

Abstract
A new, highly regio- and stereospecific SN2' substitution reaction between a zirconium oxo complex and allylic chloride has been achieved. The resulting allylic alcohol or TBS-protected allylic ether products were isolated in good to excellent yields with a wide range of E-allylic chlorides. A mechanism for the SN2' allylic substitution consistent with kinetic, stereochemical and secondary isotope effect studies was proposed.
The metal-heteroatom multiple-bonded complexes Cp* 2(pyr)Ti=S (1, Cp*=η5-C5Me5) and Cp2(THF)Zr=NTBS (2, TBS=tert-butyldimethylsilyl) react with E- and Z- allylic chlorides and trimethylsilyl allyl ethers, respectively, to selectively furnish SN2' substitution products.1 Monomeric Group IV oxometal complexes are rare compared with their sulfur and nitrogen analogues, and as a result their reactions with organic substrates remain much less studied.2 The availability of effective synthetic access to Cp* 2(L)Zr=O (where L=pyridine derivative) provides an opportunity to rectify this situation.3 In this communication, we report the reactions of oxozirconium complexes with allylic substrates that exhibit regiochemical behavior substantially different from that seen with the M=NR and M=S systems.1 However, under proper conditions, regio- and stereospecific SN2' conversion of allylic chlorides into TBS-protected allylic alcohols can be achieved.
Experiments between oxo complex Cp* 2(4-tert-butyl-pyridine) Zr=O (3), previously reported by Parkin and coworkers,3 and a variety of allylic functionality revealed that, unlike the systems involving 1 and 2, the regioselectivity of substitution was dramatically affected by the olefin geometry, leaving group and solvent. For example, reaction of 3 with E- and Z-1-bromo-2-hexene in benzene led to a 2:1 and 1:2 mixture of SN2':SN2 zirconium alkoxide products, respectively, while substitution with E-1-iodo-2-hexene proceeded with complete SN2 selectivity. In contrast to our observations with 2, allylic ethers were unreactive. Furthermore, changing the solvent from benzene to methylene chloride in the reaction between 3 and E-1-bromo-2-hexene led to complete SN2 substitution.
To eliminate the possibility of complications resulting from heterogeneity (compound 3 was not completely soluble in the solvents tested in the above experiments), we prepared zirconium oxo complex 4, possessing substantially improved solubility, and examined its reaction with allylic chlorides. We discovered that reaction of 3-chloro-1-octene (5) with 4 proceeded under mild and homogeneous reaction conditions with complete SN2' regioselectivity, and that the initially formed zirconium alkoxide could be efficiently trapped with tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) to furnish TBS ether 6 as a single E-isomer in 92% yield in a single flask (eq 1). 4
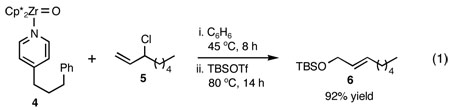
Since SN2' derived product 6 would have been expected to be favored over direct SN2 substitution based on steric effects, we next investigated the reaction of 4 with a variety of primary allylic chloride substrates to determine the scope of the SN2' regioselectivity. Reaction of 4 with E-1-chloro-2-hexene (7), followed by addition of TBSOTf, furnished SN2' derived TBS ether 8 as the sole product in 70% isolated yield (Table 1, entry 1). As we observed in our preliminary experiments with 3, reaction of the corresponding Z-isomer (i.e., Z-1-chloro-2-hexene) with 4 led to a 3.6:1 mixture of SN2':SN2 zirconium alkoxide products. However, variously substituted aliphatic (entries 2–4) and aromatic (entry 5) E-allylic chlorides reacted with 4, followed by TBSOTf, to afford the SN2' products exclusively.
Table 1.
