

Abstract
In a continuing study of curcumin analogs as potential drug candidates to treat prostate cancer at both androgen-dependent and androgen-refractory stages, we designed and synthesized over 40 new analogs classified into four series: monophenyl analogs (series A), heterocycle-containing analogs (series B), analogs bearing various substituents on the phenyl rings (series C) and analogs with various linkers (series D). These new compounds were tested for cytotoxicity against two human prostate cancer cell lines, androgen-dependent LNCaP and androgen-independent PC-3. Antiandrogenic activity was also evaluated in LNCaP cells and PC-3 cells transfected with wild-type androgen receptor. Ten compounds possessed potent cytotoxicity against both LNCaP and PC-3 cells; seven only against LNCaP; and one solely against PC-3. This study established an advanced structure-activity relationship (SAR), and these correlations will guide the further design of new curcumin analogs with better anti-prostate cancer activity.
Introduction
Prostate cancer is the most common cancer among males of Western countries,2 and is a complex heterogeneous disease that acts differently in different men. The real cause of prostate cancer is still unknown. However, androgen and the androgen receptor (AR) are postulated to play crucial roles in the development of prostate cancer.3 The current treatment for prostate cancer is a combination of surgery, radiation and chemotherapy. The therapeutic agents used clinically include steroidal antiandrogens, such as cyproterone acetate, and nonsteroidal antiandrogens, such as flutamide and bicalutamide. The steroidal antiandrogens possess partial agonistic activity and overlapping effects with other hormonal systems, leading to many complications including severe cardiovascular problems, gynecomastia, loss of libido and erectile dysfunction.4-6 The nonsteroidal antiandrogens show fewer side effects and have improved oral bioavailability; therefore, they are favored over the steroidal antiandrogens. However, antiandrogen withdrawal syndrome has been discovered in patients receiving nonsteroidal antiandrogens for several months.7,8 Long-term drug usage probably leads to mutation of the AR, and the nonsteroidal antiandrogens now exhibit agonistic activity to the mutant AR.9 In addition, the clinically available antiandrogens are unable to kill prostate cancer cells, and within one to three years of drug administration, the cancer usually develops into an androgen-refractory stage, which is not curable. Therefore, it is urgent to develop new classes of anti-prostate cancer drugs.
Prostate cancer incidence is much lower in Asian than in Western countries,10 possibly due to differences in diet. Turmeric is a spice and medicine much more highly consumed in Asian countries such as India, Thailand, China and Japan. Curcumin (1) is the major constituent in the rhizome of Curcuma longa (Zingiberaceae), commonly named turmeric. Over the last few decades, 1 has been studied fairly extensively and has been found to have various biological properties including anti-inflammatory,11 anti-oxidant,12 anti-HIV,13 chemo-preventive,14 and anticancer15 effects in several cell types. In our laboratory, we used 1 as a lead compound to design and synthesize 1-analogs as a new class of potential antiandrogenic agents for the treatment of prostate cancer.16-18 Certain 1-analogs showed antiandrogenic activity in human prostate cancer cells, and two potent antiandrogens previously developed in our laboratory, dimethylated curcumin (DMC, 2) and 4-ethoxycarbonylethyl curcumin (ECECur, 3), are currently under in vivo investigation. Most recently, we found that the curcumin analog 4 possesses potent antiandrogenic activity. From our prior studies, a preliminary antiandrogenic structure-activity relationship (SAR) was established. In a continuing study, we have now prepared four series of new 1-analogs including monophenyl curcumin analogs (series A), heterocycle-containing curcumin analogs (series B), curcumin analogs bearing various substituents on the phenyl rings (series C) and curcumin analogs with various linkers (series D). These new 1-analogs were evaluated in cytotoxicity and antiandrogenic assays in two human prostate cancer cell lines, LNCaP and PC-3. Based on the structures and anti-prostate cancer activities of the new 1-analogs, a more detailed SAR has been formulated. This advanced SAR better reveals the structural features of 1-analogs responsible for the cytotoxic as well as antiandrogenic activities in human prostate cancer cells. This information will guide our optimization of 1-analogs with better pharmacological profiles as potential drug candidates for the treatment of prostate cancer.
Chemistry
The general synthetic scheme for series A is shown in Scheme 1. To avoid the aldol condensation taking place at both terminals of 2,4-pentanedione, excess pentanedione was used in this reaction.19 Boric anhydride was first added to form a complex with 2,4-pentanedione. The aim of this complexation is to protect C-3 from Knoevenagel condensation so that the aldol condensation takes place at the terminal carbon.
Scheme 1.

The general synthetic method for monophenyl curcumin analogs
The synthetic strategies for series B (heterocycle-containing curcumin analogs) and series C (symmetric and asymmetric curcumin analogs with various substituents on the phenyl rings) are shown in Scheme 2 and Scheme 3, respectively. The symmetric compounds were synthesized by using Pederson's method.20 At least two equivalents of the aldehyde are needed to ensure aldol condensation at both terminals of the dione. To prepare asymmetric compounds having different aryl rings, a monoaryl intermediate was first prepared by using the method shown in Scheme 1, and subsequently condensed with an appropriate second aldehyde to give the target compounds.
Scheme 2.

The general synthetic strategies for symmetric curcumin analogs
Scheme 3.

The general synthetic scheme for asymmetric curcumin analogs
The syntheses of series D with various linkers between the two phenyl rings are shown in Schemes 4-9.
Scheme 4.

The general synthetic method of 1, 5-diphenyl-1, 4-pentadien-3-ones
Scheme 9.

The synthesis of 49 and 50
In the general synthesis of 1, 5-diphenyl-1, 4-pentadien-3-ones, one equivalent of acetone and two equivalents of substituted benzaldehyde were treated with 0.25 M NaOH solution and 0.25 equivalent 25% aqueous solution of cetyltrimethylammonium bromide to afford the product (Scheme 4).
Curcumin analogs with an elongated linker were prepared by employing Pederson's synthetic method.20 2,4-Pentanedione or ethyl 4-acetyl-5-oxohexanoate was reacted with 4-hydroxy-3-methoxy-cinnamaldyhyde to give 35 or 36 as the target analog (Scheme 5).
Scheme 5.

The synthesis of curcumin analogs having a long linker
Hydrogenation of DMC in the presence of 10% Pd/C gave the saturated analog 37 as the product (Scheme 6). The partially saturated analog 38 has a hydroxy group in the linker instead of an enol, and was prepared as shown in Scheme 7. 3,4-Dimethoxycinnamone and 3, 4-dimethoxycinnamaldehyde were both prepared from 3,4-dimethoxybenzaldyhyde by condensation with acetone and acetaldehyde, respectively. After treating 3,4-dimethoxycinnamone with LDA at −78 °C, 3,4-dimethoxycinnamoaldehyde was added to provide 38.21
Scheme 6.

The preparation of 37 from DMC
Scheme 7.

The synthesis of 38
To synthesize the imide curcumin analog (39), 3,4-dimethoxy cinnamic acid was first converted to the acid chloride by treatment with thionyl chloride. Then, two equivalents of the acid chloride were reacted with one equivalent of (Me3Si)2NH in the presence of triethylamine to give the target product 39.22 (Scheme 8)
Scheme 8.

The synthesis of imide analog 39
The syntheses of curcumin analogs 4, 40−45, 47 and 48, which have mono- or disubstitution at C-4, have been described and discussed elsewhere.18 The preparation of 49 and 50 is shown in Scheme 9. 4-Methyl DMC (49) was prepared by using the methodology shown in Scheme 2 and then fluorinated by SelectFluor™ under basic conditions to give 4-fluoro-4-methyl DMC (50).23
Results and Discussion
The target compounds were tested for cytotoxicity against two human prostate cancer cell lines, LNCaP and PC-3. The LNCaP cell line is an androgen-dependent human prostate cancer cell line that expresses mutant AR, and the PC-3 cell line is an androgen-independent human prostate cancer cell line that does not express functional AR. The antiandrogenic activity of these 1-analogs was examined in LNCaP cells and PC-3 cells transfected with wild type AR.
