

Abstract
Tumors frequently develop resistance to cisplatin, a platinum drug used as a cornerstone of present day chemotherapy regimens, significantly decreasing its usefulness in the clinic. Although it is known that cisplatin-resistant (CP-r) cancer cells commonly grow more slowly and exhibit reduced uptake of various compounds including nutrients, the effect of tumor metabolism on cisplatin resistance is unclear. It was found that in CP-r cells, uptake of 2-deoxyglucose was reduced due to dysfunction and altered morphology of mitochondria compared to cisplatin-sensitive (CP-s) parental cancer cells. The CP-r cells overexpressed SIRT1, a histone deacetylase that plays a central role in DNA damage response and transcriptional silencing. Incubation of drug-sensitive cells in low glucose medium induced the expression of SIRT1 and increased cellular resistance to cisplatin. Reduced SIRT1 expression by a SIRT1 SMART siRNA duplex sensitized the >20-fold resistant CP-r cells to cisplatin treatment 1.5- to 2-fold, and SIRT1 overexpression by SIRT1 cDNA transfection increased cisplatin resistance in CP-s cells by 2- to 3-fold. Our findings therefore suggest that reduced glucose use and altered mitochondrial metabolism mediated by SIRT1 is one of several alterations that contribute to cellular resistance to cisplatin.
Introduction
Tumors that recur after an initially positive response to treatment are often resistant to chemotherapeutic drugs. Cisplatin (cis-diamminedichloroplatinum II), which produces adducts between adjacent purines in DNA (1), is one of the most effective and widely used anti-cancer agents in the treatment of various human malignant solid and metastatic tumors (2). However, the development of cisplatin resistance in cancer cells is a major impediment in clinical treatment. Several mechanisms of cisplatin resistance have been postulated, including decreased drug accumulation, increased levels of intracellular thiols, and increased DNA repair (3). To date, the mechanism of clinical tumor resistance to cisplatin is not entirely understood, despite intensive research to identify the basis of resistance. It is important to explore the mechanism(s) responsible for cisplatin resistance to improve its therapeutic index and overcome the resistance to this drug that emerges in the clinic.
We found that the uptake of various compounds, including nutrients such as glucose, was reduced in CP-r cells compared to CP-s cells (4, 5). When nutrients are scarce, a successful survival strategy for tumors requires dynamic alterations in their metabolism. The molecular mechanisms that mediate these alterations are largely unknown. In general, glucose starvation induces a wide range of responses that include altered gene expression and biochemical activities. Recently, the silent information regulator 2 (Sir2) has been linked to extensive metabolic adaptations that render mitochondria dysfunctional. It functions as a histone deacetylase that critically regulates chromatin stability, transcriptional silencing, DNA damage response and aging under conditions of nutrient restriction (6). SIRT1, the mammalian ortholog of Sir2, is linked to the energy status of the cell in that the activity of SIRT1 is dependent on the NAD+/NADH+ ratio. SIRT1 plays a pivotal role in regulating cellular or organismal genome stability, metabolism and resistance to nutritional depletion, and is emerging as a key element regulating cell survival in response to various types of stress (7, 8).
To explore the mechanism of cancer resistance to cisplatin, we isolated two independent cell populations derived from human KB epidermoid adenocarcinoma cells (KB-CP) and human BEL7404 hepatoma cells (7404-CP). Previous work in our lab revealed that the growth rate and uptake of different nutrients were significantly reduced in cisplatin-resistant (CP-r) cells with increasing cisplatin resistance (9, 10). SIRT1, a known nutrient metabolism regulator, was measured in CP-r cells in the current study. Dramatic alterations in mitochondrial membrane potential and abnormal mitochondrial ultrastructure were found in CP-r cells with reduced oxygen consumption and decreased glucose uptake. SIRT1 was overexpressed in the CP-r cells with high NAD+ concentration. Regulation of SIRT1 expression by a SIRT1 SMART siRNA knock-down or SIRT1 full length cDNA transfection altered the resistance of cancer cells to cisplatin. Incubation of drug-sensitive cells in low glucose medium increased expression of SIRT1 and increased cellular resistance to cisplatin. These data indicate that SIRT1 helps mediate cisplatin resistance when glucose homeostasis is restricted.
Results
Reduced Glucose Uptake and Oxygen Consumption in CP-r Cells
With the aim of clarifying the mechanism of cisplatin resistance in cancer, our laboratory found there is a general defect in uptake of many different nutrients in human adenocarcinoma KB-3-1 and human hepatoma BEL7404 CP-r cells (9, 11). Our previous research demonstrated that the cellular proliferation rate is reduced with increasing cisplatin resistance in CP-r cells (10). The utilization of glucose has been recognized as an important feature of tumor development. Glucose uptake was first measured at different time points in KB-3-1 and BEL7404 series CP-r cells. It was found that uptake of C14-2-deoxyglucose was reduced in CP-r cells compared to sensitive KB-3-1 and BEL7404 cells (Fig. 1A). The accumulation of C14-2-deoxyglucose was 5-fold more in KB-3-1 cells than in KB-CP 20, and 4-fold more in BEL7404 than BEL7404-CP20 cells, measured after 30 minutes.
