

Abstract
The mammalian target of rapamycin (mTOR) kinase is a key regulator of several cellular functions, including cell growth and differentiation. Because hypothalamic mTOR complex 1 (mTORC1) signaling has been implicated as a target of leptin in the regulation of energy balance, we investigated its role in obesity-induced leptin resistance. In contrast to rats maintained on a low-fat (LF) diet for 3 weeks, rats maintained on a high-fat (HF)-diet had no anorexic response to intracerebroventricular leptin. Western blot analysis revealed that leptin was unable to modulate hypothalamic mTORC1 signaling in the HF group, whereas it significantly induced phosphorylation of both S6 kinase 1 (S6K1) and S6 ribosomal protein (S6) in the LF group. Similar to leptin, the cytokine ciliary neurotrophic factor (CNTF) induces hypophagia and increases signal transduction activator of transcription 3 phosphorylation. However, CNTF and its analog CNTFAx15 activate leptin-like pathways in the hypothalamus, even in leptin-resistant states, including diet-induced obesity. Intracerebroventricular CNTFAx15 decreased 24 h food intake and body weight in rats on HF or LF diets and increased the phosphorylation of hypothalamic S6K1 and S6 in a comparable way in both diets. Importantly, mice lacking the expression of S6K1 (S6K1−/−) did not respond to the anorectic action of either leptin or CNTFAx15, implying a crucial role for S6K1 in modulating the actions of these two cytokines. Finally, exposure to HF diet decreased mTORC1 signaling within the hypothalamus. Overall, these findings point strongly to the possibility that reduced hypothalamic mTORC1 signaling contributes to the development of hyperphagia, weight gain, and leptin resistance during diet-induced obesity.
Keywords: mTORC1, S6K1, hypothalamus, high-fat diet, obesity, leptin resistance
Introduction
In apparent contradiction to the normally accurate matching of caloric intake and expenditure, subjects chronically exposed to a high level of dietary fat become obese because the consumption of such a diet undermines the negative feedback system regulating body weight (Hill et al., 2003). For instance, maintenance on high-saturated-fat diets causes resistance at the level of the CNS to key feedback hormones including leptin and insulin (El-Haschimi et al., 2000; Clegg et al., 2005), which provide information about the status of stored fuel in the form of adipose tissue in the periphery (Seeley and Woods, 2003).
Leptin activates a number of key intracellular signaling pathways that are important to its anorectic effect, including signal transduction activator of transcription 3 (STAT3) (Münzberg and Myers, 2005). Similar to leptin, the cytokine ciliary neurotrophic factor (CNTF) also induces hypophagia by activating STAT3-related signaling (Lambert et al., 2001). However, unlike leptin, CNTF, as well as its analog CNTFAx15, activates leptin-like pathways in the hypothalamus even in leptin-resistant states, including diet-induced obesity (DIO) (Lambert et al., 2001; Blüher et al., 2004).
The mammalian target of rapamycin (mTOR) is an atypical serine/threonine kinase, whose activity affects several physiological functions (Wullschleger et al., 2006) including CNS regulation of energy balance (Cota et al., 2006; Ropelle et al., 2008). mTOR exists in two complexes (mTORC1 and mTORC2), which are distinguished by sensitivity to rapamycin and by the association of the mTOR kinase with specific regulatory proteins (Bhaskar and Hay, 2007). mTORC1 is a nutrient-sensitive complex, which regulates protein synthesis by inducing phosphorylation of ribosomal S6 kinase 1 (S6K1) and translation inhibitor factor 4E-binding protein 1. These proteins, in turn, control downstream targets, such as eIF4B and S6 (Bhaskar and Hay, 2007).
We have found recently that mTORC1 signaling plays a role in the hypothalamic mechanisms that respond to nutrient availability, regulating food intake and body weight (Cota et al., 2006). Moreover, similar to STAT3, mTORC1 is critically involved in mediating leptin-induced anorexia (Cota et al., 2006). A key question is the degree to which mTORC1 activity in the hypothalamus can be altered by exposure to a high-fat (HF) diet and whether changes in the activity of this intracellular signaling cascade might participate in the development of leptin resistance that occurs on an HF diet. In the present series of experiments, we therefore compared the action of leptin to that of CNTF, which is known to circumvent and hence overcome CNS resistance to an HF diet, to gain further insight into mechanistic differences characterizing the ability of these two cytokines to modulate food intake during DIO.
Materials and Methods
Animals
Adult male Long–Evans rats (Harlan), male S6K1−/− mice, and their wild-type (WT) littermates were used for the experiments. Rats and mice were housed individually in standard plastic rodent cages and maintained on a 12 h light/dark cycle. They had ad libitum access to water and pelleted rodent chow (Harlan-Teklad), a high-fat butter diet (Research Diets; 4.54 kcal/g; 40% butter), or a carefully matched low-fat butter diet (LF; Research Diets; 3.81 kcal/g; 9% butter) for differing periods of time. All animal protocols were approved by the University of Cincinnati Institutional Animal Care and Use Committee.
Feeding behavior studies
For all experiments, food intake was recorded at 1, 4, and 24 h, and body weight was measured immediately before and 24 h after the treatments.
Surgery.
Under ketamine and xylazine anesthesia, rats had 22-gauge cannulas implanted in the third cerebral ventricle (Cota et al., 2006). Correct placement of each cannula was confirmed by intra-third-ventricular (i.c.v.) administration of 10 ng angiotensin in 2 μl of 0.9% physiological saline. Rats that failed to drink a minimum of 5 ml of water within 60 min after the administration of angiotensin were removed from the studies.
Drugs.
