

Abstract
A fundamental question in cancer biology is whether cells with tumorigenic potential are common or rare within human cancers. Studies on diverse cancers, including melanoma, have indicated that only rare human cancer cells (0.1% to 0.0001%) have tumorigenic potential when transplanted into NOD/SCID mice. However, the extent to which NOD/SCID mice underestimate the frequency of tumorigenic human cancer cells has been uncertain. Here we show that modified xenotransplantation assay conditions, including the use of more highly immunocompromised NOD/SCID IL2Rγnull mice, can increase the detection of tumorigenic melanoma cells by several orders-of-magnitude. In limiting dilution assays, approximately 25% of unselected melanoma cells from 12 different patients, including cells from primary and metastatic melanomas obtained directly from patients, formed tumors under these more permissive conditions. In single cell transplants, an average of 27% of unselected melanoma cells from four different patients formed tumors. Xenotransplantation assay modifications can therefore dramatically increase the detectable frequency of tumorigenic cells, demonstrating that they are common in some human cancers.
Traditionally, many cancer cells have been considered to have tumorigenic potential even though no assay has yet demonstrated that a high percentage of single human cancer cells can form tumors. In contrast, the cancer stem cell model has suggested that only small subpopulations of cancer cells have tumorigenic potential based on experiments in which human cancer cells were xenotransplanted into NOD/SCID mice. For example, only one in a million (0.0001%) human melanoma cells is tumorigenic in NOD/SCID mice1. Indeed, the vast majority of human cancers have only rare (<0.1%) tumorigenic/leukemogenic cells (also called cancer-initiating cells or cancer stem cells) when transplanted into NOD/SCID or other highly immunocompromised mice1-11. Nonetheless, recent studies of mouse hematopoietic malignancies have raised the question of whether NOD/SCID assays underestimate the frequency of human cancer-initiating cells12-14. Indeed, human leukemias exhibit a modestly higher frequency of leukemogenic cells when assayed in mice that are more highly immunocompromised than NOD/SCID mice15,16, although leukemogenic cells still represent only 1% of cells in one such model17. The critical question is whether optimization of xenotransplantation assays could reveal that some human cancers actually have very common cells with tumorigenic potential despite only having rare tumorigenic cells in NOD/SCID mice.
The question of whether cells with tumorigenic potential are common or rare within human cancers has fundamental implications for therapy. If tumorigenic cells represent small minority populations, as suggested by the evidence supporting the cancer stem cell model, improved anti-cancer therapies may be identified based on the ability to kill these cancer stem cells rather than the bulk population of non-tumorigenic cancer cells18,19. Alternatively, if cells with tumorigenic potential are common it will not be possible to more effectively treat cancer or to better understand cancer biology by focusing on small minority subpopulations.
Melanoma-initiating cells are rare in NOD/SCID mice
Melanoma-initiating (tumorigenic) cells were reported to be rare based on the observation that only 1 in 1,090,000 human metastatic melanoma cells formed tumors within 8 weeks of transplantation into NOD/SCID mice1. To assess this, we transplanted 102 to 107 freshly dissociated melanoma cells obtained directly from 7 patients subcutaneously into NOD/SCID mice (see Suppl. Table 1 for more information on tumors). Palpable tumors were evident in some mice eight weeks after injection of cells from four of seven melanomas (Fig 1a, b). Limiting dilution analysis20 indicated that the average frequency of cells that formed tumors within 8 weeks of transplantation into NOD/SCID mice was 1 in 837,000 (Fig. 1c), confirming the published estimate1. However, most tumors took more than 8 weeks to develop (Fig. 1a). On average, tumors first became palpable after 11.4±3.8 weeks (mean±s.d.), or 14.3±7.6 weeks for tumors that arose from less than 10,000 injected cells. Variability was high, but the average frequency of cells that formed tumors within 32 weeks was 1 in 111,000 (Fig. 1c; p<0.0001). The frequency of melanoma-initiating cells is therefore significantly underestimated when tumor formation is monitored for only 8 weeks.
Figure 1. Only rare human melanoma cells form tumors in NOD/SCID mice.

a, Tumor development after subcutaneous injection of unfractionated primary melanoma cells directly from seven patients into NOD/SCID mice. Dots represent the times after injection at which individual tumors were first palpable and are colored according to cell dose. Crosses are injections that failed to form tumors. Dotted line indicates 8 weeks after injection. b, All tumors were diagnosed as metastatic melanoma by clinical pathology (see Suppl. Table 1 for more information). The tumors that formed in mice (i, arrow) became large, grew quickly once they were palpable, and were histologically similar to the patient tumors from which they were derived. Flow-cytometry demonstrated that the vast majority of tumor cells expressed human HLA (ii; dotted line represents unstained control). Some tumors were highly pigmented (iii) while others contained variable pigmentation (iv) or were amelanotic (scale bar=1cm). H&E stained sections through the same tumors showed pigmented cells (v, vi, see arrows, bars = 25μm). Cytospun cells contained melanin, as indicated by Fontana-Masson staining (vii, viii, arrows, bars = 25 μm), and showed widespread S100ß staining (ix, x), a marker used to diagnose melanoma40. c, Limiting dilution analyses of the frequency of tumorigenic melanoma cells in Fig.1a at 8 weeks or 32 weeks after transplantation (*p<0.0001).
