

Abstract
Yeast is a powerful model system for studying the action of small molecule therapeutics. An important limitation has been low efficacy of many small molecules in yeast due to limited intracellular accumulation. We used the DNA binding domain of the pleiotropic drug resistance regulator Pdr1 fused in-frame to transcription repressors to repress Pdr1 regulated genes. Expression of these chimeric regulators conferred dominant enhancement of sensitivity to a different classes of compounds and led to greatly diminished levels of Pdr1p regulated transcripts, including the yeast p-glycoprotein homologue Pdr5. Enhanced sensitivity was seen for a wide range of small molecules. Biochemical measurements demonstrated enhanced accumulation of rhodamine in yeast cells expressing the chimeric repressors. These repressors of Pdr1p regulated transcripts can be introduced into large collections of strains such as the S. cerevisiae deletion set, and enhance the utility of yeast for studying drug action and for mechanism-based drug discovery.
Introduction
Yeast cells are frequently insensitive to small molecules that target essential proteins. The lack of response is often not due to insensitivity of the yeast target to an inhibitor. Rather, yeast cells fail to accumulate small molecules to biologically effective concentrations. While the presence of the yeast cell wall may be an impediment in some cases, yeast cells are also very effective at reducing intracellular concentration of toxic small molecules using a large number of proteins homologous to transport proteins (Ernst et al., 2005; Moye-Rowley, 2003). The expression of a diverse array of proteins that can transport toxic molecules is likely an important property for the survival of yeast cells in the wild, but it creates difficulties in adapting yeast as a model for studying the action of potential therapeutic agents.
Studies of yeast cells selected for high levels of resistance to otherwise toxic molecules led to the discovery of regulatory networks that control the expression of many drug efflux proteins. Since some of these mutants were resistant to multiple classes of agents, the altered genes were termed Pdr (pleiotropic drug resistance) genes (Balzi and Goffeau, 1991). Subsequent analyses showed that the mutations conferring pleiotropic drug resistance were often within genes that regulated the expression of drug transport proteins (Balzi et al., 1987; Katzmann et al., 1994; Mahe et al., 1996). Two major regulators, Pdr1p and Pdr3p, coordinately regulate a set of proteins including several ABC transporters that are homologous to mammalian transporters such as ABCB1 (MDR1) (reviewed in (Moye-Rowley, 2003)). These ABC transporters were shown to confer drug resistance when overexpressed, and in some cases, drug hypersensitivity when the structural genes encoding the transporters were deleted. Since yeast cells express a wide range of transporters that have overlapping substrate preferences, drug hypersensitivity in mutants deleted for drug transporters was frequently limited to a narrow range of substrates. One important exception is mutants carrying deletions of PDR5 (Balzi et al., 1994; Leonard et al., 1994). Strains lacking PDR5 show hypersensitivity to a variety of small molecules including cycloheximide, herbicides and other enzyme inhibitors (Golin et al., 2003; Mitterbauer et al., 2003). However, sensitivity to many other small molecules is unaffected by deletion of PDR5 (see figure 8 for examples).
Figure 8. The Δpdr1::pdr1DBD-cyc8 insertion construct enhances the sensitivity of yeast to a wide range of anti-cancer drugs.
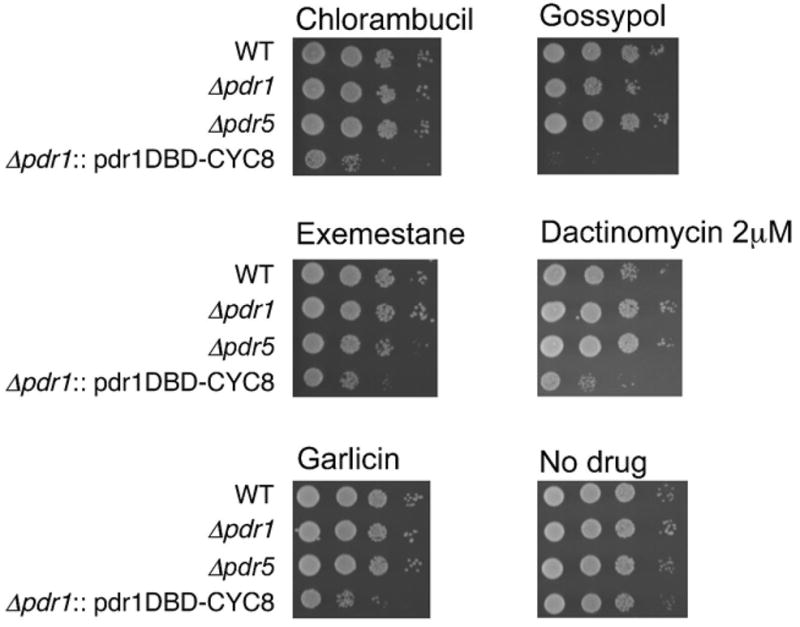
Logarithmically growing cells were spotted onto plates following 24 hr drug treatment in liquid. An assortment of small molecules/drugs as described in the text was assessed. All drugs were tested at 250μM, except dactinomycin (2μM). Five positive compounds are illustrated. Other positive compounds are not shown, and a list of test compounds is available on request.
Despite problems relating to drug accumulation, S. cerevisiae strains have been successfully used to analyze mechanisms of cytotoxicity for a variety anticancer drugs (Nitiss and Heitman, 2007). The ability to analyze drug action in yeast frequently depends on introduction of mutations enhancing drug accumulation. One strategy for overcoming this problem is to introduce multiple mutations as was done for a large screening project carried out by the National Cancer Institute. They used yeast strains harboring simultaneous deletions of PDR1, PDR3 and ERG6 to identify small molecules that specifically affected yeast cells carrying mutations in DNA repair genes (Dunstan et al., 2002).
An incentive for overcoming the insensitivity of yeast cells to small molecules is the development of a set of yeast-based tools applicable to studying drug action (Winzeler et al., 1999). Identifying reduced fitness of strains grown in the presence of a drug enables efficient genome-wide screen for drug targets, other pathways targeted by a drug, rate-limiting components of targeted pathways, and the identification of alternate or parallel biochemical pathways (Giaever et al., 1999).
In this paper, we describe chimeric transcriptional regulators using the DNA binding domain of Pdr1 fused to the transcriptional co-repressors Sin3 (Kadosh and Struhl, 1998; Wang and Stillman, 1993) or Cyc8 (Garcia-Sanchez et al., 2005; Zhang and Reese, 2004). Expression of these novel regulators renders yeast cells sensitive to a variety of small molecules. We demonstrated that expression of these fusions effectively reduces the expression of transporters regulated by Pdr/Pdr3. We showed that enhanced sensitivity to small molecules is likely due to changes in the expression of transporters, and directly demonstrate that pdr1DBD-repressor fusions increase intracellular accumulation of a small molecule. Our results show that chimeric transcription factors represent a novel effective strategy to circumvent the natural multi-drug resistance found in yeast cells, and can be used to greatly enhance the effectiveness of yeast as a system for studying drug action.
Methods
Strains and media
The S. cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and KANMX4 ORF deletion derivatives were purchased from Open Biosystems. Standard yeast lithium acetate transformation techniques were used (Gietz et al., 1992). Strains transformed with the fusion constructs were selected for on synthetic dropout SD-leu (synthetic dextrose lacking leucine) media (Nitiss et al., 1992). Drug sensitivity was performed using SD media with appropriate nutrients deleted as required for selection of plasmids. Experiments with Gal1 overexpression also used synthetic media with 2% galactose replacing dextrose. The strain JN362a is moderately hypersensitive to a variety of inhibitors including etoposide and mAMSA (Nitiss and Wang, 1988).
Vector construction
For cloning purposes, PCR primers were designed with appropriate restriction enzyme sites at the ends or with homology to adjacent sequences for second round PCR reactions (primer sequences available upon request). Yeast genomic DNA (Promega) was used as the PCR template for cloning and for generating northern blot probes.