Drugs were administered through an intracerebroventricular cannula for rats and intraperitoneally for mice (2 ml/100 g of body weight). The CNTF analog CNTFAx15 was kindly provided by Mark W. Sleeman (Regeneron Pharmaceuticals, Tarrytown, NY) and dissolved in PBS (0.1 m, pH 7.4). Leptin (EMD Bioscience) was dissolved in saline when administered intracerebroventricularly, or in PBS when given intraperitoneally.
Intracerebroventricular administration of leptin or CNTFAx15.
Before beginning the studies in HF- and LF-treated rats, two different doses of CNTFAx15 (1.5 and 3.0 μg in 2 μl) were administered just before dark onset to chow-fed rats to find behavioral effects that were comparable with those induced by central leptin administration under the same conditions.
The leptin and CNTFAx15 experiments began after 3 weeks of exposure to the HF or the LF diet. On the day of the experiment, the hoppers with HF or LF were removed from the cages 1 h before the infusion of leptin (10 μg in 2 μl) or saline, which occurred 4 h before dark onset (Air et al., 2002). CNTFAx15 (1.5 μg in 2 μl) or PBS was administered just before dark onset.
Intraperitoneal administration of leptin or CNTFAx15.
S6K1−/− mice (n = 8) and their WT controls (n = 11) were used for the leptin experiment. On the day of the experiment, the hoppers with chow were removed from the cages 1 h before the administration of leptin (5 mg/kg) or PBS, which occurred 4 h before the onset of the dark (Air et al., 2002). Ten S6K1−/− and 14 WT mice were used for the CNTFAx15 experiment. On the day of the study, hoppers were removed 3 h before the administration of the drug. CNTFAx15 (0.6 mg/kg) or PBS was administered just before dark onset.
Body composition analysis
A mouse-specific nuclear echo magnetic resonance imaging whole-body composition analyzer (EchoMedical Systems) was used to assess body fat and lean mass in conscious mice, as described previously (Strader et al., 2004).
Western blot analysis
Rats were killed 1 week after the feeding behavior studies, thus, after 4 weeks of exposure to an HF or LF diet. A new group of rats was killed after 1 d of exposure to an HF or LF diet. Brains were removed 2 h after intracerebroventricular leptin or CNTFAx15 or their respective vehicles. For the preliminary analysis of the effects of intracerebroventricular CNTFAx15 on the mTORC1 pathway, brains were removed 45 min and 2 h after the central administration of the compound in lean, chow-fed rats. After removal, brains were snap frozen, and a wedge of the mediobasal hypothalamus was excised (Cota et al., 2006). Samples were processed as described previously (Cota et al., 2006). Briefly, proteins were separated by electrophoresis and transferred to nitrocellulose membranes. The membranes were then incubated for 1 h at room temperature with 5% fat-free milk in Tris-buffered saline containing Tween 20 (TBS/T). The membranes were then incubated overnight at 4°C with one of the following primary antibodies: phospho-S6K1 (Thr 389; 1:500; Cell Signaling Technology), phospho-S6 (Ser 240/244; 1:1000; Cell Signaling Technology), or phospho-STAT3 (Tyr 705; 1:1000; Cell Signaling Technology). Notably, the phosphorylation status of S6K1 is one of the markers commonly used to evaluate mTORC1 activity in vivo (Bhaskar and Hay, 2007). After washing in TBS/T, the membranes were incubated for 1 h at room temperature with secondary antibody conjugated with horseradish peroxidase (goat anti-rabbit; 1:2000; Cell Signaling Technology). The immunoblotted proteins were detected using enhanced chemiluminescence reagents (Western lightening chemiluminescence reagent plus; PerkinElmer) and the membranes were exposed on radiographic films (Denville Scientific). After protein detection, membranes were stripped with 2-mercaptoethanol and reblotted with rabbit anti β-actin antibody (1:1000; Cell Signaling Technology), rabbit anti S6K1 (1:500; Cell Signaling Technology), and rabbit anti S6 (1:2000; Cell Signaling Technology). The optical densitometry of immunoreactive bands was measured using the National Institutes of Health program Scion Image.
Statistical analysis
All values are reported as means ± SEM. For multiple group designs, the data were analyzed by two-way ANOVA, followed by Tukey's post hoc tests. For designs with only two groups, statistical validity was assessed with unpaired t tests. p values <0.05 denote statistical significance.
Results
During exposure to an HF diet, leptin is unable to modulate either food intake or hypothalamic mTORC1 signaling
We have demonstrated previously that in rats maintained on standard rodent chow, intra-third-ventricular (i.c.v.) administration of leptin activates the mTORC1 signaling, and pharmacological inhibition of mTORC1 blunts the anorectic effect of leptin (Cota et al., 2006). Several studies have reported that, during exposure to HF diets, animals not only gain weight, but also become resistant to the anorectic action of leptin (El-Haschimi et al., 2000; Woods et al., 2003). To determine whether intracerebroventricular administration of leptin is still able to activate the hypothalamic mTORC1 pathway after exposure to an HF diet, rats were maintained on an HF or a carefully matched LF diet, respectively, for a period of 4 weeks. After 3 weeks, rats fed the HF diet weighed significantly more than the LF control group (HF group, 447.0 ± 13.1 g vs LF group, 406.7 ± 8.6 g; p < 0.05). As expected, intracerebroventricular leptin significantly decreased caloric intake and body weight over 24 h in rats consuming the LF diet, whereas rats on the HF diet developed resistance to the central anorectic action of leptin (Fig. 1A,B). Western blot analysis performed on the hypothalami of rats kept for 4 weeks on the HF or LF diet demonstrated that intracerebroventricular leptin was still able to significantly increase phosphorylation of STAT3 during exposure to either diet (Fig. 1C–F). Similarly to what has been reported during consumption of chow (Cota et al., 2006), leptin treatment significantly increased phosphorylation of mTORC1 downstream targets S6K1 and S6 in animals fed the LF diet (Fig. 1C,D). However, in rats fed the HF diet, leptin was unable to modulate the phosphorylation of these proteins (Fig. 1E,F).