Assay modifications increase tumorigenic cell detection
Some normal human hematopoietic cells engraft more efficiently in NOD/SCID mice lacking the interleukin-2 gamma receptor (NOD/SCID IL2Rγnull) as compared to NOD/SCID mice, due in part to the lack of natural killer (NK) cell activity in NOD/SCID IL2Rγnull mice21-24. NOD/SCID IL2Rγnull mice have also been used to study cancer arising from human cell lines25,26 or human leukemias15,27. We thus compared human melanoma growth in NOD/SCID mice and NOD/SCID IL2Rγnull mice to test whether more tumorigenic cells could be detected in more highly immunocompromised NOD/SCID IL2Rγnull mice. Xenografted melanoma cells (human melanomas grown in NOD/SCID mice) from 5 patients were dissociated, then live human cells were isolated by flow-cytometry (excluding mouse hematopoietic and endothelial cells; Fig. 2a) and transplanted side-by-side into NOD/SCID IL2Rγnull and NOD/SCID mice (Fig. 2b). Tumors grew faster in NOD/SCID IL2Rγnull mice (Fig. 2b and Suppl. Fig. 1), and an increased (p<0.05) frequency of melanoma-initiating cells was observed in NOD/SCID IL2Rγnull mice as compared to NOD/SCID mice in every tumor tested (Suppl. Fig. 2). Two melanoma specimens obtained directly from patients (465 and 481) also exhibited a significantly (p<0.05) higher frequency of melanoma-initiating cells in NOD/SCID IL2Rγnull mice (Suppl. Fig. 2).
Figure 2. Modifications to the xenotransplantation assay reveal that many more human melanoma cells have tumorigenic potential than detected in NOD/SCID mice.
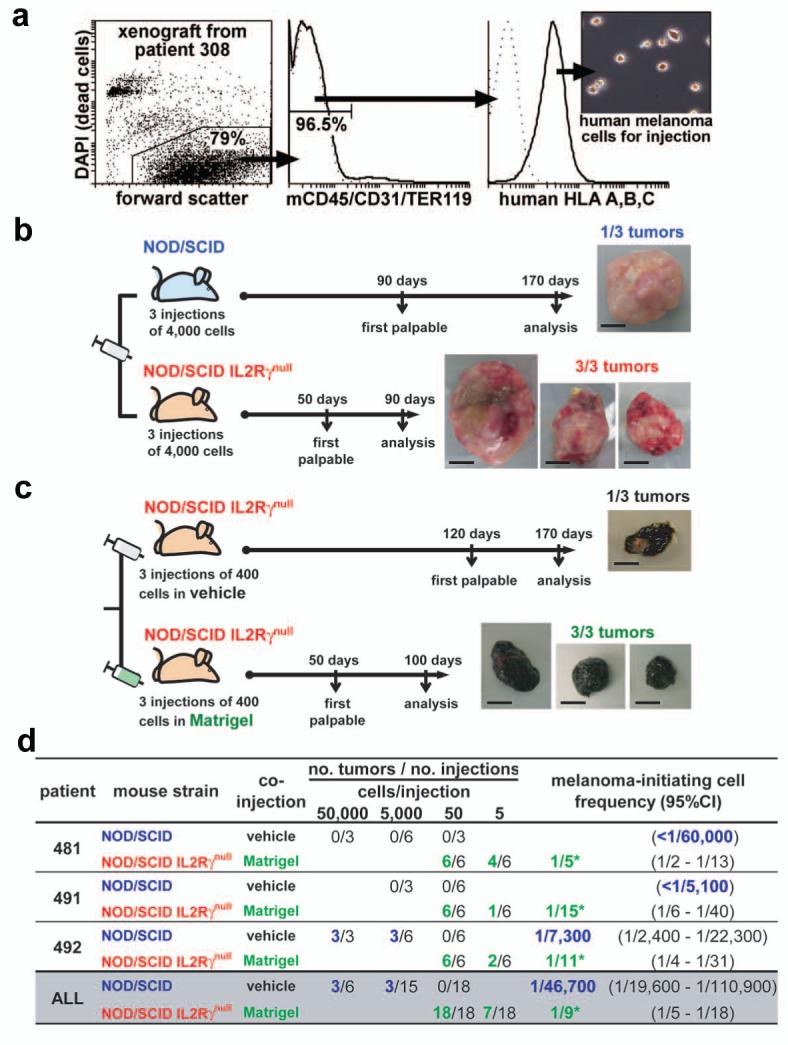
a, Live human melanoma cells were isolated from xenografted tumors by flow-cytometry. After gating to eliminate debris and dead cells, additional gates were drawn to select human HLA+ cells and to exclude mouse hematopoietic (CD45 and TER119) and endothelial (CD31) cells (middle panel). The human HLA+ cells consistently formed tumors upon transplantation into immunocompromised mice while mouse hematopoietic and endothelial cells did not (data not shown). b, Tumor development after side-by-side subcutaneous injections of 4,000 human melanoma cells from a xenograft derived from patient 308 into NOD/SCID or NOD/SCID IL2Rγnull mice. c, Tumor development after side-by-side injections of 400 human melanoma cells from a xenograft derived from patient 205 cells into NOD/SCID IL2Rγnull mice, with or without Matrigel. Photographs in b, and c, show resulting tumors at the time of analysis (bars = 1 cm). Similar experiments conducted with xenografted and non-xenografted tumors from several patients demonstrated that a significantly higher frequency of human melanoma cells formed tumors when injected into NOD/SCID IL2Rγnull mice (Suppl. Fig. 2) or with Matrigel (Suppl. Fig. 4b). The tumors were also palpable earlier and grew faster (Suppl. Figs. 1, 4a). d, Limiting dilution analyses of tumors that arose after 8 weeks in NOD/SCID or NOD/SCID IL2Rγnull mice from the side-by-side transplantation of melanoma cells (derived from xenografts) mixed with vehicle or Matrigel, respectively. In all cases, melanoma-initiating cells were significantly (*p<0.05) more frequent in the modified assay and represented 1 in 5 to 1 in 15 cells. CI: confidence interval.