FIGURE 1. Glucose uptake and oxygen consumption in CP-s and CP-r cells.

A. Accumulation of C14-2 deoxyglucose is decreased in CP-r KB-3-1 and BEL7404 cells. The numbers represent the mean of three individual experiments with standard deviations. B. Oxygen consumption measured by ESR is reduced in CP-r cells. Measurements of oxygen consumption were performed five times. Shown are the oxygen consumption rates measured at each time point relative to rates in untreated samples. Error bars represent standard derivation.
To determine the effect of reduced glucose uptake on the ability of cells to use glucose through glycolysis, the rate of oxygen consumption was examined. Consistent with the observed decrease in glucose uptake, oxygen consumption measured by ESR oxymetry was reduced in CP-r KB-3-1 and BEL7404 cells with increasing cisplatin resistance. Oxygen consumption was suppressed in CP-r KB-3-1 and BEL7404 cells by 30 to 60% (Fig. 1B). Cancer cells generally maintain a high glycolytic metabolism and utilization rate despite the presence of limited oxygen in their environment. Reduced oxygen consumption or an altered glycolytic rate in CP-r cells may be an indicator of decreasing tumor sensitivity to anticancer agents.
Dysfunction of Mitochondria in CP-r Cells
Tumors may harbor lower energy consumption through defective mitochondria after having undergone rounds of anti-cancer drug selection. Having shown that cellular cisplatin resistance results in reduced glucose uptake and oxygen utilization (Figure 1 A and B), we sought to measure mitochondrial membrane potential (Δψm), a component of the overall proton motive force, as it is important for oxidative phosphorylation through utilization of glucose and oxygen in mitochondria. The Δψm, measured with CBIC2(3) by flow cytometry, was found to be higher in the CP-r cells (Figure 2A), which was consistent with the quantitative confocal microscopic measurements that allowed direct visualization of mitochondria (Figure 2B). Both KB-3-1 and BEL 7404 cells showed increased Δψm (seen as increasing red cytoplasmic staining in Figure 2B) with increasing cisplatin resistance. The numbers under individual images in panel B indicate the value of Δψm in CP-s and CP-r cells. Combined with the measurement of oxygen consumption, these results indicated a significant difference in metabolism between cisplatin-sensitive (CP-s) and CP-r cells. The mitochondria in the CP-r cells play a different metabolic role compared to their counterparts in the CP-s cells.
FIGURE 2. Polarization of mitochondrial membrane potential as determined by flow cytometry and confocal microscopy.
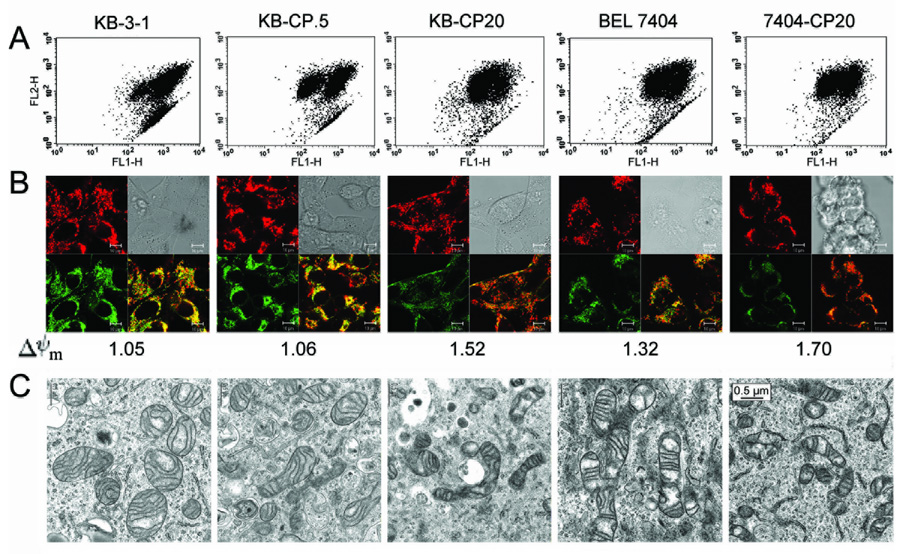
A. Mitochondrial membrane potential measured by flow cytometry. B. Mitochondrial membrane potential measured by confocal microscopy. Orange color indicates the combination of red [CBIC2(3) monomer] and green [CBIC2(3) aggregate]. Number means the relative arbitrary mitochondrial membrane potential value quantitated by confocal microscopy. C. Morphology of mitochondria in CP-s and CP-r cells observed by Electron Microcopy. Representative images from experiments performed in triplicate.
Altered Morphology of Mitochondria in CP-r Cells
Not only do mitochondria show functional alterations in CP-r cells, but electron micrographs show that the mitochondria in the CP-r cells are relatively smaller than those in CP-s cells (Figure 2C). Consistent with the Δψm measurement, mitochondria demonstrated a typical perinuclear staining pattern and appeared as fibrous, tubule-like structures in most parental cells. However, the mitochondria of CP-r cells are dispersed throughout the cell and have a disordered appearance. Also, the mitochondria of CP-r cells are much more electron dense than those in CP-s parental cells. The cristae of mitochondria in CP-r cells are thicker and more irregular (Figure 2C).