The seven monophenyl analogs (5−11) did not exhibit significant cytotoxicity in either LNCaP or PC-3 cells (Table 1A). The absence of one phenyl ring in the curcumin skeleton apparently results in decreased cytotoxicity, indicating that both phenyl rings must be present to retain cytotoxicity against human prostate cancer cells.
Table 1.
Cytotoxicity of 1-analogs against LNCaP and PC-3 human prostate cancer cells.
| A. | |||||||
|---|---|---|---|---|---|---|---|
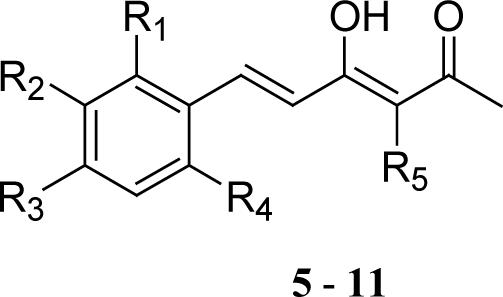 | |||||||
| Cmpd | R1 | R2 | R3 | R4 | R5 | Cytotoxicity IC50(μM)* | |
| PC-3 | LNCaP | ||||||
| 5 | H | OMe | OH | H | H | 8.0 | 6.2 |
| 6 | H | OMe | OMe | H | H | 7 | 6.6 |
| 7 | H | OH | OMe | H | H | 6.1 | 5.3 |
| 8 | OMe | H | OMe | H | H | 12.3 | 9.9 |
| 9 | OMe | OMe | OMe | H | H | 7.8 | 5.8 |
| 10 | H | OMe | OMe | OMe | H | 10.5 | 12.5 |
| 11 | H | OMe | OH | H | (CH2)2COOEt | 43.7 | 51.5 |
| B. | |||
|---|---|---|---|
| Compound | Chemical Structure | Cytotoxicity IC50 (μM)* | |
| LNCaP | PC3 | ||
| 12 | [37]** | [37] | |
| 13 | 7.3 | 9.0 | |
| 14 | 6.3 | 7.7 | |
| 15 | 13.6 | 15.4 | |
| C. | |||||||
|---|---|---|---|---|---|---|---|
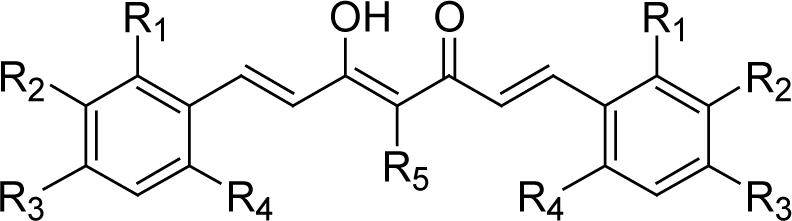 | |||||||
| Compound | R1 | R2 | R3 | R4 | R5 | PC-3 IC50(μM)* | LNCaP IC50(μM) |
| 1 (Curcumin) | H | OMe | OH | H | H | 7.7 | 3.8 |
| 2 (DMC) | H | OMe | OMe | H | H | 1.1 | 1.3 |
| 3 (ECECur) | H | OMe | OH | H | CH2CH2COOEt | 5.1 | 1.5 |
| 16 | H | OH | OMe | H | H | 8.6 | 10.9 |
| 17 | OMe | H | OMe | H | H | [45]** | 11.8 |
| 18 | OMe | OMe | OMe | H | H | 8.1 | 4.8 |
| 19 | H | OMe | OMe | OMe | H | 2.4 | 2.9 |
| 20 | H | OMe | OTHP | H | CH2CH2COOEt | 12.3 | 4.2 |
| 21 | H | OMe | OTHP | H | H | ND*** | ND |
| 22 | H | OMe | OEt | H | H | 20.3 | 6.5 |
| 23 | H | OMe |  |
H | H | >19.8 [41] | 6.1 |
| 24 |  |
14.0 | 2.6 | ||||
| 25 | H | Me | OH | H | H | 4.8 | 1.8 |
| 26 | H | Me | OMe | H | H | >27.4 [44] | 7.7 |
| D. | |||||||
|---|---|---|---|---|---|---|---|
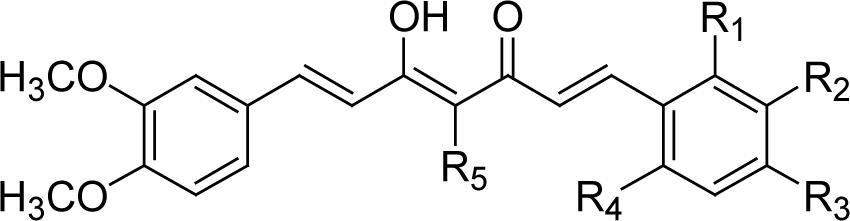 | |||||||
| Cmpd | R1 | R2 | R3 | R4 | R5 | PC-3 IC50(μM)* | LNCaP IC50(μM) |
| 27 | H | OMe | OH | H | H | 6 | 3.3 |
| 28 | H | OH | OMe | H | H | 5.5 | 4.8 |
| 29 | OMe | OMe | OMe | H | H | 9.1 | 7.7 |
| 30 | H | OMe | OMe | OMe | H | 8.2 | 8.6 |
| 31 | H | OMe | OH | H | CH2CH2COOEt | 6.3 | 2.1 |
| E. | ||||
|---|---|---|---|---|
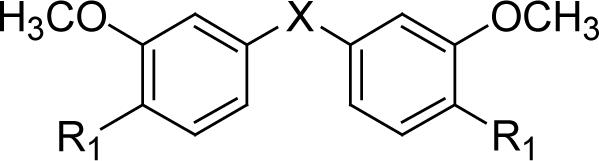 | ||||
| Compound | R1 | X | PC-3 IC50(μM)* | LNCaP IC50(μM) |
| 32 | OH | 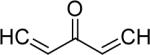 |
2.4 | 1.4 |
| 33 | OMe | 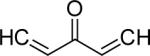 |
3.8 | 3.4 |
| 34 | 18.7 | 21.8 | ||
| 35 | OH | 10.6 | 4.4 | |
| 36 | OH | 6.4 | 3.1 | |
| 37 | OMe | [33]** | 10.8 | |
| 38 | OMe | 18.4 | 17.7 | |
| 39 | OMe | 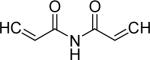 |
>10 [36] | 2.2 |
| 4 | OMe | 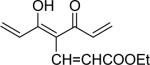 |
1.0 | 0.2 |
| 40 | OTHP | 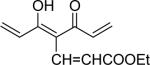 |
3.1 | 2.6 |
| 41 | OH | 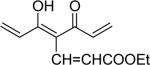 |
11.7 | 7.0 |
| 42 | OMe | 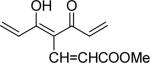 |
0.2 | 0.4 |
| 43 | OMe | 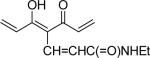 |
1.2 | 0.6 |
| 44 | OMe | 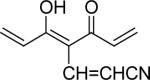 |
0.7 | 0.2 |
| 45 | OMe | 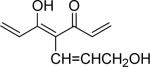 |
1.3 | 0.2 |
| 46 | OH | 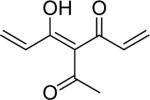 |
23.4 | 8.8 |
| 47 | OTHP | 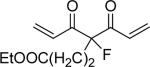 |
6.3 | 7.3 |
| 48 | OH | 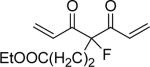 |
9.8 | 6.3 |
| 49 | OMe | 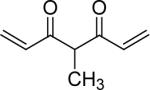 |
3.5 | 5.8 |
| 50 | OMe | 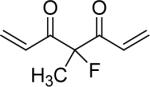 |
4.6 | 6.4 |
IC50 values are mean concentrations that inhibit growth by 50 percent and variation between replicates was less than 5 percent. The cut-off line is 4 μM. Compound with IC50 less than 4 μM is considered to be active.