To test whether NOD/SCID IL2Rγnull mice selected for growth of more aggressive clones, we transplanted cells from tumors grown in NOD/SCID IL2Rγnull mice back into NOD/SCID versus NOD/SCID IL2Rγnull recipients. The frequency of detectable tumorigenic cells in these tumors went back down in NOD/SCID mice (Suppl. Fig. 3a), demonstrating that heritable changes in the frequency of melanoma-initiating cells did not occur as a result of having grown in NOD/SCID IL2Rγnull mice. An increased xenogeneic immune response likely contributes to reduced tumorigenesis by melanoma cells in NOD/SCID mice as compared to NOD/SCID IL2Rγnull mice.
We next tested whether further improvements could be made in melanoma cell engraftment. Coinjection with Matrigel28 increases tumor formation by cancer cell lines29,30 and enhances the engraftment of primary human epithelial cancer cells in immunocompromised mice6,31. To test the effect of Matrigel on the ability to detect tumorigenic melanoma cells, we isolated live human melanoma cells by flow-cytometry (Fig. 2a) from xenografts derived from 3 patients. The same cell preparations were transplanted into NOD/SCID IL2Rγnull mice after mixing with either vehicle (see Methods) or vehicle with Matrigel. Tumor cells injected with Matrigel produced tumors that grew faster than cells injected with vehicle alone (Fig. 2c and Suppl. Fig. 4a), and limiting dilution analysis revealed that in every case more melanoma cells were tumorigenic in Matrigel (Suppl. Fig. 4b).
To quantify the combined effect of the individual assay improvements described above, we performed side-by-side transplantation of flow-cytometrically isolated live melanoma cells obtained from xenografted tumors from three different patients. The same cell preparations were either mixed with vehicle and injected into NOD/SCID mice or mixed with Matrigel and injected into NOD/SCID IL2Rγnull mice. In every case, we observed much higher frequencies of tumorigenic cells in the NOD/SCID IL2Rγnull mice. On average more than 5,000-fold more cells exhibited tumorigenic activity under the modified assay conditions, in which an average of 1 in 9 human melanoma cells formed tumors (Fig. 2d). A somewhat higher frequency of melanoma cells formed tumors in NOD/SCID mice in this experiment as compared to the experiment performed in Figure 1 because the isolation of melanoma cells by flow-cytometry in this experiment improved the ability to eliminate dying cells, debris, hematopoietic cells, and endothelial cells. These results indicate that xenotransplantation assays can be modified to detect much higher than expected frequencies of human cells with tumorigenic potential.
Tumorigenic potential is a common attribute of melanoma cells
To ensure that our modified assay conditions did not somehow confer tumorigenic capacity on human cells, we injected 10,000 primary human melanocytes and/or 100,000 primary human mesenchymal stem cells with Matrigel into NOD/SCID IL2Rγnull mice. These injections did not lead to the formation of tumors (Fig. 3a). The frequency of tumorigenic melanoma cells also did not increase with passaging in mice (Supp. Fig. 4c). Therefore, this xenotransplantation assay does not confer tumorigenic potential on human cells. Consistent with this, serial transplantation experiments demonstrated that the increased tumorigenicity of melanoma cells in NOD/SCID IL2Rγnull mice reflected an effect of the environment, and not a heritable change in the transplanted cells themselves (Suppl. Fig. 3a).
Figure 3. A high percentage of human melanoma cells are tumorigenic but normal human cells are not.
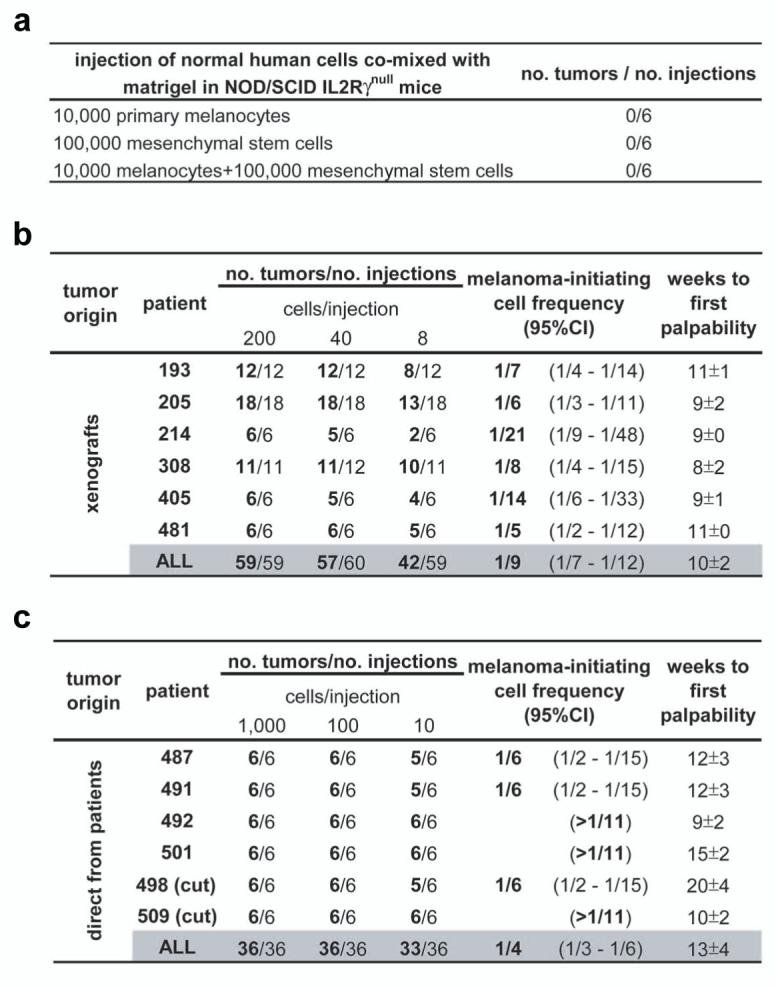
a, Large doses of primary human melanocytes and/or mesenchymal stem cells were mixed with Matrigel and transplanted into NOD/SCID IL2Rγnull mice. No tumors were palpable after 19 weeks. The frequency of melanoma-initiating cells in 12 human tumors obtained either from xenografts (b) or directly from patients (c) was calculated by limiting dilution analysis. Tumors from patients 498 and 509 were primary cutaneous lesions (cut), whereas other tumors were metastases (Suppl. Table 1). In each case, live human melanoma cells were isolated by flow-cytometry, mixed in Matrigel, and injected subcutaneously into NOD/SCID IL2Rγnull mice. 11% of melanoma cells from xenografts and 25% of melanoma cells directly from patients formed tumors within 32 weeks of transplantation. Weeks to palpability (mean±s.d.) is indicated for the lowest dose of cells from each tumor. CI: confidence interval.