SIRT1 Overexpression is Associated With Increased Cisplatin Resistance in KB-3-1 and BEL7404 Cells
In mammalian cells, SIRT1 levels may change during the progression from normal to cancerous cell growth (12). In addition, reduced energy biogenesis can promote long-term cellular survival by augmenting expression of the SIRT1 deacetylase (13, 14). Also, SIRT1 overexpression has been associated with increased MDR1 expression in drug-resistant cells and tumors (15). Consistent with reduced bioenergenesis in CP-r cells, there is more SIRT1 expression in whole cell lysates and the nuclei fraction. SIRT1 expression increases 3- and 5-fold with increasing levels of resistance in the CP-r KB-3-1 and BEL7404 cells (Fig. 3A). The expression of the SIRT1 mRNA was also increased in CP-r cells (2-fold for BEL CP-20 cells and 4-fold for KB CP-20 cells) compared to CP-s cells, as determined by semiquantitative RT-PCR (Fig. 3B). The equal loading of protein or RNA was determined by protein staining of electrophoresis gels and EB staining of RNA agarose gels. Housekeeping genes could not be used as loading controls, owing to the altered expression of standard protein (such as actin, GAPDH, tubulin) in CP-r cells compared to CP-s cells (16, 17). These SIRT1 expression levels were confirmed by confocal microscopy. The images showed higher SIRT1 expression in the nuclei of CP-r cells than in those of CP-s cells (Fig. 3C). In addition, NAD+ concentration was significantly higher in CP-r cells that overexpress SIRT1 (Fig. 3D), which indicates that SIRT1 requires the presence of NAD+ in order to function. Our results show that SIRT1 was overexpressed in CP-r KB-3-1 and BEL7404 cells, which do not express MDR1. Therefore, overexpression of SIRT1 may be a marker and/or modulator of the drug resistance phenotype in cancer with or without MDR1 expression.
FIGURE 3. SIRT1 expression in CP-s and CP-r cells KB-3-1 and BEL7404 cells.
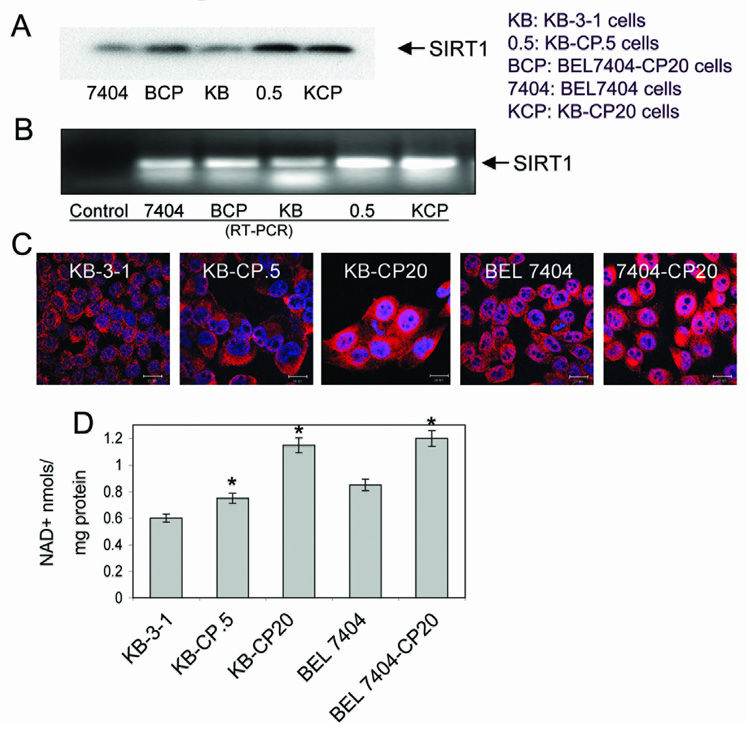
A. SIRT1 expression was detected in the whole cell lysate of CP-s and CP-r cells KB-3-1 and BEL7404 cells by Western blotting. B. SIRT1 expression was detected by semiquantitative RT-PCR. RNA was extracted from each type of cell and RT-PCR analysis was performed in triplicate using specific oligonucleotides. Representative images from experiments performed in triplicate. BCP, BEL7404-CP 20 cells; KCP, KB-CP 20 cells. C. SIRT1 expression was detected by confocal microscopy. Red staining indicates the expression of SIRT1 shown with Texas-red conjugated antibody. The nuclei labeled with DAPI and mounted in anti-fade medium appear in blue. Scale bar: 20µm. D. NAD+ concentration was increased in CP-r cells compared to CP-s cells. NAD+ concentration was measured as described in Materials and Methods. Results were expressed as mean (SD). Comparisons of each group were evaluated by one-way analysis of variance (ANOVA). When twice the S.D. was higher than the mean, a non-parameter test was used to evaluate the difference. A significant difference was assumed to exist when P < 0.05. The results shown are the mean of three individual experiments. Asterisks over bars indicate that there was a statistically significant difference between the CP-r and CP-s cells.