[]: % inhibition of cell growth at 20 μg/ml
ND, not determined.
In addition, none of the four heterocycle-containing series B analogs (12−15) showed significant cytotoxicity in either LNCaP or PC-3 cells (Table 1.B). These results suggest that replacing the phenyl ring(s) with 5-membered-heterocyle(s) will not improve the desired activity. This information will guide us to design new 1-analogs with the phenyl rings intact.
Series C contains symmetric and asymmetric curcumin analogs with various substituents on the phenyl rings. None of these newly synthesized analogs showed better cytotoxicity than DMC (2), which has IC50 values of 1.1 μM (PC-3) and 1.3 μM (LNCaP) (Table 1.C). Compound 19, which has 3’,4’,6’-dimethoxy substitution on both phenyl rings, was active against both human prostate cancer cell lines. Compounds 24 and 25, which have 3’,5’-dimethoxy-4’-hydroxy and 3’-methyl-4’-hydroxyl substituted phenyl rings, respectively, were active against androgen-dependent LNCaP cells. Among the five asymmetric analogs, 27 and 31 also exhibited activity against LNCaP cells. Both compounds have 3’,4’-dimethoxy substitution on one phenyl ring and 3’-methoxy-4’-hydroxy substitution on the other (Table 1.D). In addition, 31 has an ethoxycarbonylethyl side chain at position C-4 of the linker. Thus, symmetry is not a vital requirement for selective cytotoxic activity. The common features of the phenyl rings of these active compounds are as follows. 1) The C-2’ positions should be unsubstituted. 2) The C-3’ positions should be substituted. However, while methoxy or methyl substituents are favorable, hydroxy groups are not favorable at these positions. 3) The C-4’ positions should also be substituted. The substituents can either be two methoxy groups, two hydroxy groups or one methoxy and one hydroxy. Ethoxy and isoprenyl substituents are not favorable at these positions. 4) Except for 24, most of these synthesized compounds are unsubstituted at the C-5’ position. However, 24 has C-5’ methoxy substituents, and it was active in LNCaP cells. 5) Most of the new analogs have no substitution on the C-6’ positions. An exception is 19, which has methoxy substituents on C-6’, and retained activity in both cell lines. Additional investigation will be needed to probe what functional groups can be accommodated at the C-5’ and C-6’ positions to keep or even enhance the activity of curcumin analogs.
With 3’,4’-dimethoxy or 3’-methoxy-4’-hydroxy substituents on the phenyl rings, compounds having a 1,4-pentadien-3-one linker (e.g. 32 and 33) showed significant cytotoxicity against both human prostate cancer cell lines (Table 1E). However, 34, an analog that also possesses a 1,4-pentadien-3-one linker but has 2,4-dimethoxy substituted phenyl rings, was inactive. These results once again confirm the importance of 3’,4’-disubstitution on the phenyl rings. Curcumin analogs 35 and 36 have elongated linkers (11 carbons) connecting the 3’-methoxy-4’-hydroxy phenyl rings. Compound 35 was not cytotoxic in either prostate cancer cell line, while 36 showed cytotoxicity against LNCaP cells (IC50 3.1 μM), but activity against PC-3 cells was not significant. Overall, compared with 1 and 3, the cytotoxicity of 35 and 36 decreased. Thus, elongation of the linker did not improve the cytotoxic activity of 1-analogs against prostate cancer cells. Compounds 37 (saturated DMC: 3-hydroxy-heptane-5-one linker)and 38 (partially saturated DMC: 3-hydroxy-1,6-heptadien-5-one linker) were not cytotoxic toward prostate cancer cells, suggesting that an unsaturated and conjugated linker, such as the 3-hydroxy-1,4,6-heptatrien-5-one found in 1 and 2, is required for the bioactivity of 1-analogs. When NH replaced the CH2 between the two carbonyl groups in a similar linking group, the resulting imide analog (39) lost cytotoxicity against PC-3 cells but remained cytotoxic against LNCaP cells with an IC50 of 2.2 μM. Compounds with both fluorine and either a propionic ethyl ester or a methyl group at the middle (C-4) carbon (e.g. 47, 48, 50) were not cytotoxic against either tested cell line. However, various unsaturated (acrylic ester, acrylamide, acrylonitrile, allyl alcohol) mono-substitution at C-4 of the linker (e.g. 40, 42−45) resulted in high cytotoxicity against both human prostate cancer cell lines. These analogs were generally more potent than 1 and as potent as 2−4. From the bioassay results of Series D, we can see that it is essential to design a proper linker connecting the substituted phenyl rings in order to obtain potent anti-prostate cancer analogs. The best linkers discovered so far are 3-hydroxy-1,4,6-heptatrien-5-one (the original curcumin linker), 1,4-pentadien-3-one (a shorter linker) and several 4-mono-substituted 3-hydroxy-1,4,6-heptatrien-5-ones that are shown in the structures of ECECur (3), 4, 40 and 42−45.
New 1-analogs also were evaluated in an anti-AR assay in LNCaP cells and PC-3 cells transfected with wild-type AR. Table 2 shows the antiandrogenic activity of the most potent compounds (3, 4, 40 and 44); the remaining analogs exhibited no or weak activity in the antiandrogenic bioassay. All four potent compounds have similar phenyl substitutions, linking groups, and side chains. Because the most cytotoxic compounds (42, 43 and 45) were inactive in the anti-AR bioassay, they likely have mechanisms of action other than interrupting the AR signaling pathway in human prostate cancer cells. Either with or without antiandrogenic activity, these cytotoxic 1-analogs have potential to treat both androgen-dependent prostate cancer and androgen-refractory prostate cancer.
Table 2.
The antiandrogenic activity of compounds 3, 4, 40 and 44.
| DHT (1 nM) + tested compounds | LNCaP transactivation (%) | PC3+wt. AR transactivation (%) |
|---|---|---|
| DHT (Control) | 100 | 100 |
| DHT+ HF (5 μM) | 65 | 31 |
| DHT+ 3 (5 μM) | 4 | 13 |
| DHT+ 4 (5 μM) | 6 | 40 |
| DHT+ 40 (3 μM) | 54 | 46 |
| DHT+ 44 (5 μM) | 42 | 68 |
LNCaP and PC-3 human prostate cell lines were seeded and cotransfected with reporter MMTV-luciferase (both cell lines), wild-type AR expression plasmid (PC-3) using SuperFect. Subsequently, the transfected cells were harvested and re-plated in 10% charcoal-stripped fetal bovine serum DMEM medium. The cells were then treated with dehydrotestosterone (DHT, 1 nM), and tested compounds (3 μM or 5 μM) and harvested for detection of the luciferase activity. (cf. Experimental).
An advanced SAR has been established on the basis of the structure information and bioactivities of the newly synthesized 1-analogs discussed above. The following SAR conclusions were made. 1) Biphenyl rings are required for the cytotoxic and antiandrogenic activities. 2) The C-2’ positions of the phenyl rings should be unsubstituted. 3) The C-3’ and C-4’ positions should be substituted, with 3’,4’-dimethoxy and 3’-methoxy-4’-hydroxy substituents on the phenyl rings found to be most favorable. Substitution at the C-5’ and C-6’ positions probably will not affect the cytotoxicity. 4) The length of the linker impacts the cytotoxicity as well as antiandrogenic activity in human prostate cancer cells. Elongation of the linker results in the loss of cytotoxicity and antiandrogenic activity. 5) An unsaturated and conjugated linker is required for the cytotoxic and antiandrogenic activities. 6) Substitution at the C-4 position of the linker is very crucial for improved anti-AR activity in this compound class. Mono-substitution with proper side chains improved the cytotoxicity significantly.