A segment of the PDR1 gene including DNA binding domain (DBD) of yeast PDR1 (nucleotides 1–621) was amplified using the Expand High Fidelity PCR kit (Roche) and subcloned into the BamHI/EcoR1 sites of pBluescript II SK (Stratagene). The entire SIN3 ORF was PCR amplified and cloned in-frame downstream of the PDR1-DBD using EcoR1 and XhoI sites of pBluescript. The entire ORF of CYC8 was cloned downstream of the PDR1-DBD using the same scheme. The PDR1-GFP fusion was constructed by PCR amplification of GFP from the plasmid pIRES-EGFP (Clonetech) using primers homologous to the PDR1-DBD. PDR1-DBD amplification used primers containing homology to GFP. The products were mixed, annealed, and followed by a second round of amplification using PDR1 and GFP specific primers. The product was cloned into pBluescript using BamHI and XhoI. The PDR1-DBD-Sin3, -Cyc8, and -GFP fusions were excised using BamHI/XhoI and cloned into pYX142, a yeast centromeric plasmid, under the control of theTPI1 promoter and containing the LEU2 selectable marker. The vector overexpressing PDR5 was constructed using the plasmid pYES2 and a PCR product containing the entire PDR5 ORF. Where appropriate, pYX vectors with different selectable markers were used as empty vector control plasmids.
Integrating fusion construct
The polyA signal and LEU2 gene were amplified from pYX142 and cloned into the XhoI site downstream of the fusion constructs in pBluescript (SalI/XhoI). A 455 bp fragment containing the 3′ XhoI site within the PDR1 ORF was amplified using primers that generated SalI sites at the ends of the PCR product and cloned into the XhoI site downstream of the LEU2 marker. A 390 bp fragment 160 bp upstream of the PDR1 start codon (containing a natural XhoI site) was amplified using primers that were homologous to TPI1 promoter at the primer 3′end. The product was annealed to another PCR product containing the TPI1 promoter and a 3′ BamHI site. A second round of PCR using external primers produced a 5′pdr1Δ fragment (upstream of PDR1) fused to the TPI1 promoter. This BglII/BamHI piece was cloned into the BamHI site upstream of the fusion constructs in pBluescript. XhoI digestion releases a DNA fragment of about 300 bp of homology 5′ of the PDR1 gene, the TPI1 promoter, the pdr1:Cyc8 fusion construct, and the LEU2 selectable marker. This fragment was used for transformation and homologous integration in yeast. Correct integration at the PDR1 locus was verified by PCR.
Drug sensitivity
For growth on media containing drug, logarithmically growing cells were diluted to OD600=0.3 and 10-fold serial dilutions were spotted onto synthetic media containing the indicated drug. For all compounds added to media containing agar, drug was added when the media was at approximately 50°C, followed by immediate pouring of the agar into plastic plates. Plates were incubated at 30°C for 2–3 days and photographed. Drugs examined in this way included cycloheximide (Sigma), camptothecin (A.G. Scientific), etoposide (Bedford Laboratories), miconazole (Sigma), and Rhodamine 6G (Molecular Probes). An additional set of compounds termed “Cancer plate” containing 80 clinical and experimental anticancer drugs was obtained from Discovery Microsource http://www.msdiscovery.com/. Compounds on the plate were obtained as 10mM stock solutions in DMSO. Drug sensitivity for these compounds (Figure 8) was assessed using a different protocol than the protocol described above. An overnight culture of the appropriate yeast strains was diluted to 2*106 cells/ml. A portion of the culture (200μl) was dispensed into 96 well plates. Drug or DMSO was added and the 96 well plate was incubated without shaking for 24 hours at 30°C. After the incubation, 10 fold serial dilutions of the final cultures were spotted on drug free SD- leu media and grown for 48 hours at 30°C. Clonogenic survival assays with etoposide were carried out as previously described (Nitiss et al., 1992).
Analysis of GFP localization
GFP localization and DAPI staining of yeast nuclei was performed as described by O’Shea and colleagues (Huh et al., 2003), using the PDR1-GFP fusion described above.
Rhodamine-6-G uptake assay
Rhodamine uptake assays were performed using a modified procedure of van den Hazel and colleagues(van den Hazel et al., 1999). Approximately 2.8 × 108 logarithmically growing cells were washed 3 times and resuspended in 2ml of buffer A (50 mM HEPES/NaOH, pH 7.0). 200 μl aliquots were taken for measuring background cell fluorescence. Rhodamine-6-G was added to a final concentration of 5 mM, and 200 μl aliquots were taken every 10 min for 1 hour. For each timepoint, cells were washed 3× with ice cold buffer A, and Rhodamine-6-G fluorescence was then measured using a Cytofluor 2300 with excitation filter at 530nm and emission filter at 590nm.
Affymetrix GeneChip Analysis
Total RNA was extracted from logarithmic yeast cultures using the Ambion RiboPure-Yeast protocol according to the manufacturer’s instructions. RNA quality was confirmed by UV spectrophotometry and by analysis on an Agilent 2100 Bioanalyzer. Ten micrograms of total RNA were processed in the St. Jude microarray core facility according to the Affymetrix eukaryote target labeling protocol revision 4 (http://www.affymetrix.com/support/technical/manual/expression_manual.affx). Labeled targets were hybridized to Affymetrix YG_S98 GeneChip arrays which interrogate ~7,000 S. cerevisiae ORFs and transcripts. Signal values and detection calls were determined using the default parameters in the Affymetrix GCOS software (v1.4). Signals were scaled to a 2% trimmed mean of 500. Probeset annotations (March, 2007) were obtained from the Affymetrix website (http://www.affymetrix.com/analysis/index.affx). All microarray data have been submitted to GEO (http://www.ncbi.nlm.nih.gov/geo/) (GSE8326).
Three replicate cultures of each yeast strain were used to identify differentially expressed transcripts. Signal values were log2-transformed prior to analysis. The local pooled error (LPE) t-test (Jain et al., 2003) was used to compare transcript levels in cultures containing Pdr1-fusion constructs to those containing an empty vector (pYX142). To adjust for multiple hypothesis testing, the method of Benjamini and Hochberg (Benjamini and Y., 1995) was used to estimate the false-discovery rate (FDR). Transcripts with differential expression were defined as those with a minimum of 2-fold difference in magnitude and with an FDR<0.05. Statistical analyses were performed using the ArrayAnalyzer module in S-Plus 6.2 (Insightful).
Northern analysis
Total RNA was isolated using the same procedure used for isolating RNA for microarray analysis. Electrophoresis of RNA, transfer to nylon membranes, and hybridization was performed using standard techniques (Sambrook et al., 1989). Probes specific for PDR5, YOR1, and yeast actin (loading control) were purified by gel electrophoresis pior to labeling by random priming.
Results
The redundancy of transporters conferring pleiotropic drug insensitivity in yeast implies that repression or inhibition of several drug efflux proteins is required to enhance intracellular accumulation of small molecules. Two major regulators of the Pdr network, Pdr1p and Pdr3p, are DNA binding transcriptional activators that recognize the same DNA sequence (TTCGGCGGAA, termed a PDR response element, PDRE)(Katzmann et al., 1994; Katzmann et al., 1996). A dominant negative allele of these activators might represent an effective strategy for blocking the expression of proteins regulated by this network. We hypothesized that we could deliver a potent repressor of transcription to promoters regulated by Pdr1p and Pdr3p by fusing the DNA binding domain of Pdr1p (or Pdr3p) to potent transcriptional repressors. We chose two yeast transcriptional repressors, Cyc8p (Keleher et al., 1992) and Sin3p (Wang and Stillman, 1993). Cyc8p and Sin3p are components of distinct histone deacetylase complexes (Hda1p and Rpd3p, respectively (Davie et al., 2003; Heinzel et al., 1997)), and neither protein interacts with DNA except by interaction with other sequence specific DNA binding proteins (Kasten et al., 1997).