SIRT1 Overexpression Due to Limited Glucose Utilization Increases Cisplatin Resistance
Rodgers et al. (18) showed that impaired glucose homeostasis is associated with SIRT1 expression. Our study of KB-3-1 and BEL7404 cells shows that there is increasing expression of SIRT1 when sensitive cells are incubated with media as glucose decreases from 4.5 g/L to 0.5 g/L (Fig. 4A). These results are consistent with the increased SIRT1 levels found in PC-12 cells cultured with nutrient limitation (19). In addition, the induced overexpression of SIRT1 due to limited glucose incubation is associated with increased resistance to cisplatin (Fig. 4B).
FIGURE 4. SIRT1 expression is induced by low glucose incubation, increasing cellular resistance to cisplatin.

A. KB-3-1 and BEL7404 cells were incubated in medium with various concentrations (4.5, 2, 1 and 0.5 g/L) of glucose for 3 days. Protein extracts were normalized for Western blotting for SIRT1 expression and actin was used as a control. Representative images are shown from experiments performed in triplicate. B. Low glucose incubation increases cellular resistance to cisplatin in parental CP-s KB-3-1 and BEL7404 cells. Cells seeded in plates were treated with cisplatin at various concentrations. Results were expressed as mean (SD). Comparisons of each group were evaluated by one-way analysis of variance (ANOVA). When twice the S.D. was higher than the mean, a non-parameter test was used to evaluate the difference. A significant difference was assumed to exist when P < 0.05. The results shown are the mean of three individual experiments.
Alteration of SIRT1 Expression Mediates Cisplatin Resistance in Cancer Cells
To further measure the effect of SIRT1 expression on cisplatin resistance in cancer cells, we established CP-r cells transfected with a SIRT1 SMART siRNA and CP-s cells transfected with a pCruzHA SIRT1 full-length cDNA. It was found that SIRT1 SMART siRNA duplex transfection efficiently reduced the SIRT1 expression in CP-r KB-CP20 and 7404-CP20 cells compared with the CP-r cells transfected with non-targeting random siRNA used as a control (Figure 5A). There was a 5-fold decrease in SIRT1 expression in BEL 7404-CP20 cells and a 3-fold decrease in KB-CP20 cells. This reduction of SIRT1 expression sensitizes the CP-r KB-CP 20 cells and BEL7404-CP 20 cells approximately 1.5- to 2-fold to cisplatin treatment (Figure 5B). In addition, when SIRT1 was overexpressed in CP-s cells after pCruzHA-SIRT1 full-length cDNA transfection, as measured by Western blotting after 72 hrs (Figure 5C), the parental CP-s KB-3-1 and BEL7404 cells were 3- and 2-fold more resistant, respectively, to cisplatin treatment (Figure 5D). Similarly, PC-12 cells transfected with the full-length WT SIRT1 vector show a 3-fold increase in cisplatin resistance (data not shown). This demonstrates that the alteration of SIRT1 expression can directly mediate cellular resistance to cisplatin in KB-3-1 and BEL7404 cancer cells. However, it is known that there are multiple, simultaneous alterations frequently encountered in CP-r cells. CP-r cells are 20-fold resistant to cisplatin compared to CP-s cells. Altered expression of SIRT1, one of the variations that affect cellular resistance to cisplatin, appears to circumvent cisplatin resistance 10 to 20% in these carefully designed models.
FIGURE 5. Alteration of SIRT1-mediated cellular resistance to cisplatin.

A. SIRT1 expression was knocked down in CP-r KB-CP20 and BEL7404-CP 20 cells by SIRT1 SMART siRNA transfection. SIRT1 expression was measured by specific anti-SIRT1 antibody, with actin used as a control, by Western blotting. The experiment is representative of three individual experiments. B. Cellular viability was measured in CP-r KB-CP20 and BEL7404-CP 20 cells transfected with SIRT1 SMART siRNA. Cell counting kit-8 was used for cellular viability assay, as described as in Methods and Materials. The data presented represent the mean of at least three experiments. C. SIRT1 expression was increased in CP-s KB-3-1 and BEL7404 cells by pCruzHA SIRT1 full-length cDNA transfection. Actin was used as a control. The image is representative of three experiments. D. Cellular viability was measured in CP-s KB-3-1 and BEL7404 cells transfected with a pCruzHA SIRT1 full-length cDNA. The data presented represent the mean of at least three experiments.
Discussion
Cellular resistance to cisplatin is multifactorial and may consist of mechanisms that limit cisplatin uptake and accumulation, altered repair mechanisms, and changes that promote cell survival (20). Our previous study and the work of others has shown that altered uptake of cisplatin occurs because of reduced membrane-binding/transport proteins and reduced endocytosis (9, 21). In this work, we show that SIRT1 upregulation also contributes to cisplatin resistance in association with altered mitochondrial metabolism.