Figure 1.
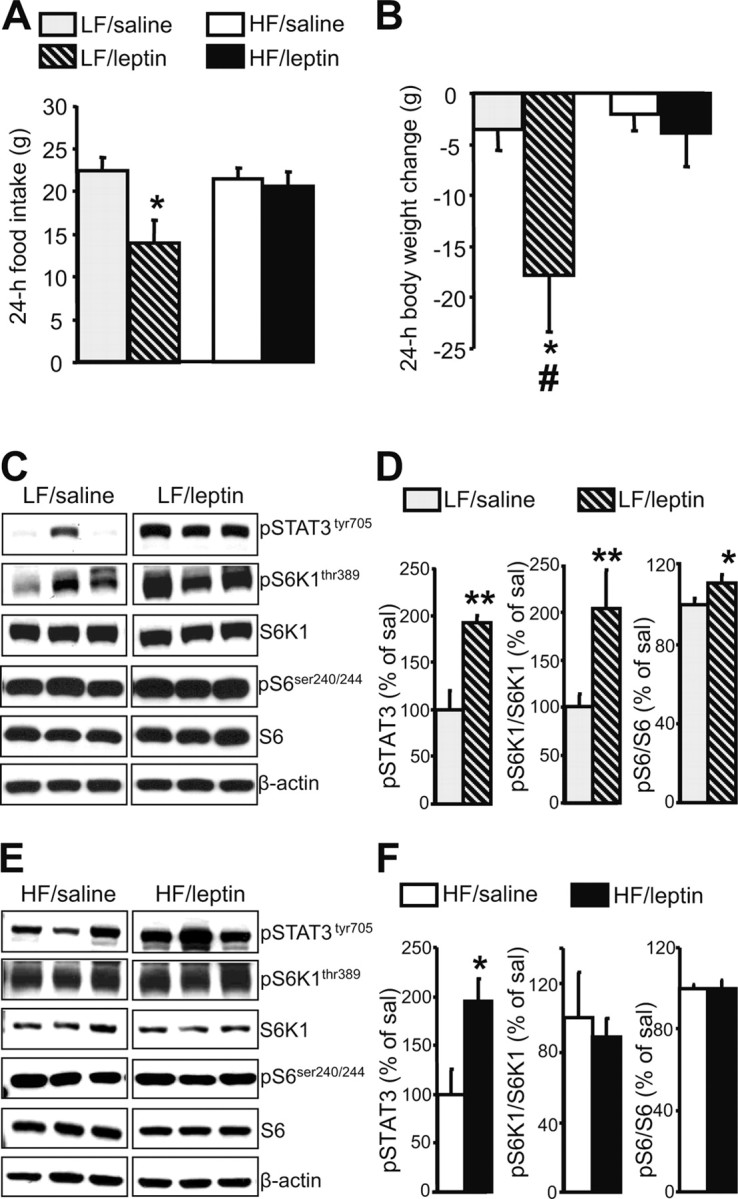
During exposure to an HF diet, leptin is unable to modulate either food intake or hypothalamic mTORC1 signaling. A, B, Effect of intracerebroventricular leptin (10 μg in 2 μl saline) on 24 h food intake (A) and body weight (B) of rats maintained on an HF or LF diet for 3 weeks (mean ± SEM of 4–7 rats used for each treatment group). *p < 0.05 versus saline-treated rats; #p < 0.05 versus HF/leptin-treated rats. C, Intracerebroventricular leptin increases hypothalamic mTORC1 signaling on an LF diet. Representative Western blots from the hypothalami of saline-treated (5) or leptin-treated (5) rats are shown. β-Actin was used as a loading control. D, Quantification by image analysis of hypothalamic STAT3, S6K1, and S6 phosphorylation. Error bars indicate SEM. *p < 0.05, **p < 0.005 versus saline condition. E, Intracerebroventricular leptin does not affect hypothalamic mTORC1 signaling on an HF diet. Representative Western blots from hypothalami of saline-treated (5) or leptin-treated (7) rats are shown. β-Actin was used as a loading control. F, Quantification by image analysis of hypothalamic STAT3, S6K1, and S6 phosphorylation. Error bars indicate SEM. *p < 0.05 versus saline condition.
S6K1−/− mice do not respond to the anorectic action of leptin
To further verify the role of the mTORC1 signaling in modulating the actions of leptin, we used mice lacking S6K1(S6K1−/−) and their WT littermates. This animal model is characterized by smaller body size caused by the lack of functional S6K1 (Shima et al., 1998). S6K1−/− mice used for this study had significantly lower body weight than age-matched WT controls (WT, 24.7 ± 0.9 g vs S6K1−/−, 21.5 ± 0.6 g; p < 0.05). However, body composition analysis revealed that both fat mass and lean mass, when expressed as a percentage of body weight, were comparable between S6K1−/− and WT mice (Fig. 2A). Whereas WT mice decreased 24 h food intake and body weight after intraperitoneal leptin administration, S6K1−/− mice did not (Fig. 2B,C). Moreover, the statistical analysis revealed a significant interaction among the evaluated factors (food intake analysis: genotype × drug, p < 0.05, F = 7.90; time × genotype × drug, p < 0.001, F = 10.19). Thus, these findings support the hypothesis that mTORC1 signaling, and S6K1 in particular, play an important role in leptin-induced anorexia.