Conclusions
In conclusion, we synthesized over 40 new 1-analogs and first examined their cytotoxicity against androgen-dependent LNCaP and androgen-independent PC-3 human prostate cancer cell lines. Ten compounds (2, 4, 19, 32, 33, 40 and 42−45) showed significant cytotoxicity against both androgen-dependent LNCaP cells and androgen-independent PC-3 cells. Seven compounds [1 (curcumin), 3 (ECECur), 24, 25, 27, 31 and 39] were cytotoxic only against androgen-dependent LNCaP cells. Only one compound (49) was active against PC-3 cells but inactive against LNCaP cells. The most potent antiandrogenic compounds were 3 (ECECur), 4, 40 and 44. Those analogs that were active in the cytotoxicity assay but inactive in the antiandrogenic assay are presumed to act by a different mechanism other than interrupting the AR signaling pathway. All active compounds are potential drug candidates for the treatment of prostate cancer at the androgen-dependent or androgen-refractory stage. The study of their mechanism(s) of action will be useful in exploring the causes of prostate cancer. In addition, an advanced SAR has been established from this work. This extensive SAR information will guide us to design optimized 1-analogs having better, selective anti-prostate cancer activity.
Experimental Section
Melting points were determined on a Fisher-Johns melting apparatus and are uncorrected. 1H NMR spectra were recorded on a Varian Gemini-300 spectrometer. The chemical shifts are presented in terms of ppm with TMS as the internal reference. MS spectra were recorded on Agilent 1100 series LC/MS trap and API-3000 LC/MS/MS spectrometer. Column chromatography was carried out on CombiFlash® Companion ™ (Isco, Inc.), and thin-layer chromatography was performed on pre-coated silica gel or aluminum plates (Aldrich, Inc.). Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA, and agreed with theoretical values to within ± 0.4%.
Preparation of mono-phenyl analogs 5−11
To a solution of 2,4-pentanedione or 4-acetyl-5-oxo-hexanoate in EtOAc (3 eq) was added boric anhydride (0.7 eq). The solution was stirred at 70 °C for 0.5 h. To the solution, the aldehyde (1 eq) and tributyl borate (1 eq) were added. The mixture was stirred for 30 min. At 85 °C, butylamine (1 eq) dissolved in EtOAc was added dropwise over 15 min. The stirring continued for 1 h at 100 °C. The mixture was then hydrolyzed by adding 1N HCl at 50 °C and stirring for 0.5 h at 50 °C. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed until neutral, dried over anhydrous sodium sulfate. After removing the solvent in vacuo, the crude products were purified by flash column chromatography eluting with a hexane-EtOAc gradient.
4-Hydroxy-6-(4-hydroxy-3-methoxyphenyl)-hexa-3,5-dien-2-one (5)
(from vanillin and 2,4-pentanedione) Yellow powder, 50% yield. mp 146 −147 °C (lit.24 146 −147 °C); ESI MS m/z 235.0 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 2.16 (3H, s), 3.94 (3H, s), 5.63 (H, s), 6.33 (H, d, J= 15.9 Hz), 6.92 (H, d, J= 8.1 Hz), 7.01 (H, d, J= 1.8 Hz), 7.10 (H, dd, J= 8.1 Hz, J= 1.8 Hz), 7.53 (H, d, J=15.9 Hz).
6-(3,4-Dimethoxyphenyl)-4-hydroxyhexa-3,5-dien-2-one (6)
(from 3,4-dimethoxybenzaldehyde and 2,4-pentanedione) 48% yield. mp 78−79 °C; ESI MS m/z 249.0 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 2.10 (3H, s), 3.92 (6H, s), 5.64 (H, s), 6.35 (H, d, J= 16.2 Hz), 6.87 (H, d, = 8.4 Hz), 7.04 (H, d, J= 1.8 Hz), 7.11 (H, dd, J= 8.4 Hz, J= 1.8 Hz), 7.55 (H, d, J=16.2 Hz).
4-Hydroxy-6-(3-hydroxy-4-methoxyphenyl)-hexa-3,5-dien-2-one (7)
(from 3-hydroxy-4-methoxybenzaldehyde and 2,4-pentanedione) 45% yield. mp 163−164 °C (lit.24 160−162 °C); ESI MS m/z 233.2 (M-1)+; 1H NMR (300 MHz, CDCl3): δ 2.16 (3H, s), 3.93 (3H, s), 5.63 (H, s), 6.32 (H, d, J= 15.6 Hz), 6.85 (H, d, J= 8.7 Hz), 7.02 (H, dd, J= 8.7 Hz, J= 2.1 Hz), 7.14 (H, d, J= 2.1 Hz), 7.51 (H, d, J=15.6 Hz).
6-(2,4-Dimethoxyphenyl)-4-hydroxyhexa-3,5-dien-2-one (8)
(from 2,4-dimethoxybenzaldehyde and 2,4-pentanedione) 44% yield. mp 92−93 °C; ESI MS m/z 249.0 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 2.15 (3H, s), 3.85 (3H, s), 3.87 (3H,s), 5.63 (H, s), 6.48 (H, d, J= 15.9 Hz), 6.50 (H, d, J= 2.1 Hz), 6.51 (H, dd, J= 8.1 Hz, J= δ 2.1 Hz), 7.46 (H, d, J= 8.4 Hz), 7.84 (H, d, J=15.9 Hz). Anal. (C14H16O4·1/8 H2O) C, H.
4-Hydroxy-6-(2,3,4-trimethoxyphenyl)-hexa-3,5-dien-2-one (9)
(from 2,3,4-trimethoxybenzaldehyde and 2,4-pentanedione) 45% yield. mp 66−67 °C; ESI MS m/z 279.2 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 2.16 (3H, s), 3.88 (3H, s), 3.90 (3H, s), 3.92 (3H, s), 5.64 (H, s), 6.48 (H, d, J= 15.9 Hz), 6.70 (H, d, J= 8.7 Hz), 7.27 (H, d, J= 8.7 Hz), 7.79 (H, d, J=15.9 Hz). Anal. (C15H18O5) C, H.
4-Hydroxy-6-(2,4,5-trimethoxyphenyl)-hexa-3,5-dien-2-one (10)
(from 2,4,5-trimethoxybenzaldehyde and 2,4-pentanedione) 48% yield. mp 107−108 °C; ESI MS m/z 279.2 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 2.15 (3H, s), 3.88 (3H, s), 3.89 (3H, s), 3.94 (3H, s), 5.66 (H, s), 6.42 (H, d, J= 15.9 Hz), 6.50 (H, s), 7.02 (H, s), 7.89 (H, d, J=15.9 Hz). Anal. (C15H18O5) C, H.
4-Acetyl-5-hydroxy-7-(4-hydroxy-3-methoxyphenyl)-hepta-4,6-dienoic acid ethyl ester (11)
(from vanillin and ethyl 4-acetyl-5-oxohexanoate) 40% yield. Yellow oil; ESI MS m/z 335.0 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 1.25 (3H, t, J= 7.2 Hz), 2.18 (3H, s), 3.96 (3H, s), 4.13 (2H, quart, J= 7.2 Hz), 5.97 (0.5H, s), 6.67 (H, d, J= 15.9 Hz), 6.94 (H, d, J= 8.1 Hz), 7.06 (H, d, J= 1.8 Hz), 7.14 (H, dd, J=1.8 Hz, J= 8.1 Hz), 7.63 (H, d, J=15.9 Hz). Anal. (C18H22O6·7/4 H2O) C, H.