It is known that cells derive up to 95% of their energy through oxidative phosphorylation carried out in mitochondria. Subtle alterations in the glycolytic metabolic pathway could affect energy biosynthetic reactions and cellular susceptibility in the face of stress. Harper et al. identified and characterized a cellular metabolic strategy that differentiates drug-resistant cells from drug-sensitive cells (22). According to this strategy, drug-resistant cells use nonglucose carbon sources (fatty acids) for mitochondrial oxygen consumption when glucose becomes limiting, and mitochondria become dysfunctional in resistant cells. Abnormal mitochondrial structure may have substantial impact on cellular metabolism, and especially on mitochondrial metabolism, which plays an important role in stress resistance, chromatin-dependent gene regulation, and genome stability. Support for mitochondria as a direct target of cisplatin toxicity comes from studies which show that intestinal epithelial IEC-6 rho(0) cells with reduced number of mitochondria are more resistant to cisplatin than normal cells (23). This is consistent with our results, which showed that uptake of 2-deoxyglucose was reduced due to dysfunction and altered morphology of mitochondria in Cp-r cells.
In addition, glucose transporter 1 (GluT1), a common molecular target of most anti-diabetic drugs, is no longer localized to the plasma membrane in CP-r cells (data not shown), which is similar to the recycling defect we have previously reported for a transmembrane protein (multidrug resistant protein 1, MRP1) in malignant CP-r cells (5). GluT1, the mediator of basal glucose uptake, is the primary glucose transporter in human cancer and is expressed by most cancer cells. Reduced 2-deoxyglucose uptake in CP-r cells is likely due to the altered localization of GluT1, the main membrane protein transporting glucose into tumor cells, by a mechanism not yet elucidated. Aft et al. found that overexpression of Glut1 transporter protein was associated with increased glucose uptake in breast cancer cells treated with 2-deoxyglucose (24). The disruption of glucose uptake in CP-r cells is associated with abnormal mitochondrial function, as measured by oxygen consumption and mitochondrial membrane potential. It was found previously that SIRT1 overexpression could reduce oxygen consumption and alter mitochondrial bioenergenesis by acting with PGC-1 in PC-12 cells (19). Given that oxygen consumption is linked to the generation of reactive oxygen species and reactive oxygen species levels correlate with cisplatin toxicity (25), these results may have important implications for how SIRT1 regulates cisplatin resistance.
Limitation of glucose utilization alone results in SIRT1 overexpression in KB-3-1 and BEL7404 cells, and increases the resistance of parental CP-s KB-3-1 and BEL7404 cells to cisplatin. The bioenergetic dysfunction in CP-r KB-CP 20 and BEL7404-CP 20 cells suggests that reducing glucose metabolism also leads to overexpression of SIRT1, which confers cisplatin resistance to KB-3-1 and BEL7404 cells. How might SIRT1 overexpression contribute to cisplatin resistance? Cisplatin perturbs nucleic acid structure and function when it incorporates into DNA or RNA, thereby causing tumor cells to be killed. SIRT1 is known to deacetylate p53 protein, which recognizes and binds to DNA modified with cisplatin (26). It has been proposed that deacetylation by SIRT1 reduces p53-dependent apoptosis in response to DNA damage (27). Interactions between p53 and SIRT1 after platination might provide a molecular link between DNA damage and p53-mediated DNA repair. The cross-resistance of the CP-r cells studied here to numerous cytotoxic agents, including heavy metals, alkylating agents, and methotrexate (5, 9) could reflect such a general effect on apoptosis mediated by SIRT1.
It is well known that SIRT1 is involved in cellular metabolism, and in order to function, SIRT1 requires the presence of NAD+. However, there is an active debate about whether NAD+ directly controls in vivo activity of SIRT1 (28, 29). In this study, we found that NAD+ concentration was higher in CP-r cells that overexpress SIRT1 (Figure 3D). If the cellular NAD+ concentration were low, SIRT1 deacetylase activity could be attenuated, thus increasing the chances of a cell becoming senescent or apoptotic through the acetylated form of p53 (30). Lai and colleagues (31) provide evidence of this, by demonstrating increased acetylation of wild-type p53 during cisplatin-induced apoptosis. In addition, the requirement for NAD+ in the deacetylase activity of SIRT1 suggests that SIRT1 might be involved in metabolism of NAD+ as a metabolic regulator. SIRT1 may function as a bridge, coordinating metabolic status with regulation of key target genes involved in cancer resistance to cisplatin. Thus, higher NAD+ and SIRT1 levels seen in CP-r cells would favor a less effective apoptotic pathway.
Processes that require energy, such as fatty acid synthesis, protein synthesis, and cell growth are curtailed in CP-r cells (9, 10, 32), possibly through the effects of SIRT1 as a central modulator. Overexpressed SIRT1 in CP-r cells might result in a higher threshold for apoptosis by targeting numerous cellular factors. It is possible that SIRT1 acts as a metabolic sensor, via its NAD+ dependence, that links energy consumption to a transcriptional program that modulates response to stress. Deacetylation of transcriptional complexes by SIRT1 is generally associated with diminished transcriptional activation by removing the acetyl group from lysines of certain transcription factors (e.g. NF-κB, FOXO and p53, etc.) (8, 30, 33). Several transcription factors, such as NF-κB, YB-1, mtTFA, Ets-1 and AP-1 are activated by cisplatin treatment, and are involved in drug resistance and DNA repair (34). SIRT1 may act as a scaffold to tether various transcriptional complexes. Brunet et al. (8) found that SIRT1 conferred resistance to etoposide by augmenting DNA repair, and thymocytes derived from SIRT1 knockout mice were found to be more sensitive to ionizing radiation. Therefore, overexpression of SIRT1 might shift the transcription-dependent response of cisplatin resistance away from cell death and toward cell survival.