SN2’ Substitution of (E)-Allylic Chlorides with 4
| Entry | Substrate | Conditionsa | Product | Yield |
|---|---|---|---|---|
| 1 | A | 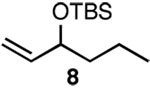 |
70% | |
| 2 | 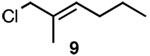 |
B | 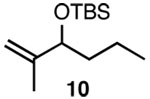 |
87% |
| 3 | 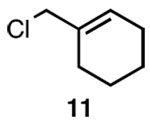 |
C | 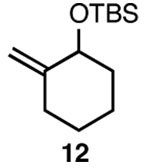 |
92% |
| 4 | 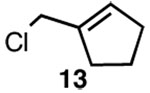 |
C | 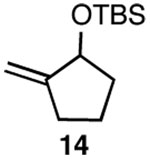 |
59% |
| 5 | 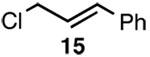 |
D | 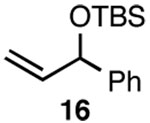 |
76% |
| 6 | A | 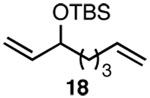 |
86% | |
| 7 | A | 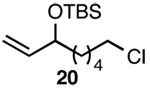 |
88% | |
| 8 | 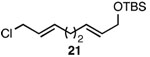 |
A | 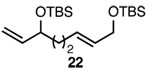 |
89% |
| 9 | 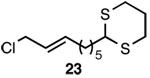 |
A | 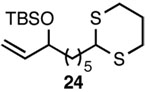 |
84% |
| 10 | E |  |
77% |
Conditions: All reactions run in C6H6 at 0.036 M with 1.3 equiv. 4, unless otherwise noted; A, i. 45 °C, 6 h, ii. 2.0 equiv. TBSOTf, 80 °C, 24 h; B, i. 75 °C, 3 h, ii. 4.0 equiv. TBSOTf, 105 °C, 14 h; C, i. 75 °C, 3 h, 0.014 M, ii. 4.3 equiv. TBSOTf, 105 °C, 14 h; D, i. 45 °C, 8 h, ii. 2.0 equiv. TBSOTf, 105 °C, 14 h; E, i. 45 °C, 6 h, ii. 2.0 equiv. 4-(trifluoromethyl)phenol, rt, 30 min.
In addition to exhibiting complete SN2' regioselectivity with E-allylic chlorides, the substitution reaction employing 4 also demonstrated excellent functional group tolerance. For example, substitution could be effectively executed in the presence of terminal alkene, alkyl chloride, allylic TBS ether, dithiane and dimethyl acetal functionality (Table 1, entries 6–10).5
Motivated to further understand the elementary steps associated with the SN2' reaction, we next initiated a kinetic study by monitoring the homogeneous reaction of oxo complex 27 with 7 by 1H-NMR spectroscopy.6 In the presence of excess 4-trifluoromethylpyridine (4-CF3pyr) and 7, the substitution exhibited pseudo-first order kinetics with no observable intermediates, indicating the overall reaction is first order in 27.7 In addition, the first order rate constant for the reaction (kobs = 1.4 ± 0.1 × 10−3 s−1 at 27 °C) was found to be independent of the initial concentration of 27, while the kobs values obtained in the presence of 7 and various concentrations of 4-CF3pyr established that the overall reaction is inverse first-order in [4-CF3pyr].6 Based on these data, and in analogy to complexes 1 and 2,1 we propose that the SN2' reaction is initiated by rapid and reversible dissociation of the pyridine ligand, followed by rate-limiting C-O bond formation (Scheme 1). Consistent with the rate law predicted by this mechanism, we observed that the substitution reaction exhibited saturation kinetics at high concentrations of 7.6 By measuring kobs at different [4-CF3pyr]/[7] ratios, we were able to extract values for k1 = 8.40 ± 0.01 × 10−4 (s−1) and k−1/k2 = 3.1 ± 0.1 at 10 °C.6
Scheme 1.

Proposed mechanism for substitution
To provide support for rate-limiting C-O bond formation we also conducted competition experiments between E-1-chloro-2-hexene (7) and deuterated analogues 29, 30 and 31 (Scheme 2). As expected based on hybridization changes,8 we observed the averaged secondary isotope effects (kH/kD) of 1.16, 1.055 and 0.571, respectively, shown in Scheme 2.6
Scheme 2.