Details of the construction of the chimeric transcriptional repressors are described in Materials and Methods. In brief, both chimeric repressors were expressed from the constitutive TPI (triose phosphate isomerase promoter) carried on the yeast single copy vector pYX142. The first 621 nucleotides of the PDR1 coding sequence was fused with either to the entire SIN3 coding sequence (yielding plasmid pdr1DBD-SIN3), or the entire CYC8 coding sequence (yielding plasmid pdr1DBD-CYC8). A control construct carried the first 621 nucleotides of the PDR1 coding sequence fused to GFP to assess the importance of the repressor for efficient blocking of Pdr gene expression. We also expressed the coding sequence of CYC8 with the TPI1 promoter to assess the effect of CYC8 overexpression (not conjugated to a DNA binding domain) on drug sensitivity.
We first tested the effects of the chimeric repressors on sensitivity to the translation inhibitor cycloheximide, a compound that is frequently used to study the Pdr network in yeast (Balzi et al., 1987; Meyers et al., 1992) (Figure 1a). Wild type cells (BY4741) carrying either pdr1DBD-SIN3 or pdr1DBD-CYC8 were significantly more sensitive to cycloheximide than cells with an empty vector. Cells carrying pdr1DBD-CYC8 were unable to grow in medium containing 20 ng/ml cycloheximide, while cells carrying pdr1DBD-SIN3 were somewhat less sensitive, but failed to grow in media containing 50 ng/ml cycloheximide. By comparison, Δpdr1 cells showed reduced growth only at 50 ng/ml cycloheximide, while Δpdr5 cells exhibited reduced growth at 20 ng/ml cycloheximide. Importantly, cells carrying pdr1DBD-GFP or cells overexpressing CYC8 showed only modest reductions in growth at 50 ng/ml cycloheximide. One reason that the pdr1DBD-GFP fusion might fail to repress genes of the Pdr network is improper localization. To rule out this possibility we showed that pdr1DBD-GFP localized to the nucleus (Figure 1C). These results indicate that the Pdr1DBD:repressor fusions can impact sensitivity to cycloheximide, and that both the Pdr1 DNA binding domain, as well as the presence of a repressor is required for enhanced cycloheximide sensitivity.
Figure 1. Dominant interference with pleiotropic drug resistance using Pdr1 repressor fusions.
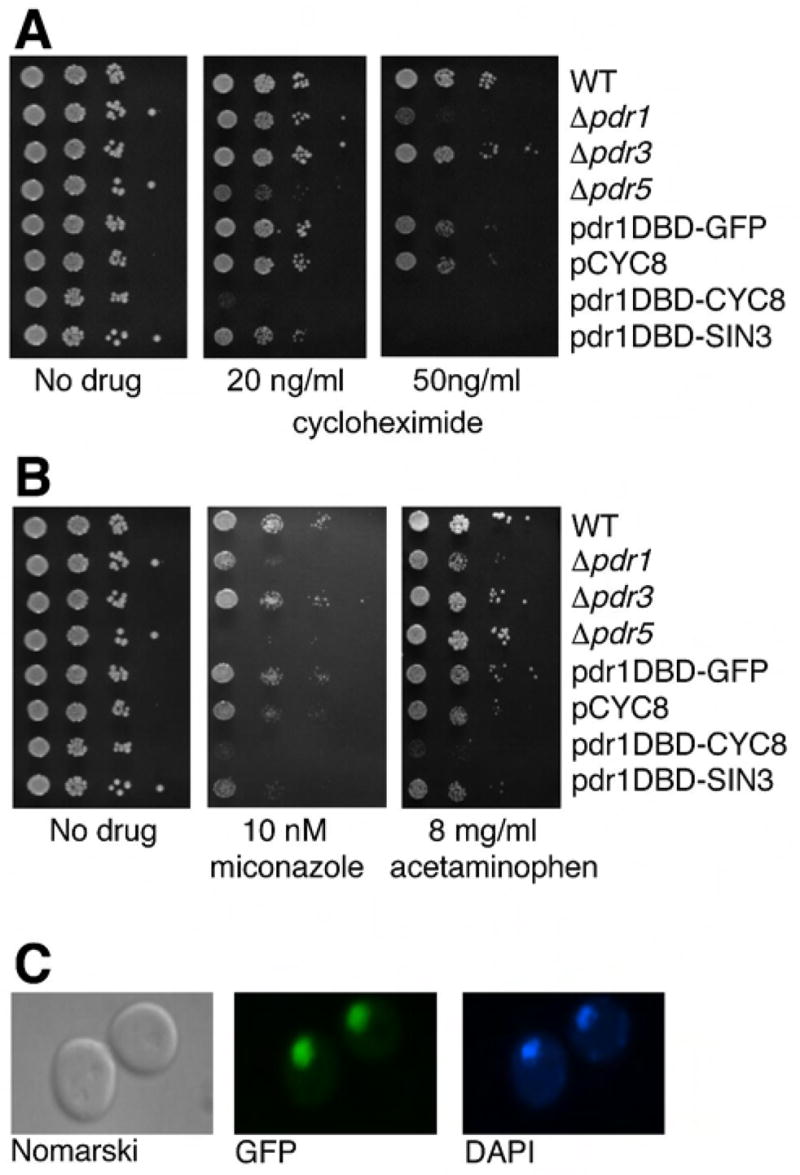
(a) Cycloheximide sensitivity of yeast carrying the pdr1DBD-repressor fusion constructs compared to yeast strains with ORF deletions of various PDR genes. Logarithmically growing yeast cultures were serially diluted and spotted onto plates containing 20 ng/ml or 50 ng/ml cycloheximide. Plates were incubated at 30°C for 2 days then photographed. (b) Sensitivity of yeast carrying pdr1DBD-repressor fusions to other drugs was determined by spotting diluted cell cultures on to agar plates containing miconazole and acetominophen at the concentrations indicated. The drug sensitivity experiments were carried out at the same time as those shown in panel 1A, and the same no drug data are displayed. (c) Pdr1DBD-GFP fusion was detected in the nucleus by fluorescent microscopy. The nucleus was also stained with DAPI.
To demonstrate that the achieved drug sensitivity is not limited to a specific compound or biochemical pathway, we tested the same strains for sensitivity to several unrelated drugs targeting different biological processes, including inhibitors of ergosterol synthesis such as miconazole, and the analgesic acetaminophen, which has been shown to inhibit the growth of yeast cells at high concentrations (Srikanth et al., 2005). Cells expressing pdr1DBD-CYC8 showed sensitivity to miconazole equivalent to that seen in Δpdr5 cells, and pdr1DBD-SIN3 bearing cells were strongly inhibited by 10 nM miconazole. The other strains tested exhibited less sensitivity at this drug concentration (figure 1b). Similarly, pdr1DBD-CYC8 strains failed to grow in media containing 8 mg/ml acetaminophen, and was the most sensitive strain tested. As was observed with cycloheximide, strains carrying pdr1DBD-GFP, or cells overexpressing Cyc8p without a Pdr1 DNA binding domain showed wild type levels of sensitivity to these two agents. Cells carrying pdr1DBD-CYC8 were also hypersensitive to other compounds, (see figure 8),
To determine if increased drug sensitivity depends on higher intracellular concentrations of inhibitors, we directly measured intracellular accumulation of Rhodamine-6G (van den Hazel et al., 1999) in yeast cells expressing pdr1DBD-SIN3 or pdr1DBD-CYC8 compared to cells carrying a pYX empty vector. Cells carrying either fusion accumulated approximately 3 fold higher levels of Rhodamine-6G compared to cells carrying a pYX empty vector (figure 2a). Exposure to 20 μg/ml Rhodamine-6G completely inhibited the growth of cells carrying pdr1DBD-CYC8 and greatly inhibited the growth of cells carrying pdr1DBD-SIN3, while growth of wild type cells was affected to a much lesser extent (figure 2b), indicating an enhanced biological effect of Rhodamine-6G on repressor fusion bearing cells. This result directly demonstrates that the pdr1DBD-repressor fusions can result in alterations in yeast cell accumulation of a small molecule.
Figure 2. Enhanced intracellular concentration of Rhodamine 6G in pdr1:repressor fusion strains.