5-Hydroxy-1,7-bis-(5-hydroxymethyl-furan-2-yl)-hepta-1,4,6-trien-3-one (12)
2,4-Pentanedione (0.2 ml, 2 mmol) and boric anhydride (100 mg, 1.4 mmol) were dissolved in 15 ml of EtOAc. The solution was stirred at 70°C for 0.5 h. 5-Hydroxymethyl-2-furaldehyde (506 mg, 4 mmol) and tributyl borate (1.08 ml, 4 mmol) were added. After stirring for 30 min, butylamine (0.3 ml, 3 mmol) dissolved in 4 ml of EtOAc was added dropwise over 15 min. The stirring continued for 5 h at 85 °C. The mixture was then hydrolyzed by adding 8 ml of 1N HCl and stirring for 0.5 h at 60 °C. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed until neutral, and dried over anhydrous sodium sulfate. The solvent was removed in vacuo, and the crude product was purified by CombiFlash® column chromatography eluting with hexane-EtOAc to give 68 mg red powder, obtained in 12% yield. mp 129−130 °C; ESI MS m/z 339.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 4.67 (4H, s), 5.74 (H, s), 6.40 (2H, d, J= 3.3 Hz), 6.53 (2H, d, J= 15.3 Hz), 6.58 (2H, d, J= 3.3 Hz), 7.43 (2H, d, J=15.6 Hz). Anal. (C17H16O6) C, H.
Preparation of heterocycle-containing curcumin analogs 13−15
Compound 5 and boric anhydride (0.7 equiv.) dissolved in EtOAc were stirred at 70 °C for 0.5 h. The appropriate benzaldehyde (1 equiv.) and tributylborate (2 equiv.) were added, and the mixture was stirred for 0.5 h. Piperidine dissolved in EtOAc was added dropwise. After increasing the temperature to 100 °C, stirring was continued for 1 h. The mixture was then hydrolyzed by adding 1N HCl, and stirring at 60 °C for 0.5 h. The organic layer was separated, and the aqueous layer was extracted with EtOAc three times. The combined organic layers were washed with water until neutral. The solvent was removed in vacuo. The crude products were purified by flash column chromatography eluting with hexane-EtOAc.
5-Hydroxy-7-(4-hydroxy-3-methoxyphenyl)-1-(5-hydroxymethyl-furan-2-yl)-hepta-1,4,6-trien-3-one (13)
(from 5-hydroxymethyl-2-furaldehyde) 32% yield. mp 140−142°C; ESI MS m/z 343.3 (M+H)+, 366.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 3.90 (3H, s), 4.64 (2H, s), 5.71 (H, s), 6.36 (H, d, J= 2.7 Hz), 6.43 (H, d, J= 7.5 Hz), 6.47 (H, d, J= 7.2 Hz), 6.53 (2H, d, J= 3.0 Hz), 6.90 (H, d, J= 8.1 Hz), 7.01 (H, s), 7.08 (H, d, J= 7.5 Hz), 7.33 (H, d, = 15.3 Hz), 7.55 (H, d, J=15.6 Hz). Anal. (C19H18O6) C, H.
5-Hydroxy-7-(4-hydroxy-3-methoxyphenyl)-1-thiophen-2-yl-hepta-1,4,6-trien-3-one (14)
(from thiophene-2-carbalydehyde) 38% yield. mp 130−132 °C; ESI MS m/z 327.1 (M-1)+; 1H NMR (300 MHz, CDCl3): δ 3.89 (3H, s), 5.28 (H, s), 5.74 (H, s), 6.37 (H, d, J= 15.6 Hz), 6.45 (H, d, J= 15.9 Hz), 6.91 (H, d, J= 7.8 Hz), 7.01 (H, d, J= 2.4 Hz), 7.02 (H, d, J= 1.2 Hz), 7.04 (H, s), 7.08 (H, dd, J= 1.2 Hz, J= 7.8 Hz), 7.21 (H, d, J= 3.6 Hz), 7.34 (H, d, J= 4.8 Hz), 7.57 (H, d, J=15.6 Hz), 7.73 (H, d, J= 15.3 Hz). Anal. (C18H16O4S) C, H.
5-Hydroxy-7-(4-hydroxy-3-methoxyphenyl)-1-(1H-pyrrol-2-yl)-hepta-1,4,6-trien-3-one (15)
(from 1H-pyrrole-2-carbaldehyde) 24% yield. mp 138−139 °C; ESI MS m/z 334.2 (M+Na)+; 1H NMR (300 MHz, CD3COCD3): δ 3.92 (3H, s), 5.86 (H, s), 6.25 (H, s), 6.47 (H, d, J= 15.9 Hz), 6.64 (H, s), 6.68 (H, d, J= 15.9 Hz), 6.88 (H, d, J= 7.8 Hz), 7.06 (H, s), 7.16 (H, d, J= 7.8 Hz), 7.32 (H, s), 7.57 (H, d, J=15.9 Hz), 7.59 (H, d, J= 15.9 Hz). Anal. (C18H17O4) C, H, N.
Preparation of symmetric curcumin analogs 16−26
Compounds 16−19 and 22−26 were prepared from corresponding benzaldehydes by using the same procedure described above for 12 from 5-hydroxymethyl-2-furaldehyde. The preparation of 20 and 21 was reported elsewhere by us.
5-Hydroxy-1,7-bis-(3-hydroxy-4-methoxyphenyl)-hepta-1,4,6-trien-3-one (16)
(from 3-hydroxy-4-methoxybenzaldehyde and 2,4-pentanedione) 45% yield. mp 181−183 °C (lit.25 190−192 °C); ESI-MS m/z 367.1 (M-1)+; 1H NMR (300 MHz, CD3COCD3): δ 4.01 (6H, s), 6.01 (H, s), 6.66 (2H, d, J= 15.6 Hz), 7.03 (2H, d, J= 8.1 Hz), 7.20 (2H, dd, J= 2.1 Hz, J= 8.1 Hz), 7.26 (2H,d, J=2.1 Hz), 7.65 (2H, d, J= 15.3 Hz).
1,7-Bis-(2,4-dimethoxyphenyl)-5-hydroxy-hepta-1,4,6-trien-3-one (17)
(from 2,4-dimethoxybenzaldehyde and 2,4-pentanedione) 48% yield. mp 135−137 °C; ESI-MS m/z 367.1 (M+1)+; 1H NMR (300 MHz, CD3COCD3): δ 3.87 (12H, s), 5.81 (H, s), 6.46 (2H,d, J=2.4 Hz), 6.52 (2H, dd, J= 2.4 Hz, J= 8.7 Hz), 6.63 (2H, d, J= 15.6 Hz), 7.49 (2H, d, J= 8.7 Hz), 7.90 (2H, d, J= 15.9 Hz). Anal. (C23H24O6·1/4 H2O) C, H
5-Hydroxy-1,7-bis-(2,3,4-trimethoxyphenyl)-hepta-1,4,6-trien-3-one (18)
(from 2,3,4-trimethoxybenzaldehyde and 2,4-pentanedione) 30% yield. mp 108−109 °C; ESI MS m/z 457.2 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 3.89 (6H, s), 3.90 (6H, s), 3.94 (6H, s), 5.83 (H, s), 6.64 (2H, d, J= 15.9 Hz), 6.71 (2H, d, J= 9 Hz), 7.31 (2H, d, J= 8.7 Hz), 7.85 (2H,d, J=16.2 Hz). Anal. (C25H28O8) C, H.
5-Hydroxy-1,7-bis-(2,4,5-trimethoxyphenyl)-hepta-1,4,6-trien-3-one (19)
(from 2,4,5-trimethoxybenzaldehyde and 2,4-pentanedione) 28% yield. mp 140−142 °C; ESI MS m/z 457.2 (M+H)+; 1H NMR (300 MHz, CDCl3): δ3.88 (6H, s), 3.94 (6H, s), 5.86 (H, s), 5.57 (2H, d, J= 15.9 Hz), 6.51 (2H, s), 7.06 (2H, s), 7.95 (2H, d, J=15.9 Hz). Anal. (C25H28O8) C, H.