Secondary kinetic isotope effect studies
Following our kinetic studies, we sought to determine the stereochemical outcome of the SN2' reaction for a chiral allylic chloride. We subjected allylic chloride (–)-35 to reaction with 4, followed by quenching with 4-(trifluoromethyl)phenol, to furnish allylic alcohol (–)-36 in 96% yield (Scheme 3). Importantly, the substitution proceeded with essentially complete syn selectivity.9 Based on this stereochemical outcome, we propose transition state 37 for C-O bond formation. Cyclic transition states such as 37 have previously been postulated to rationalize syn stereochemistry in allylic substitutions,1,10 as well as for the formation of Cp2*Zr(I)(OH) via reaction of Cp2*(pyr)Zr=O with tert-butyl iodide.3c In addition, the unfavorable steric interaction depicted between one of the Cp* ligands and axial substituent of a Z-allylic chloride (see (R) in 37) is consistent with our observation that reaction of Z-1-chloro-2-hexene with 4 was less regioselective than that of the corresponding E-isomer.
Scheme 3.

Substitution of (R)-(–)-35 with 4
In conclusion, we have discovered a new mode of reactivity for zirconium oxo complexes that results in the regio- and stereospecific SN2' substitution of E-allylic chlorides. We have also found that the oxo complexes exhibit excellent substrate scope and functional group compatibility, and that the initially formed zirconium alkoxides could be efficiently trapped with TBSOTf to furnish TBS protected allylic ethers in a single flask. Finally, we have carried out detailed kinetic, isotope labeling and stereochemical experiments that allow us to propose a mechanism for the overall reaction, involving a concerted “closed” transition state for rate-determining C-O bond formation. These results provide insight into the reactivity of zirconium oxo complexes, and may aid in the development of alternative transition metal-mediated SN2' reactions.
Supplementary Material
Experimental procedures and spectral data for products (PDF). This material is available free of charge via the Internet at http://pubs/acs.org.
Acknowledgment
This work was supported by the NIH through grant No. GM-25459 to R.G.B.
References
- 1.(a) Sweeney ZK, Polse JL, Andersen RA, Bergman RG. J. Am. Chem. Soc. 1998;120:7825. [Google Scholar]; (b) Lalic G, Blum SA, Bergman RG. J. Am. Chem. Soc. 2005;127:16790. doi: 10.1021/ja056132f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For the in situ formation and trapping of Cp*2Zr=O and Cp*2Zr=S, see Carney MJ, Walsh PJ, Hollander FJ, Bergman RG. J. Am. Chem. Soc. 1989;111:8751.Carney MJ, Walsh PJ, Bergman RG. J. Am. Chem. Soc. 1990;112:6426.Carney MJ, Walsh PJ, Hollander FJ, Bergman RG. Organometallics. 1992;11:761.
- 3.Howard WA, Waters M, Parkin G. J. Am. Chem. Soc. 1993;115:4917.Howard WA, Parkin G. J. Am. Chem. Soc. 1994;116:606.Howard WA, Trnka TM, Waters M, Parkin G. J. Organomet. Chem. 1997;528:95.. For reactions involving the corresponding titanium analogue, seePolse JL, Andersen RA, Bergman RG. J. Am. Chem. Soc. 1995;117:5393.
- 4.Both 3 and 4 furnished similar SN2':SN2 ratios with all substrates, demonstrating that the SN2' selectivity for allylic chlorides was not the result of the pyridine substituent or homogenicity.
- 5.4-(trifluoromethyl)phenol was substituted for TBSOTf in entry 10 due to incompatibility of TBSOTf with the dimethyl acetal moiety.
- 6.For details of kinetics and kinetic isotope effect experiments, see the Supporting Information.
- 7.4 was replaced with 27 for the kinetic studies since minor amounts of SN2 substitution were detected when the reaction between 4 and 7 was run in the presence of excess 4-(3-phenylpropyl)pyridine and 7. The increased electron-withdrawing nature of 4-(trifluoromethyl)pyridine presumably enhances that rate of ligand dissociation.
- 8.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. Sausalito, CA: University Science Books; 2006. Chapter 8. [Google Scholar]
- 9.The absolute stereochemistry of (-)-36 was verified by Kakisawa-Mosher ester analysis. See the Supporting Information for details.
- 10.Morrill C, Beutner GL, Grubbs RH. J. Org. Chem. 2006;71:7813. doi: 10.1021/jo061436l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for products (PDF). This material is available free of charge via the Internet at http://pubs/acs.org.