To determine whether xenografted melanomas consistently exhibit high frequencies of tumorigenic cells, we tested 5 additional tumor samples in an independent series of experiments, including some tumors that exhibited rare tumorigenic cells in NOD/SCID mice (Fig. 1). In each case, live human melanoma cells were isolated by flow-cytometry (Fig. 2a) and injected with Matrigel into NOD/SCID IL2Rγnull mice. Palpable tumors from the injection of 8 cells were first detected 10±1 weeks after injection. Every tumor exhibited a high frequency of tumorigenic cells (range 1 in 21 to 1 in 5 cells), averaging 1 in 9 cells (Fig. 3b).
To assess whether melanoma cells obtained directly from patients also contain a high frequency of cells with tumorigenic potential we assessed cells obtained from 6 patients. These included four metastatic melanomas and two primary cutaneous melanomas. Live melanoma cells were isolated by flow-cytometry, excluding human hematopoietic (CD45+ or glycophorinA+) and endothelial cells (CD31+), which collectively represented 50±33% of cells in tumors obtained directly from patients. These excluded cells were confirmed to be hematopoietic and endothelial cells rather than melanoma cells by microscopy, and were greatly depleted of tumorigenic activity when transplanted (data not shown). The flow-cytometrically-isolated melanoma cells were mixed with Matrigel and injected into NOD/SCID IL2Rγnull mice. On average, palpable tumors from the injection of 10 cells were first detected 13±4 weeks after injection. Every tumor exhibited a high frequency of tumorigenic cells, with an average of 1 in 4 injected cells forming a tumor based on limiting dilution analysis (Fig. 3c). In tumors obtained directly from patients 492, 501 and 509 (the latter a cutaneous primary tumor) every injection (6/6) of only 10 cells produced a tumor (Fig. 3c).
We have thus performed injections of small numbers of melanoma cells from 12 different patients and have not yet found a tumor that contained rare tumorigenic cells. Tumorigenic cells were common in all tumors, irrespective of whether they were derived from xenografts or directly from patients, and whether they were from primary cutaneous or metastatic melanomas.
Tumorigenesis by single, unselected melanoma cells
To our knowledge, no study has yet demonstrated that a high percentage of single cells from a spontaneously occurring human cancer have the potential to form tumors in vivo. To assess this, we flow-cytometrically sorted live human melanoma cells from xenografted tumors obtained from 4 different patients, then deposited one cell per well in Terasaki plates (well volume 10 μl). Wells were visually confirmed to contain a single cell, then mixed with Matrigel, and injected into NOD/SCID IL2Rγnull mice (Fig. 4a). Twelve to 65% of single cells formed tumors, depending on the patient (Fig. 4b). Overall, 69 tumors (27%) developed from 254 single cell injections. This demonstrates that xenotransplantation assays can be improved to the point that single human melanoma cells can engraft and form tumors in vivo, confirming that cells with tumorigenic potential are common within human melanomas.
Figure 4. Efficient tumor development from the xenotransplantation of single human melanoma cells.

a, Flow-cytometrically isolated human melanoma cells derived from xenografts from 4 patients were diluted into Terasaki microwells such that wells containing single cells could be identified by phase contrast microscopy. In control experiments, the presence of single cells was confirmed by the observation of single nuclei with Acridine Orange staining in 90 out of 90 cases. The single cells were mixed with Matrigel and injected into NOD/SCID IL2Rγnull mice. Tumors arising from the injection of single cells were confirmed to be melanoma by H&E, S100, and HMB45 staining (right panels show sections from a tumor that arose from single cells obtained from patient 214) b, The percentage of single cell injections (69/254=27%) that formed tumors within 20 weeks of transplantation. CI: confidence interval. Weeks to first palpability (mean±s.d.) is indicated for each set of tumors.