Figure 2.
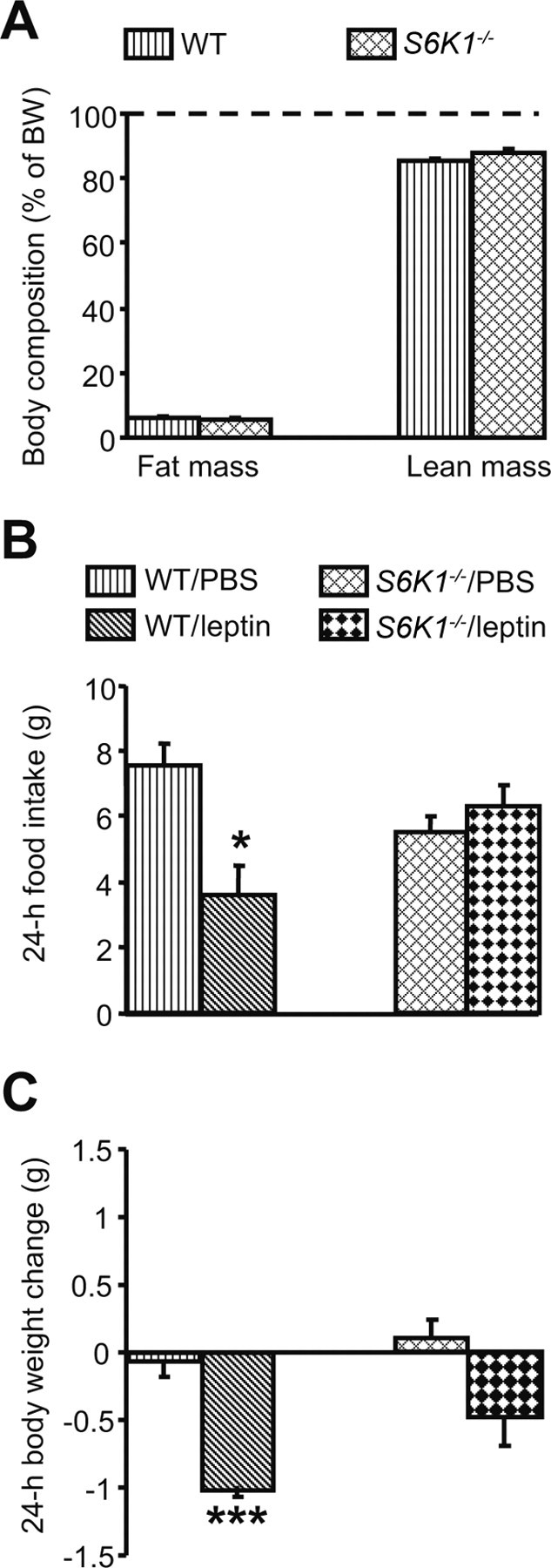
S6K1−/− mice do not respond to the anorectic action of leptin. A, S6K1−/− mice, although smaller, have a body composition comparable with WT mice when expressed as a percentage of body weight. Eight S6K1−/− mice and 11 WT were used. B, C, Leptin (5 mg/kg, i.p.) does not affect either food intake (B) or body weight change (C) in S6K1−/− mice. Four to six mice (mean ± SEM) were used for each treatment group. *p < 0.05, ***p < 0.001 versus WT/PBS condition.
During exposure to an HF diet, CNTFAx15 induces anorexia and increases mTORC1 signaling
CNTF and its analog CNTFAx15 are both capable of potently suppressing food intake by modulating leptin-like pathways in the hypothalamus (Lambert et al., 2001; Blüher et al., 2004). To evaluate the ability of CNTFAx15 to alter phosphorylation of hypothalamic S6K1 and S6, we first determined a dose of CNTFAx15 that, when administered intracerebroventricularly in rats fed a standard chow diet, would reduce food intake and body weight comparably to leptin (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). This dose (1.5 μg in 2 μl) also time-dependently increased phosphorylation of both S6K1 and S6 within the hypothalamus (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). Unlike leptin, CNTFAx15 can also reduce food intake and body weight in DIO (Blüher et al., 2004). Indeed, in rats maintained for 3 weeks on either HF or LF diets, intracerebroventricular CNTFAx15 induced comparable anorexia and body weight loss in both groups (Fig. 3A,B). Whereas the ability of leptin to modulate S6K1 and S6 is void when animals are fed an HF diet, CNTFAx15 induced phosphorylation of STAT3, S6K1, and S6 to the same degree in both diets (Fig. 3C–F).
Figure 3.