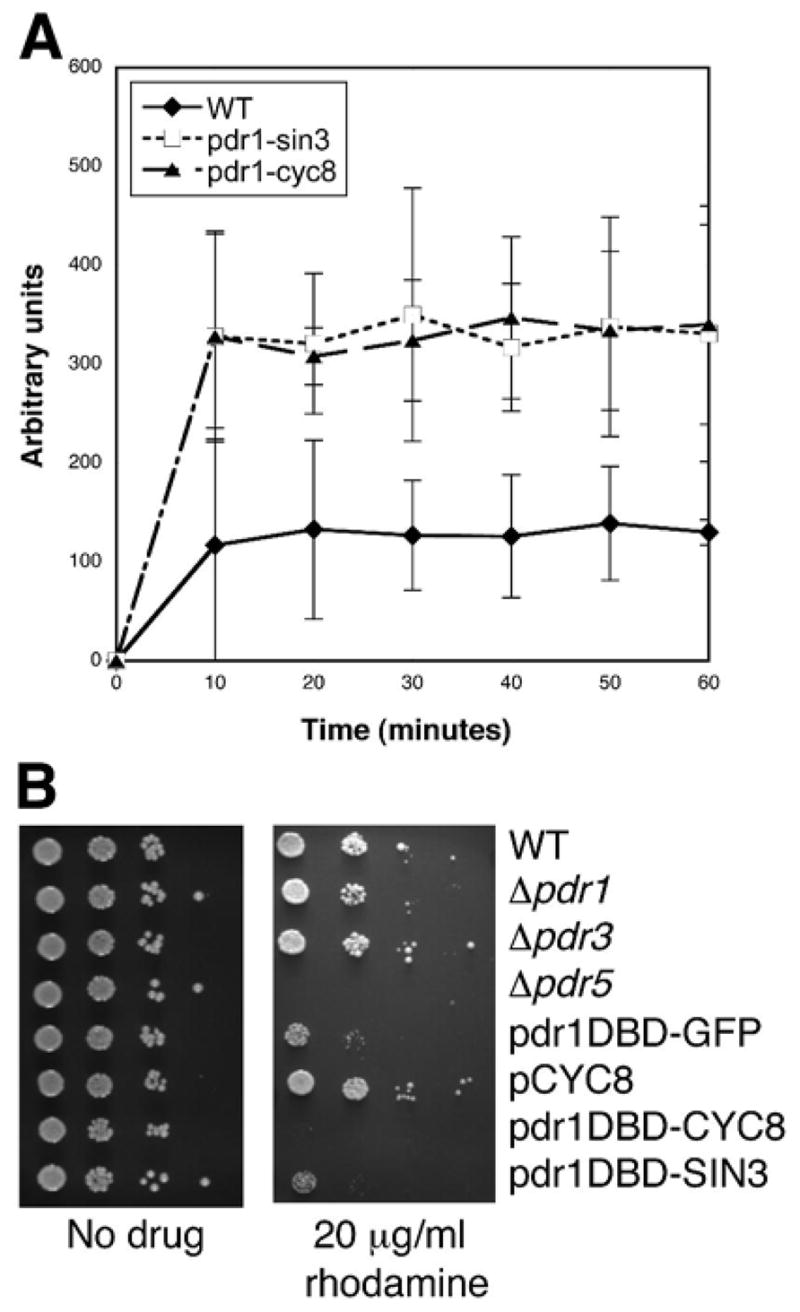
(a) Cell-associated fluorescence of Rhodamine 6G. Cells from logarithmically growing yeast cultures carrying either a vector control or plasmids expressing pdr1DBD-repressor fusions were assayed for the ability to accumulate Rhodamine-6-G. Cell associated fluorescence due to Rhodamine-6-G was measured by spectrofluorimetry. Error bars indicate ± SD of three independent experiments. (b) Sensitivity of yeast cultures carrying pdr1DBD-repressor fusions or deletions of Pdr genes was assayed by spotting diluted cell cultures on to plates containing rhodamine 6G. The drug sensitivity experiments were carried out at the same time as those shown in Figure 1A, and the same no drug data are displayed.
We predicted that expression of pdr1DBD-repressor fusions should enhance drug accumulation by down regulating the expression of Pdr1 regulated genes. Furthermore, since the experiments presented above were carried out in strains carrying a wild type PDR1 gene, we predicted that the down regulation of Pdr1 regulated genes could overcome transcriptional activation by wild type Pdr1. Since genes regulated by Pdr1 and Pdr3 show different dependence on Pdr1 or Pdr3 loss-of-function (reviewed in (Moye-Rowley, 2003)) we were interested in determining the transcriptional targets of the Pdr1DBD:repressor fusions.
We carried out microarray analysis using Affymetrix YG_S98 GeneChip arrays and assessed whether the Pdr1DBD:repressor fusions altered the expression of genes regulated by Pdr1. For this analysis, we compared the effect of the Pdr1DBD:repressor fusions with genes upregulated by the expression of a dominant allele, PDR1-3, that confers a hyper-resistant phenotype to cycloheximide and other agents, and results in overexpression of Pdr1 target genes (reviewed in (Jungwirth and Kuchler, 2005; Moye-Rowley, 2003)). As tabulated in Figure 3, three genes upregulated by PDR1-3 were significantly down regulated by both Pdr1DBD:repressor fusions: ICT1, a protein of unknown function whose deletion enhances sensitivity to Calcofluor white, PGA3, an essential protein that localizes to the endoplasmic reticulum, and the drug transporter PDR5. One gene, HXK1 (hexokinase) that was upregulated by PDR1-3 was also upregulated by both promoter fusions. Upregulation of YGR243W was also seen although the effect was not significant at a false discovery rate (FDR)<0.05. Several other genes showed reduced transcription in one but not both repressor fusions, while another set of genes showed reduced expression that was not significant at a FDR<0.05. Also shown in Figure 3 is the analysis of the effect of Pdr1DBD-CYC8 in a strain carrying a deletion of the PDR1 gene. In this case, six additional genes upregulated by PDR1-3 were downregulated by the combination of expression of Pdr1DBD-CYC8 and deletion of PDR1. Interestingly, not all of the genes identified by de Risi and colleagues that were upregulated by PDR1-3 contained a PDRE, however, all genes that were down regulated by the combination of expression of Pdr1DBD-CYC8 and deletion of PDR1 contained a PDRE. The genes that were upregulated also lack a PDRE. Taken together, these results suggested that at least some Pdr1 target genes were downregulated by the Pdr1DBD:repressor fusions, and that deletion of PDR1 enhanced the effectiveness of the Pdr1DBD:repressor fusions. The full set of expression data is available on-line (http://www.ncbi.nlm.nih.gov/geo/, (GSE8326).
Figure 3. Expression analysis in cells containing pdr1DBD-fusion constructs. Expression analysis in cells containing pdr1DBD-fusion constructs.

Gene expression profiles were obtained by hybridization to Affymetrix YG_S98 GeneChip arrays. Profiles were obtained from three independent RNA isolations for each strain tested. Data presented are the expression profiles of genes that are upregulated by the pdr1–3 allele. The last three columns show the mean fold change relative to that of empty vector in yeast strains expressing PDR1-fusion repressors. A minus sign indicates expression was decreased relative to control. Ratios highlighted in yellow were significant at a false discovery rate (FDR) <0.05. Genes highlighted in blue contain PDR response elements in their promoter regions as reported previously(DeRisi et al., 2000; Hikkel et al., 2003). Three genes, (COS10, PDR5 and YOR1) had multiple probesets on the YG-S98 array. For these genes we reported the median change relative to vector, and highlighted that ratio when at least one of the probesets achieved the FDR<0.05. In addition to the conditions reported here, we also analyzed the effect of deletion of PDR1 in cells carrying an empty vector. The full set of expression data is available on-line (http://www.ncbi.nlm.nih.gov/geo/, GSE8326).