5-Hydroxy-7-[3-methoxy-4-(tetrahydropyran-2-yloxy)-phenyl]-4-{3-[3-methoxy-4-(tetrahydropyran-2-yloxy)-phenyl]-acryloyl}-hepta-4,6-dienoic acid ethyl ester (20)
Yellow powder, 59% yield, mp 60− 61 °C; ESI MS m/z 635.2 (M-1)+; 1H NMR (300 MHz, CDCl3): δ 1.25 (3H, t), 1.57−2.17 (12H, m), 2.96 (0.57H, t), 3.62 (4H, t), 3.91 (6H, s), 4.13 (2H, q), 5.47 (2H, t), 6.72 (2H, d, J=15.6 Hz), 6.90−7.18 (6H, m), 7.44 (2H, d, J=15.6 Hz); Anal. (C30H32O8) C, H.
5-Hydroxy-1,7-bis-[3-methoxy-4-(tetrahydropyran-2-yloxy)-phenyl]-hepta-1,4,6-trien-3-one (21)
Yellow powder; 67% yield, mp 67−69 °C; ESI MS m/z 535.0 (M-1)+; 1H NMR (300 MHz, CDCl3): δ 1.57−2.17 (12H, m), 3.62 (4H, t), 3.91 (6H, s), 5.47 (2H, t), 5.83 (1H, s), 6.50 (2H, d, J=15.9 Hz), 7.09−7.16 (6H, m), 7.60 (2H, d, J=15.9 Hz); Anal. (C31H36O8·H2O) C, H.
1,7-Bis-(4-ethoxy-3-methoxyphenyl)-5-hydroxy-hepta-1,4,6-trien-3-one (22)
(from 4-ethoxy-3-methoxybenzaldehyde and 2,4-pentanedione) 31% yield, mp 139−140°C (lit.26 102 °C); ESI-MS m/z 425.1 (M+1)+; 1H NMR (300 MHz, CDCl3): δ 1.49 (6H, t, J=7.2Hz), 3.93 (6H, s), 4.15 (4H, quart, J=8.4 Hz), 5.82 (H, s), 6.49 (2H, d, J= 15.6 Hz), 6.88 (2H, d, J= 8.1 Hz), 7.08 (2H,d, J=2.1 Hz), 7.14 (2H, dd, J= 2.1 Hz, J= 8.1 Hz), 7.61 (2H, d, J= 15.6 Hz).
3-Methoxy-4-(3-methyl-but-2-enyloxy)-benzaldehyde
Vanillin (1.52 g, 10 mmol) and anhydrous potassium carbonate (1.38 g, 10 mmol) were dissolved in dry acetone (20 mL). 1-Bromo-3-methyl-but-2-ene (3.0 mL) was then added to the solution, and stirring continued for 12 h. The solvent was evaporated in vacuum, and the resulting solid was extracted with CH2Cl2 three times. The CH2Cl2 solution was washed with water and dried over anhydrous sodium sulfate. The organic solvent was removed in vacuum. A colorless liquid (418 mg) was obtained by flash column chromatography eluting with a gradient hexane-EtOAc solvent. 1H NMR (300 MHz, CDCl3): δ1.76 (3H, s), 1.80 (3H, s), 3.94 (3H, s), 4.68 (2H, d, J=6.6 Hz), 5.52 (H, t, J=6.6 Hz), 6.98 (H, d, J= 8.4 Hz), 7.41 (H,d, J=2.1 Hz), 7.44 (H, dd, J= 2.1 Hz, J= 8.4 Hz), 9.85 (H, s).
5-Hydroxy-1,7-bis-[3-methoxy-4-(3-methyl-but-2-enyloxy)-phenyl]-hepta-1,4,6-trien-3-one (23)
(from 3-methoxy-4-(3-methyl-but-2-enyloxy)-benzaldehyde and 2,4-pentanedione) 25% yield, mp 124−125°C; ESI MS m/z 527.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 1.75 (6H, s), 1.79 (6H, s), 3.92 (6H, s), 4.63 (4H, d, J=6.6 Hz), 5.52 (2H, t, J=6.6 Hz), 5.82 (H, s), 6.50 (2H, d, J= 15.6 Hz), 6.88 (2H, d, J= 8.4 Hz), 7.08 (2H,d, J=1.8 Hz), 7.12 (2H, dd, J= 1.8 Hz, J= 8.4 Hz), 7.61 (2H, d, J= 15.9 Hz). Anal. (C31H36O6) C, H.
5-Hydroxy-1,7-bis-(4-hydroxy-3,5-dimethoxyphenyl)-hepta-1,4,6-trien-3-one (24)
(from 4-hydroxy-3,5-dimethoxybenzaldehyde and 2,4-pentanedione) 17% yield, mp 198−199 °C (lit.27 188−190 °C); ESI MS m/z 451.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): 3.94 (6H, s), 5.80 (H, s), 6.49 (2H, d, J= 15.3 Hz), 6.80 (4H, s), 7.58 (2H, d, J= 15.3 Hz).
5-Hydroxy-1,7-bis-(4-hydroxy-3-methylphenyl)-hepta-1,4,6-trien-3-one (25)
(from 4-hydroxy-3-methylbenzaldehyde and 2,4-pentanedione) 52% yield, mp 217−218 °C; ESI MS m/z 337.2 (M+1)+; 1H NMR (300 MHz, CDCl3): δ 2.26 (6H, s), 5.81 (H, s), 6.50 (2H, d, J= 15.9 Hz), 6.86 (2H, d, J= 8.4 Hz), 7.27 (2H,d, J=8.4 Hz), 7.36 (2H, s), 7.58 (2H, d, J= 15.9 Hz). Anal. (C21H20O4·1/4H2O) C, H.
5-Hydroxy-1,7-bis-(4-methoxy-3-methylphenyl)-hepta-1,4,6-trien-3-one (26)
(from 4-methoxy-3-methylbenzaldehyde and 2,4-pentanedione) 55% yield, mp 131−132 °C; ESI MS m/z 365.1 (M+1)+; 1H NMR (300 MHz, CDCl3): δ 2.24 (6H, s), 3.87 (6H, s), 5.78 (H, s), 6.49 (2H, d, J= 15.6 Hz), 6.83 (2H, d, J= 8.7 Hz), 7.37 (2H,d, J=8.7 Hz), 7.38 (2H, s), 7.60 (2H, d, J= 15.6 Hz). Anal. (C23H24O4·1/8H2O) C, H.
Preparation of asymmetric curcumin analogs 27−31
By using the same procedure described above for 13−15 from 5, compounds 27−30 were prepared from 6, and 31 from 11.
1-(3,4-Dimethoxyphenyl)-5-hydroxy-7-(4-hydroxy-3-methoxyphenyl)-hepta-1,4,6-trien-3-one (27)
(from 6 and vanillin) 55% yield, mp 83−84 °C (lit.17 89−91 °C); ESI-MS m/z 381.1 (M-1)+; 1H NMR (300 MHz, CDCl3): δ 3.94 (9H, s), 5.81 (H, s), 6.48 (2H, d, J= 15.6 Hz), 6.87− 7.14 (6H, m), 7.60 (2H, d, J= 15.6 Hz).
7-(3,4-Dimethoxyphenyl)-5-hydroxy-1-(3-hydroxy-4-methoxyphenyl)-hepta-1,4,6-trien-3-one (28)
(from 6 and 3-hydroxy-4-methoxybenzaldehyde) 45% yield, mp 157−158°C; ESI-MS m/z 405.3 (M+ Na)+; 1H NMR (300 MHz, CDCl3): δ 3.93 (3H, s), 3.94 (6H, s), 5.80 (H, s), 6.48 (H, d, J= 16.2 Hz), 6.51 (H, d, J= 15.9 Hz), 6.86 (H, d, J= 8.4 Hz), 6.89 (H, d, J= 8.4 Hz), 7.06 (H, dd, J= 8.4 Hz, J= 1.8 Hz), 7.09 (H, d, J= 1.8 Hz), 7.15 (H, dd, J= 8.4 Hz, J= 1.8 Hz), 7.18 (H, d, J= 1.8 Hz), 7.58 (H, d, J= 15.9 Hz), 7.61 (2H, d, J= 15.6 Hz).