Additional modifications to xenotransplantation assays may further improve the detection of human cells able to form tumors, such that 25% could remain an underestimate of the percentage of melanoma cells with tumorigenic potential. Since some melanoma cells are fated to undergo cell death or senescence as a result of deleterious genetic changes or proximity to necrotic areas, it is possible that most melanoma cells that are not fated to undergo cell death or senescence have tumorigenic potential. It also remains possible that some melanomas, particularly early stage primary tumors, may contain less frequent tumorigenic cells than observed in our studies.
Tumorigenic melanoma cells are phenotypically heterogeneous
To assess whether tumorigenic melanoma cells are phenotypically distinct from melanoma cells that fail to form tumors, we examined the expression of more than 50 surface markers on melanomas derived from several patients (Suppl. Table 2). These included markers of other cancer stem cell populations, melanocytes, melanoma, neural crest derivatives, and other cell types. Fifteen of these markers (A2B5, c-kit, CD44, CD49B, CD49D, CD49f, CD54, CD133, CD166, E-cadherin, HNK-1, L1CAM, MCAM, N-cadherin, and p75) were heterogeneously expressed by melanoma cells from multiple patients and were tested for the ability to distinguish tumorigenic from non-tumorigenic melanoma cells in vivo (Suppl. Table 2). In each case, melanoma cells were fractionated by flow-cytometry (except CD133, which was sometimes fractionated using magnetic beads as in prior studies10,32) and cells that expressed different levels of the indicated markers were injected into NOD/SCID IL2Rγnull mice. In every case, tumors arose from all fractions of cells. We found no marker that distinguished tumorigenic from non-tumorigenic cells (Suppl. Table 2). Detailed results are shown for CD49f (a6 integrin, a marker expressed by many different stem cells33) and L1CAM (which is associated with CD133 expression in glioma stem cells34) in Suppl. Fig. 5.
A prior study found that 1 in 120,735 (0.00083%) ABCB5+ metastatic melanoma cells formed tumors in NOD/SCID mice, a 10-fold enrichment over unfractionated cells1. Since ABCB5 expression has been shown to correlate with the expression of CD166 and CD13335, we tested whether CD166 or CD133 could enrich tumorigenic melanoma cells in the modified xenotransplantation assay (Suppl. Fig. 6). The frequency of CD133+ cells in tumors from 12 different patients was consistently lower (almost always lower than 5%) than the frequency of tumorigenic cells in the same tumors (5 to 20%; Suppl. Fig. 6b). Moreover, CD133+ cells were not enriched for tumorigenic melanoma cells. Both CD133+ and CD133- fractions from two different melanomas exhibited very high frequencies of tumorigenic cells (Suppl. Fig. 6c). Flow cytometric analyses indicated that all of the resulting tumors contained both CD133+ and CD133- cells, irrespective of whether they were derived from CD133+ or CD133- cells (data not shown). Both CD166+ and CD166- fractions also contained very high frequencies of tumorigenic melanoma cells (Suppl. Fig. 6f). We have therefore been unable to identify any phenotypic differences that distinguish tumorigenic from non-tumorigenic melanoma cells. These results raise the possibility that markers that enrich rare cells with tumorigenic potential in NOD/SCID mice may fail to distinguish tumorigenic from non-tumorigenic cells in assays that detect much higher frequencies of tumorigenic cells.
Discussion
Our data demonstrate that modifications in xenotransplantation assays can dramatically increase - by several orders-of-magnitude - the detectable frequency of cells with tumorigenic potential. This means that some cancers that appear to have rare tumorigenic cells in NOD/SCID mice actually have very common cells with tumorigenic capacity under other conditions. Other cancers may still have infrequent tumorigenic cells, even when studied under optimised conditions, but the frequency and phenotypic diversity of these cells may be considerably greater than currently thought. Efforts to optimise the xenotransplantation of human cancer cells will be necessary to identify and study the full spectrum of human cancer cells capable of contributing to disease progression.
It is important to note that neither our study nor prior cancer stem cell studies address the question of what cells actually contribute to tumor growth and disease progression in patients. These studies address the potential of cancer cells to proliferate extensively and to form tumors/leukemias, not their actual fate within patients. Depending on how different the tumor environment is within patients as compared to mouse models, it is possible that different cancer cells form tumors in mouse models as compared to human patients. Thus, although we observe a high percentage of melanoma cells that have the potential to proliferate extensively and form new tumors, it is possible that an even greater, or a much smaller, fraction of melanoma cells actually contributes to disease progression in patients.
Although most cancer stem cell studies have detected only rare cells with tumorigenic capacity, it has recently been pointed out that leukemogenic/tumorigenic cells do not necessarily have to be rare for cancers to follow a cancer stem cell model17. Our observation that NOD/SCID xenotransplantation can dramatically underestimate the frequency of tumorigenic cells in at least some human cancers does not necessarily mean that such cancers will not have intrinsically different populations of tumorigenic and non-tumorigenic cells. Having said that, the frequency of tumorigenic cells in human melanoma is much higher than reported for any cancer previously suggested to follow a cancer stem cell model, and we have not yet been able to identify phenotypic differences between melanoma cells that form tumors as compared to those that do not. If markers that distinguish tumorigenic from non-tumorigenic melanoma cells in optimised xenotransplantation assays are identified in future, it will be important to test whether these markers reflect epigenetic differences between cells (as envisioned under the cancer stem cell model) or genetic/environmental differences between cells.