We confirmed some of the results obtained with the microarray studies by Northern analysis using probes for two target genes, PDR5, and YOR1. Both genes are well-established PDR1 targets. The microarray analysis showed that PDR5 expression was reduced by both Pdr1DBD:repressor fusions, while YOR1 was only significantly reduced by Pdr1DBD-SIN3. However, by Northern analysis, Pdr1DBD-CYC8 clearly reduced the expression of both PDR5 and YOR1 (Figure 4). Taken together, these results indicate that multiple PDR1 targets are downregulated by Pdr1DBD:repressor fusions. The Northern analysis also demonstrated efficient downregulation in the strain carrying both Pdr1DBD-CYC8 and ΔPDR1. Given the low level of expression of these two target genes in the Pdr1DBD-CYC8 strain, it was not possible to determine whether deletion of PDR1 enhanced the silencing of these two target genes.
Figure 4. Northern analysis of strains carrying pdr1DBD-fusion constructs confirms repression of PDR1 target genes.
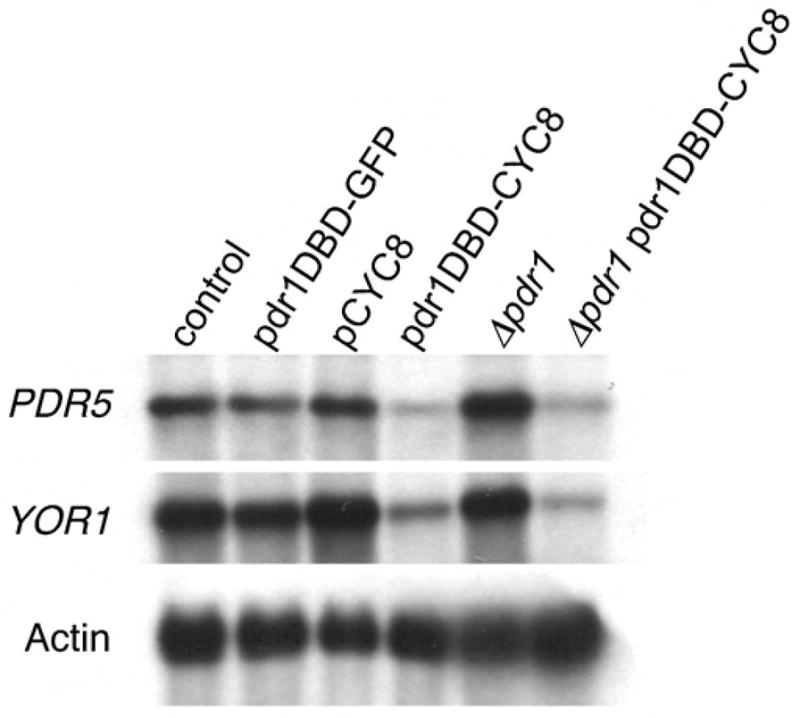
Northern blot analysis of PDR5 and YOR1 gene expression in strains carrying vector control (lane 1), pdr1DBD-GFP fusion (lane 2), CYC8 over-expression (no fused DNA binding domain) (lane 3) pdr1DBD-CYC8 fusion (lane 4), Δpdr1 strain (lane 5), Δpdr1::pdr1DBD-CYC8 fusion strain (lane 6). mRNA from the yeast actin gene was used as loading control.
Although the Pdr1DBD:repressor fusions are clearly dominant, deletion of PDR1 might confer an additional advantage in conferring drug sensitivity when coupled with Pdr1DBD:repressor fusions. To assess this possibility, we examined the sensitivity of Pdr1DBD:repressor fusion bearing strains to etoposide, a Top2 targeting drug. Etoposide does not readily enter yeast cells, and our previous results indicated that single mutations such as Δerg6 or Δpdr5 failed to result in etoposide sensitivity. Figure 5a shows that strains carrying a pYX empty vector, Δpdr1, or Δpdr5, are not sensitive to etoposide. Deletion of PDR1 combined with expression of Pdr1DBD-CYC8 completely blocks growth on 100 μg/ml etoposide, while deletion of PDR5 along with Pdr1DBD-CYC8 exerts a lesser effect. This result clearly shows that deletion of PDR1 greatly enhances the effectiveness of the Pdr1DBD:repressor fusions.
Figure 5. Deletion of PDR1 enhances the sensitivity of pdr1DBD-repressor fusions to etoposide.
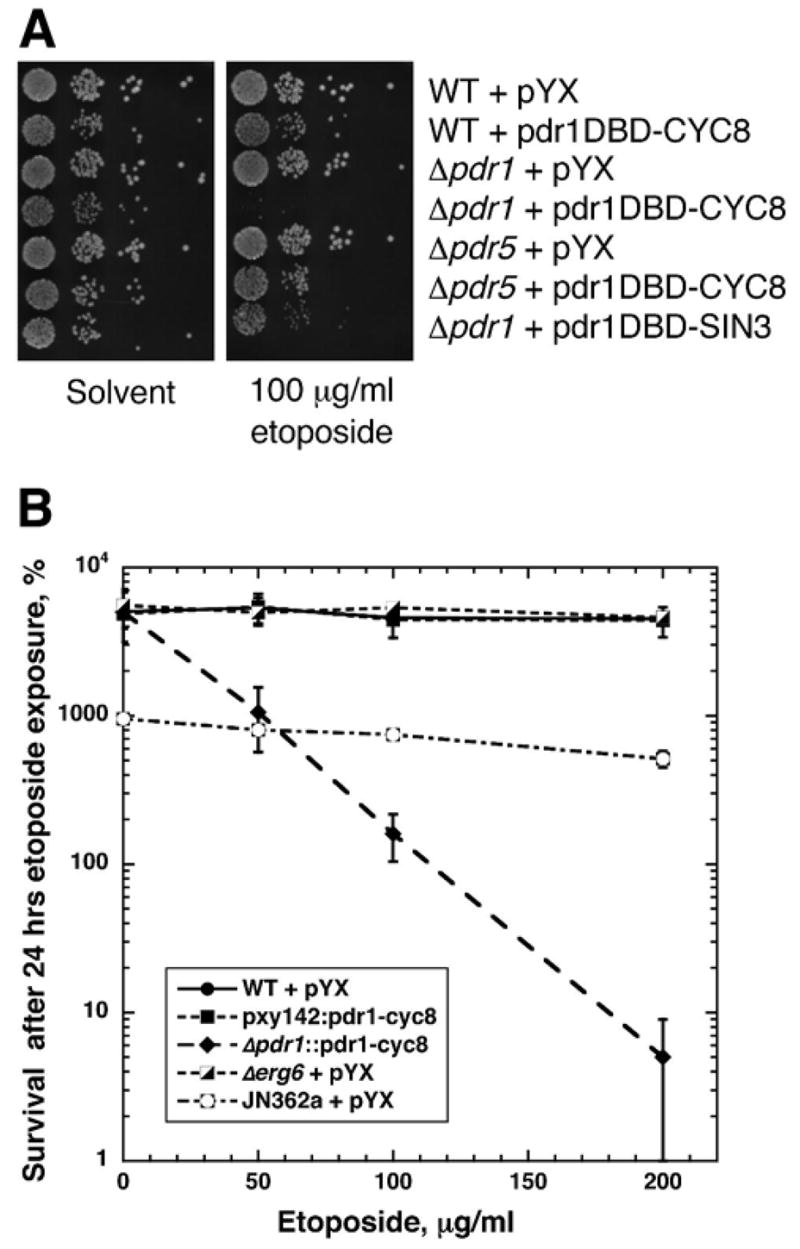
(a) Cell sensitivity was carried out similar to the experiments shown in Figure 1a using either a solvent control or 100 μg/ml etoposide containing media. The strains used are indicated on the figure. The results of this experiment show that deletion of either PDR1 or PDR5 alone is insufficient to confer etoposide sensitivity, and that strong synergistic sensitivity is seen with pdr1DBD-CYC8 and deletion of PDR1 but not deletion of PDR5. For etoposide, pdr1DBD-CYC8 appears superior to pdr1DBD-SIN3. (b) Clonogenic surival assays were carried out using the strains indicated in the legend. Cells were treated in SD-leu medium containing indicated concentrations of etoposide for 24 hrs. After incubation, aliquots were removed, diluted, and plated to SD-leu plates to determine viable titer. WT cells were strain BY4741, and all strains shown (except JN362a are BY4741 derivatives. Survival is shown relative to the viable titer at the time of etoposide addition. Cytotoxicity is indicated in this experiment by a viability below 100%.