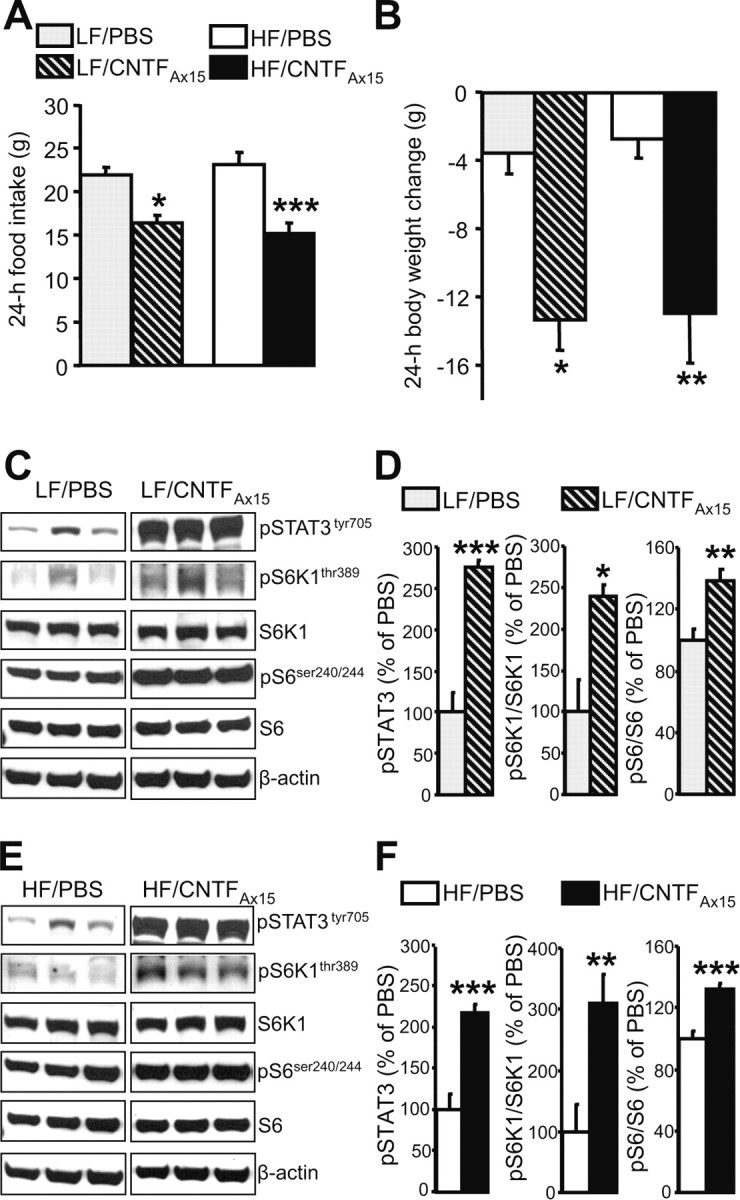
During exposure to an HF diet, CNTFAx15 induces anorexia and increases hypothalamic mTORC1 signaling. A, B, Effect of intracerebroventricular CNTFAx15 (1.5 μg in 2 μl of PBS) on food intake (A) and body weight (B) of rats maintained on an HF or LF diet for 3 weeks. Six to seven rats (mean ± SEM) were used for each treatment group. C, E, Intracerebroventricular CNTFAx15 increases hypothalamic mTORC1 signaling on the LF and HF diets. Representative Western blots from PBS- or CNTFAx15-treated rats are shown. Seven brains were examined in each condition. β-Actin was used as a loading control. D, F, Quantification by image analysis of hypothalamic STAT3, S6K1, and S6 phosphorylation. Error bars indicate SEM. *p < 0.05, **p < 0.005, ***p < 0.001 versus PBS condition.
The ability of CNTFAx15 to reduce food intake depends on functional S6K1
As described previously for leptin, to further determine whether mTORC1 signaling represents an important intracellular target for the effects of CNTFAx15 on energy balance, we tested the ability of CNTFAx15 to reduce food intake in S6K1−/− mice. As expected, the administration of CNTFAx15 significantly decreased 24 h food intake and body weight in WT mice (Fig. 4A,B). Conversely, the anorectic response of S6K1−/− mice was significantly blunted compared with that of WT controls and was associated with a nonsignificant decrease in body weight (Fig. 4A,B). Thus, intact mTORC1 signaling seems to be required for the full ability of CNTFAx15 to reduce food intake.
Figure 4.
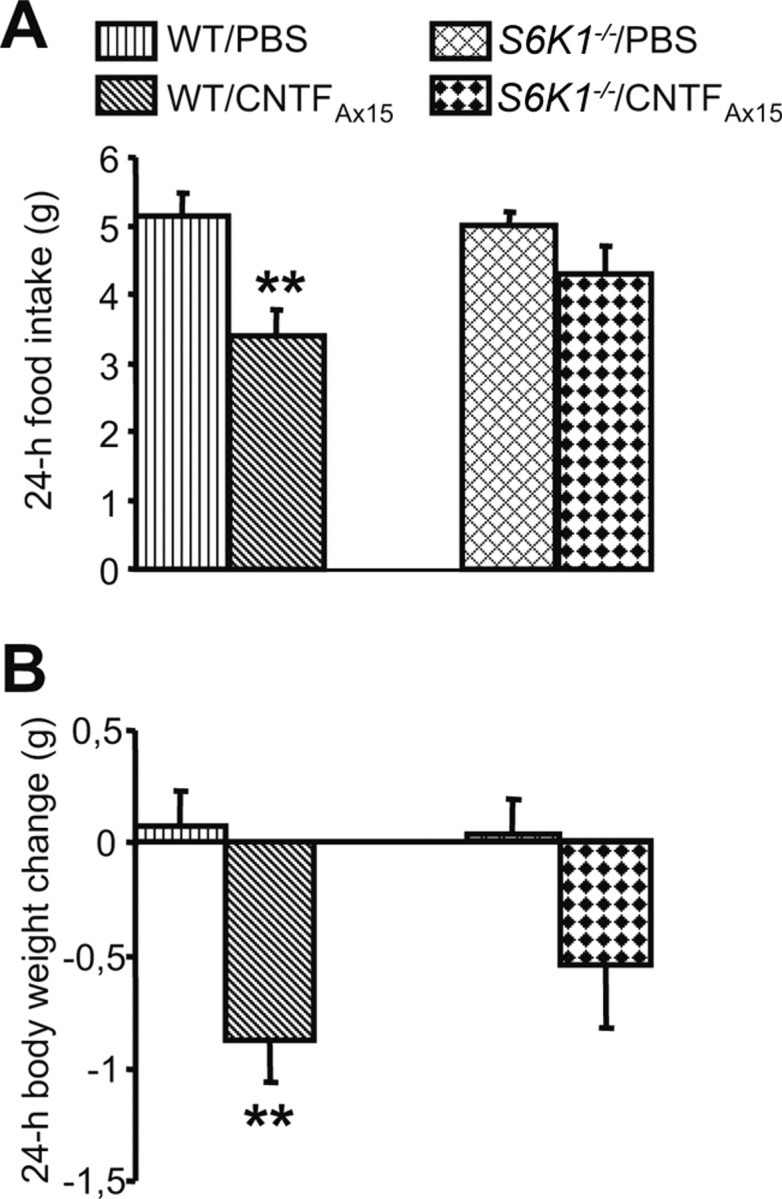
The ability of CNTFAx15 to reduce food intake depends on functional S6K1. A, B, CNTFAx15 (0.6 mg/kg, i.p.) does not affect either food intake (A) or body weight change (B) in S6K1−/− mice. Five to seven mice (mean ± SEM) were used for each treatment group. **p < 0.005 versus WT/PBS condition.