Although cells with tumorigenic potential are likely to be much more frequent in most human cancers than estimated based on NOD/SCID transplantation, we believe the available evidence continues to support the conclusion that some human cancers follow a cancer stem cell model. For example, data from the syngeneic transplantation of mouse AML36 and the transplantation of human AMLs into improved mouse models17, continue to suggest that many AMLs have small, intrinsically distinct subpopulations of AML-initiating cells. Extensive clinical experience with germ cell cancers proves that therapies that eliminate the undifferentiated subset of cancer cells can cure patients, even if differentiated cancer cells are left behind37,38. Nonetheless, careful ptimisation of xenotransplantation assays will likely yield a more nuanced view of cancer in which some cancers contain small subpopulations of cancer-initiating cells, while other cancers contain common tumorigenic cells with little evidence of hierarchical organization. In both cases it will be critical to identify all cancer cells that have the potential to contribute to disease in patients in order to develop more effective therapies.
METHODS SUMMARY
Tumor cell preparation
Melanoma specimens were obtained from patients according to protocols approved by the Institutional Review Board of the University of Michigan Medical School (IRBMED approval # 2004-1058 and 2000-0713). Fresh tumor tissue was mechanically dissociated, enzymatically digested and filtered to obtain a single cell suspension.
Cell labeling and sorting
Cells were stained with directly conjugated antibodies to human CD45, human CD31 and Glycophorin A (to eliminate hematopoietic and endothelial cells from tumors obtained directly from patients) or mouse CD45, mouse CD31, Ter119 and human HLA-A,B,C (to eliminate mouse hematopoietic and endothelial cells and select human cells from xenografted tumors). Cells were resuspended in 10 μg/ml DAPI to label dead cells and sorted on FACSVantage SE or FACSAria flow cytometers. Some samples were labeled with anti-CD133 antibody and separated magnetically using a CD133 Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). When testing other markers, cells were usually labeled with unconjugated primary antibodies (see Supp Table 2) and then with directly-conjugated secondary antibodies prior to staining with the directly conjugated antibodies described above.
Transplantation of human melanoma cells
After sorting, cells were counted and resuspended in staining medium (L15 medium containing 1mg/ml BSA, 1% penicillin/streptomycin and 10mM HEPES (pH7.4)), with or without 25% Matrigel (BD Biosciences). Subcutaneous injections of human melanoma cells were performed in NOD.CB17-Prkdcscid/J (NOD/SCID) and NOD.CB17-Prkdcscid Il2rgtm1Wjl/SzJ (NOD/SCID IL2Rγnull) mice (Jackson Laboratories) according to protocols approved by the Committee on the Use and Care of Animals at the University of Michigan (protocol #9055).
Statistics
Limiting dilution analyses were performed based on Bonnefoix et al39, using the limdil function of the “statmod” package (author G.K. Smyth, http://bioinf.wehi.edu.au/software/limdil/), part of the R statistical software project (http://www.r-project.org). Melanoma-initiating cell frequencies were compared using likelihood ratio tests.
METHODS
Cell preparation
Tumors were mechanically dissociated with a McIlwain tissue chopper (Mickle Laboratory Engineering Co., Guilford, U.K.) prior to sequential enzymatic digestion in 200 U/mL collagenase IV (Worthington, Lakewood, NJ) for 20 min followed by 0.05% trypsin-EGTA for 5 min, both at 37°C. DNase (50-100 U/mL) was added to reduce clumping of cells during digestion. Cells were filtered (40μm cell strainer) to obtain a single cell suspension. Dead cells and debris were reduced by density centrifugation (1.1g/ml Optiprep™, Sigma, St Louis, MO) and/or magnetic bead separation (Dead Cell Removal Kit; Miltenyi Biotec, Auburn, CA) as necessary. Primary human melanocytes41 and mesenchymal stem cells42 were cultured as described.
Cell labeling and sorting
All antibody labeling of cells was performed for 20 min on ice, followed by washing and centrifugation. Secondary antibodies were conjugated to phycoerythrin (goat anti-mouse IgG or IgM, goat anti-rat IgG or goat anti-rabbit IgG; Jackson ImmunoResearch, West Grove, Pennsylvania). Primary isotype controls followed by the same secondary antibodies were used to set background. Cells were subsequently stained with directly conjugated antibodies to human CD45 (HI30-APC, BD Biosciences, San Jose, CA), human CD31 (WM59-APC, eBiosciences, San Diego, CA) and Glycophorin A (HIR2-APC, Biolegend, San Diego, CA) (for tumors obtained directly from patients) or mouse CD45 (30-F11-APC, eBiosciences), mouse CD31 (390-APC, Biolegend), Ter119 (TER-119-APC, eBiosciences) and human HLA-A,B,C (G46-2.6-FITC, BD Biosciences) (for xenograft tumors) to select live human melanoma cells and to exclude endothelial and hematopoietic cells. Cells were resuspended in 10μg/ml DAPI (Sigma) and sorted on a FACSVantage SE-dual laser, three line flow cytometer or a FACSAria Cell Sorter (Becton-Dickinson, San Jose, CA). After sorting, an aliquot of sorted cells was always reanalyzed to check for purity, which was usually >95%. Magnetic cell separation using the CD133 Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) was performed according to the manufacturer’s instructions.
Identification of single melanoma cells
Sorted cells were diluted and aliquoted into 10 μL microwells (Thermo Fisher Scientific, Roskilde, Denmark). Plates were centrifuged at 450xg for 30 seconds and wells containing single cells were identified by phase microscopy. In control experiments, the presence of microscopically-identified single cells was correlated by Acridine Orange staining, with a single nucleus seen in 90/90 cells identified as single cells by phase contrast microscopy. Cell doublets could be identified easily, were rare (7 of 312 wells examined) and were discarded. Wells containing single cells were transferred to a syringe containing Matrigel prior to injection. Cell transfer to the syringe was confirmed by observing the absence of a cell in the well it came from.