We further confirmed the etoposide sensitivity by carrying out clonogenic survival assays with various BY4741 derivatives. Wild type BY4741 is insensitive to etoposide, as are Δerg6 derivatives. Expression of Pdr1DBD-CYC8 from a plasmid (with a wild type chromosomal copy of PDR1 is also etoposide insensitive, consistent with the spot tests shown in figure 5A. The integrated Pdr1DBD-CYC8 construct that deletes the chromosomal copy of PDR1 is very sensitive to etoposide, with concentrations above 100 μg/ml reducing viability below the viable titer at the time of drug addition. Strain JN362a is etoposide sensitive when it also carries a mutation compromising DNA repair pathways, but is insensitive when it is repair proficient. The integrated Pdr1DBD-CYC8 strain is the first well-defined S. cerevisiae strain we have tested that can be killed by etoposide even in the absence of any mutations conferring DNA repair defects.
Since the PDR1 gene appears to regulate the expression of genes that may affect membrane trafficking, as well as drug transporters, the effectiveness of the Pdr1DBD:repressor fusions may affect the disposition of drug transport proteins and other membrane proteins. To test this possibility, we constructed a vector that expressed the PDR5 transporter from the GAL1 promoter. If mis-localization of membrane proteins is an important effect of the Pdr1DBD:repressor fusions, expression of PDR5 from a different promoter would be insufficient to restore wild type drug resistance. However, we found that cells that express PDR5 from the GAL1 promoter become cycloheximide resistant even when Pdr1DBD-CYC8 or Pdr1DBD-SIN3 is expressed (figure 6). This result suggests that the major effect of the repressor fusions is on the expression of drug transport proteins rather than their stability or localization.
Figure 6. Ectopic expression of pdr5 from a heterologous promoter restores cycloheximide resistance.

Strains expressing pdr1DBD-repressor fusions were co-transformed with a plasmid expressing the PDR5 gene under the control of the GAL1 promoter. Cells were spotted on glucose or galactose plates containing 100 ng/ml cycloheximide. The plasmids carried by each strain are shown in the figure.
We expect that a major use of the Pdr1DBD:repressor fusions will be to analyze gene deletions affecting sensitivity to a drug of interest of the gene deletions using the yeast deletion collection. For example, genetic screen have been carried out using the yeast deletion set to identify genes required for surviving DNA damage from ionizing radiation or simple alkylating agents (Bennett et al., 2001; Chang et al., 2002; Westmoreland et al., 2004). Similar experiments using anti-cancer agents such as etoposide, or anti-metabolites such as aphidicolin require sufficient drug accumulation to elicit a biological effect. These screening experiments requires alteration of a large number of strains, therefore the construct needs to be introduced into strains by simple procedures. It would be desirable to introduce the Pdr1DBD:repressor fusion, and delete the endogenous PDR1 gene in a single step. To achieve this, we designed a construct with the TPI1 promoter expressing pdr1DBD-CYC8, LEU2 as a selectable marker, flanked by sequences homologous to the sequences flanking PDR1 genomic locus. Integration of the construct simultaneously deletes the PDR1 ORF and expresses the pdr1DBD-CYC8 fusion. We tested this construct in a series of deletions that we had previously found to confer hypersensitivity to Top2 targeting agents (Nitiss et al., 2006). Figure 7 shows the sensitivity of strains to different etoposide concentrations. The strains carry an empty vector, pdr1DBD-SIN3, pdr1DBD-CYC8, or the construct that deletes PDR1 and also expresses pdr1DBD-CYC8 (Δpdr1:: pdr1DBD-CYC8, LEU2). Figure 7 shows that the construct that deletes PDR1 clearly can be used to show the etoposide sensitivity of strains lacking MUS81, CTF8, or SAE2. The SAE2 deletion had the greatest effect, and sensitivity could be seen even without deletion of PDR1. By contrast, the sensitivity of Δmus81 strains can be partly seen in the strain carrying pdr1DBD-CYC8, but is most clear in the ΔPDR1 strain that carries pdr1DBD-CYC8.
Figure 7. Applications of the pdr1DBD-repressor fusions: sensitive detection of etoposide hypersensitivity in yeast deletion strains.

The pdr1DBD-fusion constructs enhance the sensitivity of various deletion strains to etoposide. Cells carrying a pYX empty vector, pdr1DBD-SIN3, pdr1DBD-CYC8, or Δpdr1::pdr1DBD-CYC8 were tested for sensitivity to etoposide. The strains used were wild type BY4741, BY4741 Δmus81, BY4741 Δctf8, or BY4741 Δsae2. Enhanced sensitivity for a fusion construct is noted by comparing wild type with the three mutants at a fixed etoposide concentration. For example, at 50 μg/ml etoposide, enhanced sensitivity of all three mutants is seen with Δpdr1::pdr1DBD-CYC8. Enhanced sensitivity of the Δctf8 strain is also seen with pdr1DBD-CYC8. At 100 μg/ml etoposide, enhanced sensitivity of the Δctf8 and Δsae2 strains can be clearly discerned.
The approach described here with Pdr1DBD:repressor fusions depends on the ability of the construct to enhance the accumulation of a large number of compounds with differing chemical structures. The data presented in Figures 1 and 2 illustrate that cells carrying pdr1DBD-CYC8 are sensitive to growth inhibition by small molecules acting against several different targets. To further test the suitability of our strategy for enhancing drug accumulation, we tested the sensitivity of a panel of 80 approved or experimental anti-cancer agents obtained from Discovery Microsource. This set of compounds includes DNA damaging agents, topoisomerase inhibitors, as well as compounds acting by unknown mechanisms such as garlicin, and agents that do not damage DNA such as gossypol (Cheng et al., 2002; Qiu et al., 2002). The set of compounds also include agents that would not act in yeast (such as nucleoside analogs) due to the absence of appropriate activating enzymes. For cells carrying only a pYX empty vector, 8/80 compounds conferred some growth inhibition. Single deletion of either PDR1 or PDR5 did not notably increase the number of compounds that inhibited growth. By contrast, 37/80 compounds showed growth inhibition in strains carrying a PDR1 deletion along with pdr1DBD-CYC8 (i.e., the pdr1DBD-CYC8 construct integrated at PDR1. Examples of growth inhibition of five compounds, along with Δpdr1 and Δpdr5 controls are shown in figure 8. These results indicate that this strategy should be usable for studying a broad range of compounds in yeast that are not readily assayable using standard approaches.
Discussion
A wide variety of genetic tools have been developed with Saccharomyces cerevisiae to explore fundamental aspects of cell biology in a eukaryotic system. A notable tool has been the development of a set of yeast strains that delete each of the open reading frames found in the yeast genome. This tool has been of fundamental importance for identifying genes required for a variety of basic cellular processes. For example, large numbers of genes important for surviving DNA damage, fitness under various growth conditions, and cell morphology have been identified using the yeast deletion set (Scherens and Goffeau, 2004). The deletion set has also been applied to study the action of small molecules of therapeutic interest. Several powerful approaches have been devised based on identification of gene deletions conferring drug hypersensitivity (Baetz et al., 2004; Deng et al., 2005) or drug resistance (Huang et al., 2005). While most of the experimental approaches with the S. cerevisiae deletion strains have been restricted to genes that are not essential for viability, it is also possible to include essential genes by screening for drug sensitivity (or resistance) using phenotypes observed in heterozygous diploids (phenotypes arising from haploinsufficiency), or by examination of phenotypes conferred by gene overexpression. These approaches also have great potential in identifying mechanisms of action of small molecules, but are limited to compounds that accumulate in yeast cells at levels that can produce significant biological effects. In this work, we describe a simple strategy applicable to large strain collections that can enhance accumulation of diverse small molecules.