Exposure to an HF diet directly affects CNS mTORC1 signaling
Recent reports have described increased phosphorylation of mTOR and S6K1 in the liver and muscle of diet-induced obese rats (Khamzina et al., 2005). Thus, we investigated whether exposure to an HF diet might also modulate the pathway within the hypothalamus. After 4 weeks on the HF or LF diet, phosphorylation of S6K1 was significantly decreased within the hypothalamus of rats consuming the HF diet, relative to rats on the LF diet (Fig. 5A,B). HF-diet exposure resulted in a small but statistically significant reduction in the phosphorylation of hypothalamic S6 (pS6/S6 LF diet, 100.0 ± 3.9% vs pS6/S6 HF diet, 83.8 ± 4.2%; p < 0.05). Conversely, in rats maintained on an HF diet, extrahypothalamic areas such as the hippocampus were characterized by a significant increase in S6K1 phosphorylation (pS6K/S6K LF diet, 100 ± 24.9% vs pS6K/S6K HF diet, 245.3 ± 19.9%; p < 0.001), although no significant changes occurred in the levels of phosphorylation of S6 (data not shown). In fact, even 1 d exposure to an HF diet, which is associated with a significant increase in caloric intake when compared with intake on the LF diet (1 d consumption: HF diet, 127.41 ± 11 kcal vs LF diet, 93.8 ± 5.5 kcal; p < 0.05), causes a decrease in hypothalamic levels of phosphorylation of S6K1 (Fig. 5C,D) and is associated with a strong trend toward decreased S6 phosphorylation (pS6/S6 1 d LF diet, 100 ± 7.8% vs pS6/S6 1 d HF diet, 83.44 ± 1.7%; p = 0.06).
Figure 5.
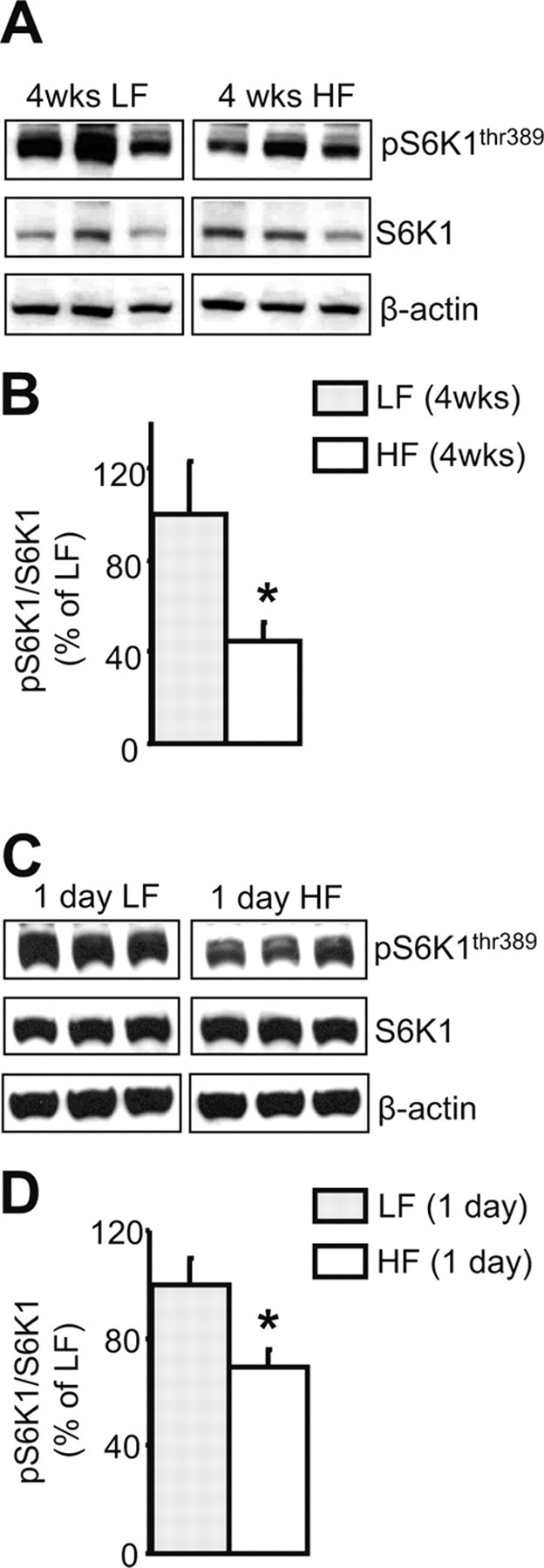
Exposure to an HF diet for either 4 weeks or 1 d dampens hypothalamic S6K1 phosphorylation. A, C, Representative Western blots from animals fed the HF or LF diet. B, D, Quantification by image analysis of hypothalamic S6K1 phosphorylation. Five to seven brains were examined in each condition. β-Actin was used as a loading control. Error bars indicate SEM. *p < 0.05 versus LF.