1-(3,4-Dimethoxyphenyl)-5-hydroxy-7-(2,3,4-trimethoxyphenyl)-hepta-1,4,6-trien-3-one (29)
(from 6 and 2,3,4-trimethoxybenzaldehyde) 55% yield, mp 138−139 °C; ESI-MS m/z 427.2 (M+H)+, 449.3 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 3.89(3H, s), 3.91 (3H, s), 3.93 (3H, s), 3.94 (6H, s), 5.83 (H, s), 6.51 (H, d, J= 15.9 Hz), 6.63 (H, d, J= 15.9 Hz), 6.71 (H, d, J= 8.7 Hz), 6.89 (H, d, J= 8.4 Hz), 7.09 (H, d, J= 2.1 Hz), 7.15 (H, dd, J= 1.8 Hz, J= 8.7 Hz), 7.31 (H, d, J= 8.7 Hz), 7.61 (H, d, J= 15.9 Hz), 7.85 (H, d, J= 16.2 Hz). Anal. (C24H26O7·1/2H2O) C, H.
1-(3,4-Dimethoxyphenyl)-5-hydroxy-7-(2,4,5-trimethoxyphenyl)-hepta-1,4,6-trien-3-one (30)
(from 6 and 2,4,5-trimethoxybenzaldehyde) 48% yield, mp 128−129 °C; ESI-MS m/z 427.2 (M+H)+, 449.3 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 3.89(3H, s), 3.90 (3H, s), 3.93 (3H, s), 3.94 (3H, s), 3.95 (3H, s), 5.84 (H, s), 6.50 (H, d, J= 15.6 Hz), 6.51(H, s), 6.57 (H, d, J= 16.2 Hz), 6.88 (H, d, J= 8.1 Hz), 7.06 (H, s), 7.08 (H, d, J= 2.1 Hz), 7.14 (H, dd, J= 2.1 Hz, J= 8.1 Hz), 7.60 (H, d, J= 15.9 Hz), 7.96 (H, d, J= 15.9 Hz). Anal. (C24H26O7·1/4H2O) C, H.
4-[3-(3,4-Dimethoxyphenyl)-acryloyl]-7-(4-hydroxy-3-methoxyphenyl)-5-oxo-hept-6-enoic acid ethyl ester (31)
(from 11 and 3,4-dimethoxybenzaldehyde) 38% yield, mp 67−68 °C; ESI-MS m/z 483.4 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 1.25 (3H, t, J= 7.2 Hz), 2.32−2.37 (2H, m), 2.55 (2H, t, J = 7.8 Hz), 2.95 (0.5H, t, J= 7.8 Hz), 3.91−3.97 (9H, m), 4.14 (2H, q, J = 7.2 Hz), 6.70 (H, dd, J= 4.2 Hz, J=15.6 Hz), 6.84−7.19 (7H, m), 7.62−7.76 (2H, m); Anal. (C27H30O8·1/4H2O) C, H.
Preparation of 32, 33 and 34
To a solution of acetone (0.36 ml, 5 mmol) and the appropriate benzaldehyde (10 mmol) in 50 ml of a 0.25 M solution of aqueous NaOH was added 1.5 ml of a 25% w/w aqueous solution of cetyltrimethylammonium bromide. The mixture was allowed to stir vigorously at room temperature for 20 h, diluted with brine and extracted with EtOAc. The EtOAc solution was concentrated and then subjected to column chromatography to obtain the target product.
1,5-Bis-(4-hydroxy-3-methoxyphenyl)-penta-1,4-dien-3-one (32)
50% yield. mp 83−84 °C (lit.25 84−86 °C); ESI MS m/z 327.3 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 3.94 (6H, s), 6.90 (2H, d, J = 8.4 Hz), 6.95 (2H, d, J= 15.6 Hz), 7.11 (2H, d, J= 1.8 Hz), 7.20 (2H, dd, J= 1.8 Hz, J= 8.4 Hz), 7.68 (2H,d, J=15.6 Hz).
1,5-Bis-(3,4-dimethoxyphenyl)-penta-1,4-dien-3-one (33)
Bright yellow powder. 76% yield; mp 74−75 °C (lit.25 72−75 °C); ESI-MS m/z 355.2 (M+H)+; 1H NMR (300 MHz, CDCl3); δ 3.86 (3H, s), 3.90 (3H, s), 6.47 (2H, d, J = 2.4 Hz), 6.53 (2H, dd, J= 2.4 Hz, J= 8.4 Hz), 7.09 (2H, d, J= 15.9 Hz), 7.57 (2H, d, J= 8.4 Hz), 7.69 (2H,d, J=15.9 Hz).
Preparation of 35 and 36
Compounds 35 and 36 were prepared from 4-hydroxy-3-methoxy-cinnamaldehyde by using the same procedure described above for 12 from 2,4-pentanedione and 5-hydroxymethyl-2-furaldehyde.
7-Hydroxy-1,11-bis-(4-hydroxy-3-methoxyphenyl)-undeca-1,3,6,8,10-pentaen-5-one (35)
(from 4-hydroxy-3-methoxy-cinnamaldehyde and 2,4-pentanedione) 15% yield; mp 162−164 °C; ESI-MS m/z 421.4 (M+1)+; 1H NMR (300 MHz, CDCl3): † 3.95 (6H, s), 5.78 (H, s), 6.13 (2H, d, J= 15.6 Hz), 6.78−7.04 (10H, m), 7.41 (H, d, J= 15.0 Hz), 7.44 (H, d, J=15.3 Hz). Anal. (C25H24O6) C, H.
5-Hydroxy-9-(4-hydroxy-3-methoxyphenyl)-4-[5-(4-hydroxy-3-methoxyphenyl)-penta-2,4-dienoyl]-nona-4,6,8-trienoic acid ethyl ester (36)
(from 4-hydroxy-3-methoxycinnamaldehyde and ethyl 4-acetyl-5-oxohexanoate) 22% yield; mp 143−144 °C; ESI-MS 519.3 (M-1)−; 1H NMR (300 MHz, CD3COCD3): δ 1.24 (3H, t, J= 7.2 Hz), 2.80−2.86 (4H, m), 3.89 (6H, s), 4.11 (2H, quart, J= 7.2 Hz), 6.78 (2H,d, J=14.4 Hz), 6.85 (2H, d, J= 8.1 Hz), 7.02 (2H, d, J= 15.0 Hz), 7.07 (2H, dd, J= 1.8 Hz, J= 8.1 Hz), 7.26 (2H, d, J= 1.8 Hz), 7.52 (H, d, J= 15.0 Hz), 7.55 (H, d, J= 14.4 Hz). Anal. (C30H16O4·3/8H2O) C, H.
1,7-Bis-(3,4-dimethoxyphenyl)-5-hydroxy-heptan-3-one (37)
Dimethyl curcumin (2) (80 mg, 0.2 mmol) was dissolved in 3 mL of EtOAc. 10% Pd/C (80 mg) was added to the solution. After hydrogenation for 24 h at 45 psi, the solution was filtrated, and the filtrate was concentrated. Flash column chromatography (hexane/EA= 2:1) afforded white product in 25% yield. mp 93−94 °C (lit.17 94−95 °C). ESI-MS m/z 425.1 (M+Na)+; 1H NMR (300 MHz, CDCl3): 1.61−1.85 (2H, m), 2.56−2.87 (8H, m), 3.85 (6H, s), 3.86 (6H, s), 4.07 (H, m), 6.69−6.90 (6H, m).