Strategies for enhancing drug accumulation in yeast can be successful, but typically rely on introducing multiple mutations affecting efflux of small molecules and possibly small molecule influx as well (Emter et al., 2002). While multiple mutations can be used with a small set of strains, it is impractical to introduce more than a single mutation to enhance drug accumulation. Of the candidate single mutations available, the most commonly used are deletions of ERG6. This mutant has significant growth defects, is incompatible with trypotophan auxotrophy, and has very poor transformation efficiency. Neither PDR1 nor PDR5 single deletions give a broad spectrum sensitivity. As shown in figure 1a, the fusions described here give superior drug sensitivity compared to PDR1 or PDR5 deletions. The data presented here demonstrate that the pdr1:repressor fusions result in enhanced sensitivity to a several model compounds. We have also examined a broad range of other agents, including other experimental anti-cancer agents, and have found that the repressor fusions confer hypersensitivity to agents acting against diverse cellular targets.
We also examined the ability of the DNA binding domains of other Pdr regulators to affect drug sensitivity. We constructed fusions containing the DNA binding domains of PDR3 and YRR1 along with the coding sequence of SIN3. Since the Pdr3 DNA binding domain recognizes the same nucleotide sequence as the Pdr1 DNA binding domain (Katzmann et al., 1994; Moye-Rowley, 2003), we anticipated that the pdr3DBD-Sin3 fusion would confer cycloheximide sensitivity. By contrast, Yrr1 regulates the expression of Snq2, but not Pdr5, and loss-of-function mutants of Yrr1 do not confer cycloheximide sensitivity (Moye-Rowley, 2003). As expected, the pdr3DBD-Sin3 fusion conferred cycloheximide sensitivity, while the yrr1DBD-Sin3 fusion did not confer cycloheximide sensitivity (A.S and J.L.N unpublished data). While the fusions using either the Pdr3 or Yrr1 DNA binding domains were not extensively characterized, they may be useful in studying compounds unaffected by expression of the Pdr1 repressor fusions.
We anticipated that the pdr1:repressor fusion constructs would confer dominant drug sensitivity. While this expectation was partly correct, we found that deletion of PDR1 along with introduction of the PDR1 fusion resulted in greater sensitivity than expression of the fusion in strain carrying the wild type PDR1 gene. The microarray data presented in figure 3 also suggests that we achieve much greater repression of PDR1 regulated genes using the pdr1:repressor fusions when PDR1 is also deleted. This does not make the construct more difficult to use for most applications, since some of our pdr1:repressor fusions are targeted to delete the wild type PDR1 gene. The pdr1:repressor is then introduced into the deletion set by mating (Tong et al., 2001). One application where the targeting of the pdr1:repressor construct to the PDR1 locus is impractical is introducing the construct into the set of strains where the targeted ORF deletions are heterozygous. This set of strains is important because it includes (heterozygous) deletions of essential yeast genes. Several robust approaches have been described for transforming large numbers of strains using robotic platforms, and substantial drug sensitivity can be seen even when wild type Pdr1 is expressed. Furthermore, there is no practical way to introduce any (recessive) mutation affecting drug accumulation into the heterozygous diploid collection.
An additional strength of our approach is the demonstration that multiple pdr1:repressor fusions are capable of repressing the expression of Pdr1 regulated genes. SIN3 and CYC8 share some mechanisms of gene repression, but also require different complements of proteins to effect repression. The availability of two different constructs allows investigators to minimize effects that are independent of the repression of genes of the Pdr network.
We envision that the pdr1:repressor fusions can be applied to a variety of problems relating to analysis of drug action. For example, as shown in figure 7, we used the pdr1:repressor fusion to test the importance of several repair genes in sensitivity to etoposide. Little sensitivity to etoposide is seen for any of the repair deficient strains when they carry an empty vector, but sensitivity was clearly seen for both the pdr1:Sin3 and pdr1:Cyc8 fusions. Similar analyses can be performed with drugs with known targets (such as etoposide) as well as drugs with more poorly defined targets.
We have also demonstrated that ectopic expression of Pdr5p reverses the cycloheximide sensitivity of cells carrying a pdr1:repressor fusion. The ability to extinguish the expression of several yeast transport proteins will allow the development of yeast strains expressing heterologous drug transport proteins. This may represent a particularly efficient system for determining substrate specificity and inhibitor profiles for transport proteins of therapeutic interest.
In conclusion, we have developed a novel approach to enhance drug accumulation in Saccharomyces cerevisiae. We have demonstrated specific repression of yeast genes that are regulated as part of the Pdr1/Pdr3 network, resulting in enhanced drug accumulation and drug efficacy. This approach opens up yeast to the study of much broader range of small molecules than was previously possible.
Acknowledgments
We thank the Hartwell Center Core laboratory at St. Jude Children’s Research Hospital for carrying out microarray hybridization.
This work was supported by a grant from the National Cancer Institute (CA82313), Core grant CA21765, and the American Lebanese Syrian Associated Charities (ALSAC)
Abbreviations
- SD
synthetic dextrose
References cited
- Baetz K, McHardy L, Gable K, Tarling T, Reberioux D, Bryan J, Andersen RJ, Dunn T, Hieter P, Roberge M. Yeast genome-wide drug-induced haploinsufficiency screen to determine drug mode of action. Proc Natl Acad Sci U S A. 2004;101:4525–4530. doi: 10.1073/pnas.0307122101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzi E, Chen W, Ulaszewski S, Capieaux E, Goffeau A. The multidrug resistance gene PDR1 from Saccharomyces cerevisiae. J Biol Chem. 1987;262:16871–16879. [PubMed] [Google Scholar]
- Balzi E, Goffeau A. Multiple or pleiotropic drug resistance in yeast. Biochim Biophys Acta. 1991;1073:241–252. doi: 10.1016/0304-4165(91)90128-4. [DOI] [PubMed] [Google Scholar]
- Balzi E, Wang M, Leterme S, Van Dyck L, Goffeau A. PDR5, a novel yeast multidrug resistance conferring transporter controlled by the transcription regulator PDR1. J Biol Chem. 1994;269:2206–2214. [PubMed] [Google Scholar]
- Benjamini YYH. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JRStatSocB. 1995;57:289–300. [Google Scholar]
- Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, Snipe JR, Resnick MA. Genes required for ionizing radiation resistance in yeast. Nat Genet. 2001;29:426–434. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci U S A. 2002;99:16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JS, Liu CP, Lo YK, Chou KJ, Lin MC, Su W, Law YP, Chou KJ, Wang JL, Chen WC, Jan CR. Gossypol, a component in cottonseed, induced increases in cytosolic Ca2+ levels in Chang liver cells. Toxicon. 2002;40:851–856. doi: 10.1016/s0041-0101(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Davie JK, Edmondson DG, Coco CB, Dent SY. Tup1-Ssn6 interacts with multiple class I histone deacetylases in vivo. J Biol Chem. 2003;278:50158–50162. doi: 10.1074/jbc.M309753200. [DOI] [PubMed] [Google Scholar]