Discussion
Although genetic factors are known to participate in the propensity of an individual to become obese (Farooqi and O'Rahilly, 2007), diet composition seems to be particularly important in determining metabolic changes that might lead to weight gain, insulin resistance, and development of diabetes and cancer (Popkin, 2007). Obesigenic diets are therefore thought to override CNS regulatory circuits otherwise assuring the maintenance of energy balance. A phenomenon usually associated with DIO is the development of CNS leptin resistance. Although leptin itself might promote the development of leptin resistance (Zhang and Scarpace, 2006), the characterization of the underlying molecular mechanisms and the temporal changes that lead to this event are not yet completely understood.
Here, we confirm that rats on an HF diet become resistant to the central anorectic action of leptin, and that this resistance is associated with the inability of the hormone to increase the activity of hypothalamic mTORC1 signaling. In fact, the ability of leptin to modulate food intake and body weight is dependent on the presence of functional S6K1, because S6K1−/− mice are resistant to the anorectic action of leptin. Noteworthy, after 4 weeks on the HF diet, intracerebroventricular leptin, although not modulating hypothalamic mTORC1 signaling, was still able to induce phosphorylation of STAT3. Leptin-induced STAT3 activation within the hypothalamus was comparable between HF- and LF-fed rats (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This last finding thus seems to suggest that STAT3 and mTORC1 signaling might work in a parallel manner and that the latter changes its level of activity more readily than the former during DIO.
Several studies have identified the hypothalamic signaling through STAT3 as a critical player for the ability of leptin to regulate energy balance (Bates et al., 2003). However, data illustrating the action of leptin in modulating hypothalamic STAT3 on an HF diet have been controversial. Some reports indicate that after extended (several weeks) exposure to an HF diet, the ability of leptin to induce STAT3 phosphorylation is impaired (El-Haschimi et al., 2000; Metlakunta et al., 2008). One study reported that leptin fails to induce STAT3 phosphorylation specifically in the arcuate nucleus of the hypothalamus as soon as 6 d after exposure to an HF diet (Münzberg et al., 2004). However, several other reports have found that short-term exposure to an HF diet resulted in no alteration in the ability of leptin to induce STAT3 phosphorylation (El-Haschimi et al., 2000; Pocai et al., 2005). The present data agree with these last studies. We observed that central leptin administration is able to increase hypothalamic STAT3 phosphorylation after a relatively short period of HF feeding, although, at the same time, it fails to modulate mTORC1 signaling and reduce food intake.
The results from the experiments with CNTFAx15 present an important contrast to those with leptin. Like leptin, CNTFAx15 potently reduces food intake and increases phosphorylation of STAT3, S6K1, and S6 in the hypothalamus of rats fed an LF diet. Like leptin, CNTFAx15 is less effective at reducing food intake in S6K1−/− mice, implicating an important role for mTORC1 signaling in the anorectic effect of CNTFAx15. However, despite these important similarities, one of the interesting things about CNTFAx15 is that its ability to reduce food intake is maintained even under conditions of obesity after exposure to an HF diet, a time when resistance to leptin has developed (Lambert et al., 2001; Blüher et al., 2004). Thus, although CNTF is known to modulate intracellular signaling (namely STAT3), similarly to leptin, its action clearly differs from that of leptin when rats are maintained on an HF diet. We confirmed these observations in the present study. Interestingly, the anorectic effect of CNTFAx15 during DIO is associated with a sustained ability to increase phosphorylation of both S6K1 and S6 within the hypothalamus. Hence, hypothalamic mTORC1 signaling activity, although reduced by the exposure to an HF diet, can still be induced by CNTFAx15 treatment. Conversely, when the mTORC1 signaling is irreversibly shut down (i.e., in S6K1−/− mice), CNTFAx15 is unable to modulate food intake. It is currently unknown how CNTFAx15 might induce hypothalamic mTORC1 signaling. Our findings point to a component of the mTORC1 pathway, or to an as-yet unidentified factor(s) interacting with this signaling cascade, as possible cellular targets whose activity can be directly modulated via administration of CNTFAx15.