Cellular mechanisms of resistance to cisplatin are multifactorial and contribute to severe limitation in the use of cisplatin in the clinic. Our research shows that cisplatin resistance reflects a reduced bioenergenesis associated with decreased glucose uptake in CP-r KB-CP20 and BEL7404-CP20 cells. CP-r KB-CP20 cells originally demonstrated 1152-fold more resistance to cisplatin than parental cells. Interestingly, the same cells showed 330-fold more resistance to cisplatin after incubation for 94 days without cisplatin selection pressure, which reduced SIRT1 expression (Shen, DW, Gottesman MM, unpublished data). SIRT1 overexpression allowed the parental CP-s KB-3-1 and BEL7404 cells to survive through limited nutrient incubation, and caused cellular resistance to cisplatin treatment in CP-s cells. This is consistent with the effect of reduced SIRT1 by siRNA, which sensitizes the CP-r cells to cisplatin treatment. Our present data shed light on how SIRT1, regulated by glucose homeostasis, may contribute to the development of tumor cell resistance to cisplatin, and provide insight that may help in the development of treatments to overcome drug resistance in cancer patients.
Materials and Methods
Cell Lines
KB-3-1 (epidermoid adenocarcinoma) and BEL7404 cells (hepatoma cells) were the parental CP-s cells used in this study. KB-CP.5 cells were selected by single-step exposure of parental KB-3-1 cells to 0.5µg/ml cisplatin. KB-CP 20 cells were derived from KB-3-1 cells by stepwise increases in cisplatin concentration up to 20µg/ml, as were BEL7404-CP 20 cells. The cell lines were all grown as monolayer cultures at 37°C in 5% CO2 using DMEM with 10% premium FCS (lot 0S010F; BioWhittaker Inc., CAMBREX Bioproducts, Walkersville, MD), l-glutamine, penicillin (50 units/mol), and streptomycin (50 µg/ml; Quality Biological Inc., Gaithersburg, MD). BEL7404-CP 20 and KB-CP20 cells were maintained in medium containing 5 µg/ml cisplatin for these experiments.
Measurement of 14C-2-deoxyglucose Uptake
The uptake of 14C-2-deoxyglucose by cell monolayers was measured in glucose-free Hanks’ buffered saline solution (HBSS) containing tracer amounts of 14C-2-deoxyglucose. After washing, the monolayers were harvested in 2 ml 0.1N NaOH for measurement of radioactivity by liquid scintillation counting and for protein determination.
Oxygen Consumption of CP-s and CP-r KB and BEL7404 Cells as Detected by Electron Spin Resonance (ESR) Oxymetry
The ESR oxymetry measurement is based on bimolecular collision of O2 with a spin label, causing shorter spin-lattice relaxation time and broadening the line width. Consequently, decreased line width indicates continuous oxygen consumption. In this work, the spin label 4-oxo-2,2,6,6-tetramethylpiperidine-d16-1-15N-oxyl (N-PDT) (Cambridge Isotope Laboratories, Inc., Andover, MA) was used, as the line width of this spin probe is very sensitive to O2 concentration. By measuring line widths obtained in a time span, one can assess respiration of cells positioned in the ESR instrument. For this purpose, an ESR chamber was filled with a suspension of 5 × 105/ ml cells and 0.2 mM N-PDT and the chamber was sealed. Decreasing oxygen concentration due to oxygen consumption by cells results in a decreasing line width of N-PDT in the closed chamber system. ESR spectra were recorded at 2 min intervals for 20 min with a Varian E-109 X-band spectrometer with a variable temperature controller accessory. Signals were obtained with 0.5 mW incident microwave power and with 100 KHz modulation. All ESR spectra were recorded at the low field line of N-PDT and at 37°C.
Measurement of Cellular Sensitivity to Cisplatin
Cellular viability was determined by a sensitive colorimetric assay (Cell Counting Kit-8, CCK-8, Dojindo Molecular Technologies, Gaithersburg, MD). 5000 cells were plated in 96 wells with 100 µl medium containing serially diluted cisplatin (Sigma Co, St. Louis, MO). After incubation for about 3 days, the cellular viability at various concentrations of cisplatin was measured at 450nm absorbance using a microplate reader after 10 µl of the CCK-8 solution added to each well. The IC50 for each cell line was calculated based on the drug concentration that reduces the cell viability to 50% of those in the control, drug-free medium. A relative resistance factor for each cell line was determined by dividing the IC50 value of cisplatin for the cisplatin-resistant cell lines by that for the appropriate cisplatin-sensitive cell line. The values are means of at least triplicate determinations.