Transplantation of melanoma cells
Subcutaneous injections were performed into each flank and the interscapular region of each mouse. Tumor formation was evaluated regularly by palpation of injection sites and tumor diameters were measured with calipers. Mice were monitored for up to 32 weeks after injection. In cases where a tumor became palpable at only one injection site and was causing the mouse distress, that tumor was surgically removed to allow continued evaluation of other injection sites. Tumors were only evident at injection sites and we never observed subcutaneous metastases. To confirm this, we performed a single subcutaneous injection of 200 cells from tumor 205 into each of six NOD/SCID IL2Rγnull mice after mixing in Matrigel and observed only one tumor per mouse at the injection site. The presence of human melanomas was always confirmed at necropsy based on gross examination and marker expression.
Histopathology and immunostaining
Portions of melanoma tumors used in experiments were fixed in 10% neutral buffered formalin, paraffin embedded, sectioned and stained with hematoxylin and eosin for histopathology analysis. Paraffin-embedded tumors were confirmed as melanomas by staining for S100 and HMB45 expression after quenching endogenous peroxidase activity. Binding of anti-S100 antibody (DAKO, Glostrup, Denmark) was performed for 30 min at room temperature, detected by anti-rabbit secondary (30 min at room temperature) and revealed using DAB chromagen. Binding of HMB45 antibody (DAKO) was performed for 30 min at room temperature after antigen retrieval with proteinase K, detected using the M.O.M. immunodetection kit (Vector Laboratories, Burlingame, CA) and revealed using DAB. S100- and HMB45-stained slides were counterstained with hematoxylin. For staining of sorted cells, cells from tumors were cytospun (18xg for 6 min) onto slides after fixation with 4% paraformaldehyde for 5 min at room temperature. For immunofluorescence, slides were rinsed in PBS and blocked in goat serum solution (GSS) (PBS containing 5% goat serum, 1%BSA and 0.3% Triton X-100) for 1 hour to reduce non-specific antibody binding. Incubation with S-100β antibody (Sigma, diluted 1:200 in GSS,) was performed overnight at 4°C, followed by secondary goat anti mouse IgG1 Alexa 488 (Invitrogen, Carlsbad, CA) for 2 hours at room temperature. Slides were counter stained with DAPI for 10 minutes at room temperature, and then mounted in fluorescent mounting solution (DAKO). For detection of melanin pigment, the Fontana Masson method was used43. Slides were microwaved for 1 min in 2.5% Fontana silver nitrate solution (Sigma), prior to rinsing and toning with 0.2% gold chloride for 2 min. After rinsing, slides were incubated in 5% sodium thiosulfate (Sigma) for 2 min, washed and nuclei were stained with DAPI. Slides were mounted in DAKO fluorescent mounting solution.
Statistics
Differences between mean times to tumor palpability were compared using unpaired t-tests. Tumor growth rates were determined by calculating averaged linear regression slopes of the diameters of each tumor at each time point, monitored for at least 10 days, and displayed with dot points representing the mean (±s.d.) of the diameters of all tumors palpable at the indicated time points.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Howard Hughes Medical Institute and by the Allen H. Blondy Research Fellowship. The University of Michigan (UM) Melanoma Bank was supported by a gift from Lewis and Lillian Becker. Flow-cytometry was partially supported by the UM-Comprehensive Cancer NIH CA46592. Thanks to David Adams, Martin White and the UM Flow Cytometry Core Facility for support, Nancy McAnsh and the UM Cancer Centre Histology Core for histological studies, and G.K. Smyth for assistance with statistics and Zalmy Azizan for support with tissue collection. Antibody production was supported in part by the National Institute of Diabetes, Digestive, and Kidney Diseases Grant NIH5P60-DK20572 to the Michigan Diabetes Research and Training Center. Some antibodies were provided by Caltag or by eBioscience to screen for cancer stem cell markers. Human primary melanocyte cultures were provided by Dr. Marisol Soengas. Human mesenchymal stem cells were provided by Drs. Zhuo Wang and Paul Krebsbach. Elsa Quintana was supported by the Spanish Ministry of Education and the Marie Curie Outgoing International Fellowship from the European Commission. Mark Shackleton was supported by the Australian National Heath and Medical Research Council, the Human Frontiers Science Program, and Australia Post.
Footnotes
AUTHOR CONTRIBUTION STATEMENT
Elsa Quintana, Mark Shackleton, and Sean J. Morrison planned the project. E.Q. and M.S. performed the experiments, and analyzed data with S.J.M. Michael S. Sabel and Timothy Johnson consented the patients and surgically obtained many of the melanoma specimens. Douglas Fullen performed all pathology and diagnosed the tumors with T.J. T.J. banked the melanomas, and provided clinical information. E.Q., M.S. and S.J.M. wrote the paper.