In addition to the disparate actions of central leptin and CNTFAx15 on hypothalamic mTORC1 signaling during DIO, exposure to an HF diet dampens S6K1 phosphorylation within the hypothalamus. Given that we can observe this phenomenon in as few as 24 h after exposure to the diet, it would appear not to be secondary to increased body weight or circulating leptin levels, events both associated with prolonged HF feeding. More likely is that there are direct effects of nutrients or nutrient overload which might downregulate the activity of the mTORC1 pathway within the hypothalamus. Particularly relevant is that this is an event restricted to this brain area, because other brain regions such as the hippocampus have increased S6K1 phosphorylation during exposure to an HF diet, similar to what is observed in peripheral tissues of obese rats (Khamzina et al., 2005). Thus, reduced activity of mTORC1 signaling within the hypothalamus points to the unique nature of the pathway and of its regulation within this brain region. How the diet might directly affect mTORC1 signaling in vivo and why these region-specific changes occur within the brain is currently unclear. However, what we have observed is consistent with the recent observation that the activity of hypothalamic phosphoinositide 3-kinase (PI3K), which is upstream from mTOR, is reduced after 4 weeks on an HF diet (Metlakunta et al., 2008). Thus, modulation of hypothalamic mTORC1 signaling possibly happens in conjunction with alteration of other intracellular pathways, such as PI3K (Metlakunta et al., 2008) and AMPK (Martin et al., 2006). Overall, given that hypothalamic activation of the mTORC1 pathway results in decreased food intake and body weight (Cota et al., 2006; Ropelle et al., 2008), the current findings strongly point to the possibility that reduced hypothalamic mTORC1 signaling contributes to the development of hyperphagia, weight gain, and leptin resistance during exposure to an HF diet, thus, further emphasizing the crucial role played by this pathway in the CNS regulation of energy balance as well as in the development of disease.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant DK 17844, NIH–National Institute of Diabetes and Digestive and Kidney Diseases Grant 5P01 DK 56863, and Department of Defense Grant W81XWH-06-2-0019. We thank Drs. Sara C. Kozma and George Thomas for providing the S6K1−/−mice, and Mark W. Sleeman (Regeneron Pharmaceuticals) for providing the CNTFAx15. We are also grateful to Joyce Sorrell for expert technical assistance.
References
- Air EL, Benoit SC, Clegg DJ, Seeley RJ, Woods SC. Insulin and leptin combine additively to reduce food intake and body weight in rats. Endocrinology. 2002;143:2449–2452. doi: 10.1210/endo.143.6.8948. [DOI] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Blüher S, Moschos S, Bullen J, Jr, Kokkotou E, Maratos-Flier E, Wiegand SJ, Sleeman MW, Mantzoros CS. Ciliary neurotrophic factorAx15 alters energy homeostasis, decreases body weight, and improves metabolic control in diet-induced obese and UCP1-DTA mice. Diabetes. 2004;53:2787–2796. doi: 10.2337/diabetes.53.11.2787. [DOI] [PubMed] [Google Scholar]
- Clegg DJ, Benoit SC, Reed JA, Woods SC, Dunn-Meynell A, Levin BE. Reduced anorexic effects of insulin in obesity-prone rats fed a moderate-fat diet. Am J Physiol Regul Integr Comp Physiol. 2005;288:R981–R986. doi: 10.1152/ajpregu.00675.2004. [DOI] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–1832. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, O'Rahilly S. Genetic factors in human obesity. Obes Rev. 2007;8(Suppl 1):37–40. doi: 10.1111/j.1467-789X.2007.00315.x. [DOI] [PubMed] [Google Scholar]
- Hill JO, Wyatt HR, Reed GW, Peters JC. Obesity and the environment: where do we go from here? Science. 2003;299:853–855. doi: 10.1126/science.1079857. [DOI] [PubMed] [Google Scholar]
- Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473–1481. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- Lambert PD, Anderson KD, Sleeman MW, Wong V, Tan J, Hijarunguru A, Corcoran TL, Murray JD, Thabet KE, Yancopoulos GD, Wiegand SJ. Ciliary neurotrophic factor activates leptin-like pathways and reduces body fat, without cachexia or rebound weight gain, even in leptin-resistant obesity. Proc Natl Acad Sci U S A. 2001;98:4652–4657. doi: 10.1073/pnas.061034298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. 2006;281:18933–18941. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- Metlakunta AS, Sahu M, Sahu A. Hypothalamic phosphatidylinositol 3-kinase pathway of leptin signaling is impaired during the development of diet-induced obesity in FVB/N mice. Endocrinology. 2008;149:1121–1128. doi: 10.1210/en.2007-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzberg H, Myers MG., Jr Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. 2005;8:566–570. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Pocai A, Morgan K, Buettner C, Gutierrez-Juarez R, Obici S, Rossetti L. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes. 2005;54:3182–3189. doi: 10.2337/diabetes.54.11.3182. [DOI] [PubMed] [Google Scholar]
- Popkin BM. Understanding global nutrition dynamics as a step towards controlling cancer incidence. Nat Rev Cancer. 2007;7:61–67. doi: 10.1038/nrc2029. [DOI] [PubMed] [Google Scholar]
- Ropelle ER, Pauli JR, Fernandes MF, Rocco SA, Marin RM, Morari J, Souza KK, Dias MM, Gomes-Marcondes MC, Gontijo JA, Franchini KG, Velloso LA, Saad MJ, Carvalheira JB. A central role for neuronal AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in high-protein diet-induced weight loss. Diabetes. 2008;57:594–605. doi: 10.2337/db07-0573. [DOI] [PubMed] [Google Scholar]
- Seeley RJ, Woods SC. Monitoring of stored and available fuel by the CNS: implications for obesity. Nat Neurosci. 2003;4:901–909. doi: 10.1038/nrn1245. [DOI] [PubMed] [Google Scholar]
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strader AD, Reizes O, Woods SC, Benoit SC, Seeley RJ. Mice lacking the syndecan-3 gene are resistant to diet-induced obesity. J Clin Invest. 2004;114:1354–1360. doi: 10.1172/JCI20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, Seeley RJ, Rushing PA, D'Alessio D, Tso P. A controlled high-fat diet induces an obese syndrome in rats. J Nutr. 2003;133:1081–1087. doi: 10.1093/jn/133.4.1081. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Scarpace PJ. The role of leptin in leptin resistance and obesity. Physiol Behav. 2006;88:249–256. doi: 10.1016/j.physbeh.2006.05.038. [DOI] [PubMed] [Google Scholar]