Immunoblotting Detection of Protein Expression
1 × 107 cells were harvested at log phase and washed twice with cold PBS. The cells were sedimented by centrifugation at 1,400 × g for 10 min and suspended in ice-cold TD solution buffer (2mM DTT, 1% aprotinin, 1mM AEBSF and DNase) for 5 min on ice. Cells were disrupted by sonication three times. Samples were checked under a phase-contrast microscope and showed more than 80% of cells broken. Protein electrophoresis and immunoblotting with antibodies directed to SIRT1 were performed. Anti-SIRT1 antibody was purchased from Upstate Biotechnology (Lake Placid, NY). Actin was used as protein loading control detected by monoclonal anti-β-actin (Sigma-Aldrich, St Louis, MO). Dr. Samuel W. Cushman kindly provided the Anti-GluT (GluT1, GluT2, GluT3, GluT4, GluT5) polyclonal antibodies. Briefly, the samples were separated by SDS-PAGE on a 4–12% gradient gel and transferred onto PVDF membranes. Subsequently, membranes were subjected to immunostaining with mAbs against human SIRT1 (1:1000), actin (1:10,000), GluTs(1:500) for 1 h at room temperature. Enhanced chemiluminescence reagents were used for developing signals as described by the manufacturer (Pierce Chemical Co., Rockford, IL).
Transfection of SIRT1 SMART siRNA or pCruzHA SIRT1 cDNA into Cells
The SIRT1 SMART siRNA duplex was designed by Dharmacon RNA technologies (Chicago, IL). Diluted CP-r cells in antibiotic-free medium were plated for transfection with SIRT1 SMART siRNA based on the Dharmacon protocol. The siCONTROL non-targeting pool was used as siRNA control, which is chemically modified for increased stability and optimized with no perfect matches to known human, mouse or rat genes. The transfection mixture was prepared by combing the SIRT1 siRNA diluent with GeneSilencer siRNA transfection reagent (Genlantis, San Diego, CA). This mixture was added directly to plated CP-r cells at 60% confluence at transfection time. SIRT1 expression was measured at different time points. Non-targeting random siRNA was used as a control. In addition, a SIRT1 full-length cDNA was cloned into a pCruzHA mammalian expression vector (Santa Cruz Biotechnology, Inc, Santa Cruz, CA). FuGene 6 (Roche Diagnostics, Indianapolis, IN) was used as the transfection reagent for pCruzHA-SIRT1 plasmid transfection into CP-s cells. SIRT1 expression was measured by Western blotting after 72 hrs transfection.
Immunofluorescence Detection of SIRT1 Expression by Confocal Microscopy
Cells were incubated on sterile 18-mm cover glasses for 3 days, then fixed with 3.5% formaldehyde in PBS for 10 min and followed by 0.1% Triton X-100 treatment for 5 min for permeabilization. After washing, cells were treated with 3% BSA in PBS for 30 min and subsequently treated with the primary Anti-SIRT1 antibody for 1 h. After three washings, cells were incubated with Texas red-conjugated affinity purified secondary Ab (1:100 dilution) (Jackson Immuno-research Laboratory). The coverslips with the treated cells were mounted on microscope slides with fluorescence mounting medium containing DAPI for nuclei staining (Dako, Carpinteria, CA). Background fluorescence was determined from cells treated only with the secondary Ab, but otherwise treated the same way as described with the primary Ab. Confocal images were acquired using a Bio-Rad MRC 1024 confocal scan head mounted on a Nikon Optiphot microscope with a 60 × planapochromat lens (Bio-Rad, Hercules, CA).
NAD+ Content Assay
NAD+ concentration was determined by conversion of NAD+ to NADH using the alcohol dehydrogenase reaction as previously described (16).
Statistical Analysis
The experiments were performed in triplicate, and all counts obtained from assays were analyzed, averaged, and expressed as mean ± SD. The two-way ANOVA test was used to compare multiple groups of data, while the unpaired Student’s t-test was used to compare two groups of data. The level of significance, unless otherwise indicated, was P<0.05.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute. We thank George Leiman for his assistance with editing this manuscript and Drs. Susan H. Garfield and Stephen M. Wincovitch for their technical assistance. We also thank Dr. Samuel W. Cushman for technical assistance regarding 2-DG uptake. We acknowledge Dr. Kunio Nagashima for electron microscopy technical assistance. The protocols for NAD+ and NADH measurements were obtained from S.-J. Lin, University of California. We are also thankful for the support of the Chinese Academy of Sciences (CAS) “Hundred Talents Program” (07165111ZX).
