi J, Csizmadia F. A method for calculating the pKa values of small and large molecules. American Chemical Society Spring meeting; Chicago, IL. 2007. [Google Scholar]
- 36.DeLano WL. PyMOL. DeLano Scientific LLC; Palo Alto: 2007. [Google Scholar]
- 37.Molecular Operating Environment. Chemical Computing Group Inc.; Montreal: 2007. [Google Scholar]
- 38.Pipeline Pilot, Accelrys: San Diego, 2007.
- 39.Pearlman RS, Smith KM. Novel software tools for chemical diversity. Perspect Drug Discovery Des. 1998;9-11:339–353. [Google Scholar]
- 40.Diverse Solutions. Tripos; St. Louis: 2005. [Google Scholar]
- 41.Bostrom J. Reproducing the conformations of protein-bound ligands: a critical evaluation of several popular conformational searching tools. J Comput Aided Mol Des. 2001;15(12):1137–52. doi: 10.1023/a:1015930826903. [DOI] [PubMed] [Google Scholar]
- 42.Open Eye Scientific Software, Inc.: Santa Fe, 2007.
- 43.Grant JA, Gallardo MA, Pickup BT. A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J Comp Chem. 1996;17(14):1653–1666. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. S1P1 agonists S1P (1), FTY720 (2), SEW2871 (3), and AFD-R (4).
Figure S2. Similarity comparison of the MLSMR and MBHF libraries; for both libraries the number of samples is shown for which the most similar compound of the other library corresponds to given similarity value; using extended connectivity fingerprints with a path length of 6 (ECFP-6) and the Tanimoto metric.
Figure S3. Coverage of chemical space of the MLSMR (blue) and the MBHF (red) libraries; illustrated in an optimized two-dimensional BCUTS space; figures 1 and 2 illustrate the same space.
Figure S4. S1P1 (A) and S1P3 (B) receptors with S1P (copper) and FTY720-P (green) docked into the binding site.
Figure S5. Detailed binding sites of S1P receptors with agonists S1P (copper) docked into S1P1 (A) and S1P3 (C) and FTY720-P (green) docked into S1P1 (B) and S1P3 (D). Docking scores correspond to reported EC50 values and binding affinities with S1P-S1P3 < S1P-S1P1, and FTY720P-S1P1 < FTY720P-S1P3; note that in D, FTY720-P is shifted lower in the S1P3 pocket compared to the poses in A-C due to interaction with Phe263, this removes the interaction of the phosphate ester with Leu279.
Figure S6. Overlay of the docked MLSMR diarloxadiazole agonists in the S1P1 receptor model (showing secondary structure): 6 green, 5 yellow, 7 cyan, 8 copper. The S1P3 Phe263 interferes with these ligand poses explaining the observed selectivity.
Figure S7. S1P3-selective dicyclohexylamides docked into the S1P3 receptor model (showing secondary structure): green SID 20, copper 28. Docking scores of these poses in the corresponding S1P1 receptor model are 0.
Figure S8. Potential pose of oxadizole 6 in the S1P3 receptor (green) in comparison to the pose in S1P1 (white).
Figure S9. Potential pose of dicyclohexylamide 28 in the S1P1 receptor (copper) in comparison to the pose in S1P3 (gray). Pose obtained by optimizing 28 in S1P1 starting from the docked orientation in S1P3.
Figure S10. Number of identified compounds (MLSMR plus MBHF) by shape similarity (blue triangle S1P1 selective diaryloxadiazole 6; red circle, S1P3 selective dicyclohexylamine 20).
Figure S11. Population density (occupancy) of the regions of chemistry space that include identified S1P1 or S1P3 agonists. The optimized 6D BCUTS chemistry space characterizing the MLSMR and MBHF libraries is split into 46,656 cells (6 bins per dimension), 6,438 of which are occupied. Red: Cells with S1P1 agonists, green, cells with S1P3 agonists, blue, cells with both S1P1 and S1P3 agonists, size by pEC50 of the most active agonist in the cell.
Figure S12. ERK phosphorylation induced by S1P (■) or 20(▲). Results are an average of two independent experiments and are normalized to vehicle (0 percent of control) and 1 uM S1P (100 percent of control).
Table S1. Summary of Screening Campaigns.
Table S2. Most active S1P1 and S1P3 agonist series based on initial data (MLSMR library; # - number of cluster members).
Table S3. Reconfirmed S1P1 agonists representing including the clusters shown in table 2 above.
Table S4. Additional selected S1P1 actives identified from the 16K MBHF library
Table S5. Selected S1P3 actives identified from the 16K MBHF library.
Table S6. S1P receptor subtype selectivity data for S1P1 oxadiazole agonist 6 and S1P3 dicyclohexylamide agonist 20.