REFERENCES
- 1.Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer research. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 3.Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu C, Wei Q, Utomo V, et al. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer research. 2007;67:8216–8222. doi: 10.1158/0008-5472.CAN-07-0999. [DOI] [PubMed] [Google Scholar]
- 5.Wang JC, Lapidot T, Cashman JD, et al. High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase. Blood. 1998;91:2406–2414. [PubMed] [Google Scholar]
- 6.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox CV, Evely RS, Oakhill A, et al. Characterization of acute lymphoblastic leukemia progenitor cells. Blood. 2004;104:2919–2925. doi: 10.1182/blood-2004-03-0901. [DOI] [PubMed] [Google Scholar]
- 8.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 9.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 11.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 12.Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes & development. 2007;21:2283–2287. doi: 10.1101/gad.1588607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly PN, Dakic A, Adams JM, et al. Science. Vol. 317. New York, N.Y: 2007. Tumor growth need not be driven by rare cancer stem cells; p. 337. [DOI] [PubMed] [Google Scholar]
- 14.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer cell. 2006;10:257–268. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 15.Agliano A, Martin-Padura I, Mancuso P, et al. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. International journal of cancer. 2008;123:2222–2227. doi: 10.1002/ijc.23772. [DOI] [PubMed] [Google Scholar]
- 16.Feuring-Buske M, Gerhard B, Cashman J, et al. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia. 2003;17:760–763. doi: 10.1038/sj.leu.2402882. [DOI] [PubMed] [Google Scholar]
- 17.Kennedy JA, Barabe F, Poeppl AG, et al. Comment onTumor growth need not be driven by rare cancer stem cells Science 20073181722; author reply 1722 New York, N.Y: [DOI] [PubMed] [Google Scholar]
- 18.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nature Cancer Reviews. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 19.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Shackleton M, Vaillant F, Simpson KJ, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 21.Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 22.Ito M, Hiramatsu H, Kobayashi K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 23.McKenzie JL, Gan OI, Doedens M, et al. Human short-term repopulating stem cells are efficiently detected following intrafemoral transplantation into NOD/SCID recipients depleted of CD122+ cells. Blood. 2005;106:1259–1261. doi: 10.1182/blood-2005-03-1081. [DOI] [PubMed] [Google Scholar]
- 24.Shultz LD, Banuelos SJ, Leif J, et al. Regulation of human short-term repopulating cell (STRC) engraftment in NOD/SCID mice by host CD122+ cells. Exp Hematol. 2003;31:551–558. doi: 10.1016/s0301-472x(03)00076-6. [DOI] [PubMed] [Google Scholar]
- 25.Hamada K, Monnai M, Kawai K, et al. Liver metastasis models of colon cancer for evaluation of drug efficacy using NOD/Shi-scid IL2Rgammanull (NOG) mice. Int J Oncol. 2008;32:153–159. [PubMed] [Google Scholar]
- 26.Suemizu H, Monnai M, Ohnishi Y, et al. Identification of a key molecular regulator of liver metastasis in human pancreatic carcinoma using a novel quantitative model of metastasis in NOD/SCID/gammacnull (NOG) mice. Int J Oncol. 2007;31:741–751. [PubMed] [Google Scholar]
- 27.Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nature biotechnology. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 28.Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15:378–386. doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Pretlow TG, Delmoro CM, Dilley GG, et al. Transplantation of human prostatic carcinoma into nude mice in Matrigel. Cancer research. 1991;51:3814–3817. [PubMed] [Google Scholar]
- 30.Sweeney TM, Kibbey MC, Zain M, et al. Basement membrane and the SIKVAV laminin-derived peptide promote tumor growth and metastases. Cancer Metastasis Rev. 1991;10:245–254. doi: 10.1007/BF00050795. [DOI] [PubMed] [Google Scholar]
- 31.Fridman R, Kibbey MC, Royce LS, et al. Enhanced tumor growth of both primary and established human and murine tumor cells in athymic mice after coinjection with Matrigel. J Natl Cancer Inst. 1991;83:769–774. doi: 10.1093/jnci/83.11.769. [DOI] [PubMed] [Google Scholar]
- 32.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 33.Iwashita T, Kruger GM, Pardal R, et al. Science. Vol. 301. New York, N.Y: 2003. Hirschsprung disease is linked to defects in neural crest stem cell function; pp. 972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bao S, Wu Q, Li Z, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank NY, Margaryan A, Huang Y, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer research. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 36.Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 37.Horwich A, Shipley J, Huddart R. Testicular germ-cell cancer. Lancet. 2006;367:754–765. doi: 10.1016/S0140-6736(06)68305-0. [DOI] [PubMed] [Google Scholar]
- 38.Kleinsmith LJ, Pierce GB. Multipotentiality of single embryonal carcinoma cells. Cancer research. 1964;24:1544–1551. [PubMed] [Google Scholar]
- 39.Bonnefoix T, Bonnefoix P, Verdiel P, et al. Fitting limiting dilution experiments with generalized linear models results in a test of the single-hit Poisson assumption. J Immunol Methods. 1996;194:113–119. doi: 10.1016/0022-1759(96)00077-4. [DOI] [PubMed] [Google Scholar]
- 40.Nakajima T, Watanabe S, Sato Y, et al. Immunohistochemical demonstration of S100 protein in malignant melanoma and pigmented nevus, and its diagnostic application. Cancer. 1982;50:912–918. doi: 10.1002/1097-0142(19820901)50:5<912::aid-cncr2820500519>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez Y, Verhaegen M, Miller TP, et al. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer research. 2005;65:6294–6304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, Song J, Taichman RS, et al. Stem cells. Vol. 24. Dayton, Ohio: 2006. Ablation of proliferating marrow with 5-fluorouracil allows partial purification of mesenchymal stem cells; pp. 1573–1582. [DOI] [PubMed] [Google Scholar]
- 43.Bancroft JD, Stevens A. Theory and practice of histological techniques. Churchill Livingstone; New York: 1990. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.