Abbreviations
- CP
cis-diamminedichloroplatinum II or cisplatin
- CP-r
cisplatin-resistant
- CP-s
cisplatin-sensitive
- SIRT1
silent information regulator two ortholog 1
References
- 1.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson ER, Lippard SJ. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 3.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 4.Shen DW, Su A, Liang XJ, Pai-Panandiker A, Gottesman MM. Reduced expression of small GTPases and hypermethylation of the folate binding protein gene in cisplatin-resistant cells. Br J Cancer. 2004;91:270–276. doi: 10.1038/sj.bjc.6601956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang XJ, Shen DW, Garfield S, Gottesman MM. Mislocalization of membrane proteins associated with multidrug resistance in cisplatin-resistant cancer cell lines. Cancer Res. 2003;63:5909–5916. [PubMed] [Google Scholar]
- 6.Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 8.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 9.Shen D, Pastan I, Gottesman MM. Cross-resistance to methotrexate and metals in human cisplatin-resistant cell lines results from a pleiotropic defect in accumulation of these compounds associated with reduced plasma membrane binding proteins. Cancer Res. 1998;58:268–275. [PubMed] [Google Scholar]
- 10.Ying YL, Shen DW, Liang XJ, Gottesman MM. Codominance of cisplatin resistance in somatic cell hybrids. J Cell Physiol. 2003;196:63–69. doi: 10.1002/jcp.10320. [DOI] [PubMed] [Google Scholar]
- 11.Shen DW, Goldenberg S, Pastan I, Gottesman MM. Decreased accumulation of [14C]carboplatin in human cisplatin-resistant cells results from reduced energy-dependent uptake. J Cell Physiol. 2000;183:108–116. doi: 10.1002/(SICI)1097-4652(200004)183:1<108::AID-JCP13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457–10463. doi: 10.1158/0008-5472.CAN-05-1923. [DOI] [PubMed] [Google Scholar]
- 13.Cohen HY, Miller C, Bitterman KJ, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 14.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 15.Chu F, Chou PM, Zheng X, Mirkin BL, Rebbaa A. Control of multidrug resistance gene mdr1 and cancer resistance to chemotherapy by the longevity gene sirt1. Cancer Res. 2005;65:10183–10187. doi: 10.1158/0008-5472.CAN-05-2002. [DOI] [PubMed] [Google Scholar]
- 16.Shen DW, Liang XJ, Gawinowicz MA, Gottesman MM. Identification of cytoskeletal [14C]carboplatin-binding proteins reveals reduced expression and disorganization of actin and filamin in cisplatin-resistant cell lines. Mol Pharmacol. 2004;66:789–793. doi: 10.1124/mol.66.4.. [DOI] [PubMed] [Google Scholar]
- 17.Shen DW, Akiyama S, Schoenlein P, Pastan I, Gottesman MM. Characterisation of high-level cisplatin-resistant cell lines established from a human hepatoma cell line and human KB adenocarcinoma cells: cross-resistance and protein changes. Br J Cancer. 1995;71:676–683. doi: 10.1038/bjc.1995.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 19.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 20.Andrews PA, Howell SB. Cellular pharmacology of cisplatin: perspectives on mechanisms of acquired resistance. Cancer Cells. 1990;2:35–43. [PubMed] [Google Scholar]
- 21.Safaei R, Howell SB. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit Rev Oncol Hemat. 2005;53:13–23. doi: 10.1016/j.critrevonc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Harper ME, Antoniou A, Villalobos-Menuey E, et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002;16:1550–1557. doi: 10.1096/fj.02-0541com. [DOI] [PubMed] [Google Scholar]
- 23.Qian W, Nishikawa M, Haque AM, et al. Mitochondrial density determines the cellular sensitivity to cisplatin-induced cell death. Am J Physiol. 2005;289:C1466–C1475. doi: 10.1152/ajpcell.00265.2005. [DOI] [PubMed] [Google Scholar]
- 24.Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer. 2002;87:805–812. doi: 10.1038/sj.bjc.6600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rybak LP, Whitworth CA. Ototoxicity: therapeutic opportunities. Drug Disc Today. 2005;10:1313–1321. doi: 10.1016/S1359-6446(05)03552-X. [DOI] [PubMed] [Google Scholar]
- 26.Pivonkova H, Brazdova M, Kasparkova J, Brabec V, Fojta M. Recognition of cisplatin-damaged DNA by p53 protein: critical role of the p53 C-terminal domain. Biochem Biophys Res Com. 2006;339:477–484. doi: 10.1016/j.bbrc.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 27.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 29.Anderson RM, Latorre-Esteves M, Neves AR, et al. Yeast life-span extension by calorie restriction is independent of NAD fluctuation. Science. 2003;302:2124–2126. doi: 10.1126/science.1088697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 31.Lai MD, Lin WC, Sun YM, Chang FL. Phosphorylated and hypoacetylated mutant p53 enhances cisplatin-induced apoptosis through caspase-9 pathway in the absence of transcriptional activation or translation. Int J Mol Med. 2005;15:725–734. [PubMed] [Google Scholar]
- 32.Liang XJ, Mukherjee S, Shen DW, Maxfield FR, Gottesman MM. Endocytic recycling compartments altered in cisplatin-resistant cancer cells. Cancer Res. 2006;66:2346–2353. doi: 10.1158/0008-5472.CAN-05-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brabec V, Kasparkova J. Molecular aspects of resistance to antitumor platinum drugs. Drug Resist Update. 2002;5:147–161. doi: 10.1016/s1368-7646(02)00047-x. [DOI] [PubMed] [Google Scholar]