- Deng C, Brown JA, You D, Brown JM. Multiple Endonucleases Function to Repair Covalent Topoisomerase I Complexes in Saccharomyces cerevisiae. Genetics. 2005 doi: 10.1534/genetics.104.028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi J, van den Hazel B, Marc P, Balzi E, Brown P, Jacq C, Goffeau A. Genome microarray analysis of transcriptional activation in multidrug resistance yeast mutants. FEBS Lett. 2000;470:156–160. doi: 10.1016/s0014-5793(00)01294-1. [DOI] [PubMed] [Google Scholar]
- Dunstan HM, Ludlow C, Goehle S, Cronk M, Szankasi P, Evans DR, Simon JA, Lamb JR. Cell-based assays for identification of novel double-strand break-inducing agents. J Natl Cancer Inst. 2002;94:88–94. doi: 10.1093/jnci/94.2.88. [DOI] [PubMed] [Google Scholar]
- Emter R, Heese-Peck A, Kralli A. ERG6 and PDR5 regulate small lipophilic drug accumulation in yeast cells via distinct mechanisms. FEBS Lett. 2002;521:57–61. doi: 10.1016/s0014-5793(02)02818-1. [DOI] [PubMed] [Google Scholar]
- Ernst R, Klemm R, Schmitt L, Kuchler K. Yeast ATP-binding cassette transporters: cellular cleaning pumps. Methods Enzymol. 2005;400:460–484. doi: 10.1016/S0076-6879(05)00026-1. [DOI] [PubMed] [Google Scholar]
- Garcia-Sanchez S, Mavor AL, Russell CL, Argimon S, Dennison P, Enjalbert B, Brown AJ. Global roles of Ssn6 in Tup1- and Nrg1-dependent gene regulation in the fungal pathogen, Candida albicans. Mol Biol Cell. 2005;16:2913–2925. doi: 10.1091/mbc.E05-01-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21:278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golin J, Ambudkar SV, Gottesman MM, Habib AD, Sczepanski J, Ziccardi W, May L. Studies with novel Pdr5p substrates demonstrate a strong size dependence for xenobiotic efflux. J Biol Chem. 2003;278:5963–5969. doi: 10.1074/jbc.M210908200. [DOI] [PubMed] [Google Scholar]
- Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- Hikkel I, Lucau-Danila A, Delaveau T, Marc P, Devaux F, Jacq C. A general strategy to uncover transcription factor properties identifies a new regulator of drug resistance in yeast. J Biol Chem. 2003;278:11427–11432. doi: 10.1074/jbc.M208549200. [DOI] [PubMed] [Google Scholar]
- Huang RY, Eddy M, Vujcic M, Kowalski D. Genome-wide screen identifies genes whose inactivation confer resistance to cisplatin in Saccharomyces cerevisiae. Cancer Res. 2005;65:5890–5897. doi: 10.1158/0008-5472.CAN-04-4093. [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Jain N, Thatte J, Braciale T, Ley K, O’Connell M, Lee JK. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics. 2003;19:1945–1951. doi: 10.1093/bioinformatics/btg264. [DOI] [PubMed] [Google Scholar]
- Jungwirth H, Kuchler K. Yeast ABC transporters - A tale of sex, stress, drugs and aging. FEBS Lett. 2005 doi: 10.1016/j.febslet.2005.12.050. [DOI] [PubMed] [Google Scholar]
- Kadosh D, Struhl K. Targeted recruitment of the Sin3-Rpd3 histone deacetylase complex generates a highly localized domain of repressed chromatin in vivo. Mol Cell Biol. 1998;18:5121–5127. doi: 10.1128/mcb.18.9.5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten MM, Dorland S, Stillman DJ. A large protein complex containing the yeast Sin3p and Rpd3p transcriptional regulators. Mol Cell Biol. 1997;17:4852–4858. doi: 10.1128/mcb.17.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Burnett PE, Golin J, Mahe Y, Moye-Rowley WS. Transcriptional control of the yeast PDR5 gene by the PDR3 gene product. Mol Cell Biol. 1994;14:4653–4661. doi: 10.1128/mcb.14.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Hallstrom TC, Mahe Y, Moye-Rowley WS. Multiple Pdr1p/Pdr3p binding sites are essential for normal expression of the ATP binding cassette transporter protein-encoding gene PDR5. J Biol Chem. 1996;271:23049–23054. doi: 10.1074/jbc.271.38.23049. [DOI] [PubMed] [Google Scholar]
- Keleher CA, Redd MJ, Schultz J, Carlson M, Johnson AD. Ssn6-Tup1 is a general repressor of transcription in yeast. Cell. 1992;68:709–719. doi: 10.1016/0092-8674(92)90146-4. [DOI] [PubMed] [Google Scholar]
- Leonard PJ, Rathod PK, Golin J. Loss of function mutation in the yeast multiple drug resistance gene PDR5 causes a reduction in chloramphenicol efflux. Antimicrob Agents Chemother. 1994;38:2492–2494. doi: 10.1128/aac.38.10.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahe Y, Parle-McDermott A, Nourani A, Delahodde A, Lamprecht A, Kuchler K. The ATP-binding cassette multidrug transporter Snq2 of Saccharomyces cerevisiae: a novel target for the transcription factors Pdr1 and Pdr3. Mol Microbiol. 1996;20:109–117. doi: 10.1111/j.1365-2958.1996.tb02493.x. [DOI] [PubMed] [Google Scholar]
- Meyers S, Schauer W, Balzi E, Wagner M, Goffeau A, Golin J. Interaction of the yeast pleiotropic drug resistance genes PDR1 and PDR5. Curr Genet. 1992;21:431–436. doi: 10.1007/BF00351651. [DOI] [PubMed] [Google Scholar]
- Mitterbauer R, Weindorfer H, Safaie N, Krska R, Lemmens M, Ruckenbauer P, Kuchler K, Adam G. A sensitive and inexpensive yeast bioassay for the mycotoxin zearalenone and other compounds with estrogenic activity. Appl Environ Microbiol. 2003;69:805–811. doi: 10.1128/AEM.69.2.805-811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moye-Rowley WS. Transcriptional control of multidrug resistance in the yeast Saccharomyces. Prog Nucleic Acid Res Mol Biol. 2003;73:251–279. doi: 10.1016/s0079-6603(03)01008-0. [DOI] [PubMed] [Google Scholar]
- Nitiss J, Wang JC. DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc Natl Acad Sci U S A. 1988;85:7501–7505. doi: 10.1073/pnas.85.20.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL, Heitman J. Yeast as a Tool in Cancer Research. Springer; Dordrecht: 2007. [Google Scholar]
- Nitiss JL, Liu YX, Harbury P, Jannatipour M, Wasserman R, Wang JC. Amsacrine and etoposide hypersensitivity of yeast cells overexpressing DNA topoisomerase II. Cancer Res. 1992;52:4467–4472. [PubMed] [Google Scholar]
- Nitiss KC, Malik M, He X, White SW, Nitiss JL. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc Natl Acad Sci U S A. 2006;103:8953–8958. doi: 10.1073/pnas.0603455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Levin LR, Buck J, Reidenberg MM. Different pathways of cell killing by gossypol enantiomers. Exp Biol Med (Maywood) 2002;227:398–401. doi: 10.1177/153537020222700605. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual 1989 [Google Scholar]
- Scherens B, Goffeau A. The uses of genome-wide yeast mutant collections. Genome Biol. 2004;5:229. doi: 10.1186/gb-2004-5-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth CV, Chakraborti AK, Bachhawat AK. Acetaminophen toxicity and resistance in the yeast Saccharomyces cerevisiae. Microbiology. 2005;151:99–111. doi: 10.1099/mic.0.27374-0. [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- van den Hazel HB, Pichler H, do Valle Matta MA, Leitner E, Goffeau A, Daum G. PDR16 and PDR17, two homologous genes of Saccharomyces cerevisiae, affect lipid biosynthesis and resistance to multiple drugs. J Biol Chem. 1999;274:1934–1941. doi: 10.1074/jbc.274.4.1934. [DOI] [PubMed] [Google Scholar]
- Wang H, Stillman DJ. Transcriptional repression in Saccharomyces cerevisiae by a SIN3-LexA fusion protein. Mol Cell Biol. 1993;13:1805–1814. doi: 10.1128/mcb.13.3.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland TJ, Marks JR, Olson JA, Jr, Thompson EM, Resnick MA, Bennett CB. Cell cycle progression in G1 and S phases is CCR4 dependent following ionizing radiation or replication stress in Saccharomyces cerevisiae. Eukaryot Cell. 2004;3:430–446. doi: 10.1128/EC.3.2.430-446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Reese JC. Ssn6-Tup1 requires the ISW2 complex to position nucleosomes in Saccharomyces cerevisiae. Embo J. 2004;23:2246–2257. doi: 10.1038/sj.emboj.7600227. [DOI] [PMC free article] [PubMed] [Google Scholar]