3,4-Dimethoxycinnamone
3,4-Dimethoxybenzaldehyde (1.5 g, 9 mmol) was dissolved in 10 mL of acetone. After 10 mL of sodium hydroxide solution (0.5 g NaOH in 10 mL of H2O) was added, the mixture was stirred for 24 h. Then the excess acetone was removed in vacuo. Upon acidification with 1 N HCl, a green precipitate was obtained. The precipitate was extracted with EtOAc. The organic layer was dried over sodium sulfate and solvent removed in vacuo. Flash column chromatography gave the desired product (1.5 g in 81% yield). 1H NMR (300 MHz, CD3COCD3): 2.27 (3H, s), 3.87 (6H, s), 6.67 (H, d, J= 16.2 Hz), 7.00 (H, d, J= 8.1 Hz), 7.04 (H, d, J= 0.9 Hz), 7.22 (H, dd, J= 2.1 Hz, J= 8.1 Hz), 7.32 (H, d, J= 2.1 Hz), 7.55 (H, d, J= 16.2 Hz).
3, 4-Dimethoxycinnamaldehyde
To a solution of acetaldehyde (0.84 ml, 15 mmol) in EtOH (10 ml), 3 M NaOH (5ml, 15 mmol) was added at 0 °C. The solution was stirred for an additional 20 min. After 3, 4-dimethoxybenzaldehyde (2.5 g, 15 mmol) in EtOH (5 ml) was added to the stirring solution dropwise, the reaction was brought to room temperature and stirred for 2 h. Then the mixture was poured into water and adjusted to pH 7 by adding 1N HCl. After extraction with EtOAc, the organic layer was washed with water three times and dried over anhydrous sodium sulfate. After removal of the solvent under vacuum, the crude product was purified with flash column chromatography. Pale yellow product (1.1 g) was obtained in 38% yield. 1H NMR (300 MHz, CDCl3): δ 3.93 (3H, s), 3.94 (3H, s), 6.62 (H, dd, J= 7.8 Hz, J= Hz), 6.91 (H, d, J= 8.1 Hz), 7.08 (H, d, J= 2.1 Hz), 7.17 (H, dd, J= 1.8 Hz, J= 8.1 Hz), 7.42 (H, d, J= 15.9 Hz), 9.67 (H, d, J= 7.8 Hz).
1,7-Bis-(3,4-dimethoxyphenyl)-5-hydroxy-hepta-1,6-dien-3-one (38)
To a stirring solution of lithium diisopropylamine (0.29 ml, 0.58 mmol) in THF (3 ml), a THF (3 ml) solution of 3, 4-dimethoxycinnamone (100 mg, 0.48 mmol) was added at −78 °C. After 15 min, 3,4-dimethoxycinnamaldehyde (85 mg, 0.44 mmol) in THF (3 ml) was added. After stirring for an additional 20 min at −78 °C, the mixture was quenched with saturated NH4Cl solution. The solution was allowed to warm to ambient temperature and extracted with EtOAc. The organic layer was washed with water and saturated NaCl solution and dried over anhydrous sodium sulfate. The crude product was purified by flash column chromatography to give 22 mg pure product in13% yield. mp 88−89 °C; ESI MS m/z 399.3 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 2.93 (2H, d), 3.80 (6H, s), 3.85 (6H, s), 4.13 (H, d), 6.25 (H, dd, J= 6Hz, J= 15.9 Hz), 6.58 (H, d, J= 15.9 Hz), 6.80 (H, d, J= 16.2 Hz), 6.88 (H, d, J=8.7 Hz), 6.90 (H, dd, J= 0.9 Hz, J= 8.7 Hz), 7.00 (H, d, J= 8.4 Hz), 7.04 (H, d, J= 0.9 Hz), 7.25 (H, dd, J= 0.9 Hz, J= 8.7 Hz), 7.34 (H, d, J= 0.9 Hz), 7.61 (1H, d, J= 16.2 Hz). Anal. (C23H26O6) C, H.
3-(3,4-Dimethoxyphenyl)-N-[3-(3,4-dimethoxyphenyl)-acryloyl]-acrylamide (39)
3,4-Dimethoxycinnamic acid (624 mg, 3 mmol) was dissolved in 15 mL of dry methylene chloride. Thionyl chloride (0.3 mL, 3.6 mmol) was added at 0 °C. The solution was stirred under reflux for 5 h. The solvent was removed under vacuum to give a yellow solid. In the same flask, 10 mL of anhydrous THF was added, and the mixture heated to reflux. HMDA (0.3 mL) was added very slowly to the refluxing solution, followed by the addition of triethylamine (0.4 mL). The solution was stirred under reflux overnight. The solvent was then removed in vacuo. The solid was extracted with CH2Cl2 three times. The combined CH2Cl2 solution was washed with water three times and brine once, and then dried over anhydrous sodium sulfate. The crude product was obtained after flash column chromatography. 78 mg (13% yield), pale yellow powder. mp 220−221 °C; ESI MS m/z 420.2 (M+Na)+; 1H NMR (300 MHz, DMSO): 3.82 (12H, s), 7.04 (2H, d, J= 8.7 Hz), 7.10 (2H, d, J= 15.9 Hz), 7.23 (2H, s), 7.24 (2H, d, J= 8.7 Hz), 7.66 (2H,d, J=15.9 Hz), 10.51 (0.5 H, s). Anal. (C22H23NO6·3/2H2O) C, H; N: 2.61.
The preparation of compounds 4, 40−45, 47 and 48 was reported by us in a recent publication.18
7-(3,4-Dimethoxyphenyl)-4-[3-(3,-dimethoxyphenyl)-acryloyl]-5-hydroxy-hepta-2,4,6-trienoic acid ethyl ester (4)
Red powder, 54% yield; mp 170 − 171°C; ESI MS m/z 494.6 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 1.34 (3H, t), 3.95 (12H, s), 4.29 (2H, quart), 5.98 (2H, d, J=15.6 Hz), 6.95 (2H, d, J= 8.4 Hz), 7.00 (1H, d, J= 15.6 Hz), 7.08 (2H, d, J= 1.8 Hz), 7.22 (2H, dd, J=8.4 Hz, J= 1.8 Hz), 7.77 (2H, d, J= 15.3 Hz), 7.91 (1H, d, J= 15.6Hz). Anal. (C28H30O8·1/4H2O) C, H.
5-Hydroxy-7-[3-methoxy-4-(tetrahydropyran-2-yloxy)-phenyl]-4-{3-[3-methoxy-4-(tetrahydropyran-2-yloxy)-phenyl]-acryloyl}-hepta-2,4,6-trienoic acid ethyl ester (40)
Orange powder; 62% yield; mp 72− 73 °C; ESI MS m/z 634.7 M+; 1H NMR (300 MHz, CDCl3): δ 1.34 (3H, t), 1.5−2.2 (12H, m), 3.62 (4H, t), 3.92 (6H, s), 4.28 (2H, q), 5.49 (2H, t) 5.96 (H, d, J=15.6 Hz), 7.00 (2H, d, J= 15.6 Hz), 7.08−7. 16 (6H, m), 7.76 (2H, d, J= 15.3 Hz), 7.83 (H, d, J= 15.9Hz); Anal. (C36H42O10) C, H.
5-Hydroxy-7-(4-hydroxy-3-methoxyphenyl)-4-[3-(4-hydroxy-3-methoxyphenyl)-acryloyl]-hepta-2,4,6-trienoic acid ethyl ester (41)
Orange powder, 93% yield; mp 106− 106.5 °C; ESI MS m/z 465.2 (M-1)+; 1H NMR (300 MHz, CDCl3): δ 1.34 (3H, t), 3.95 (6H, s), 4.29 (2H, quart), 5.96 (2H, d, J=15.6 Hz), 6.95 (2H, d, J= 8.2 Hz), 6.96 (1H, d, J= 15.6 Hz), 7.05 (2H, d, J= 2.1 Hz), 7.17 (2H, dd, J=8.2 Hz, J= 2.1 Hz), 7.75 (2H, d, J= 15.3 Hz), 7.90 (1H, d, J= 15.9Hz). Anal. (C26H26O8·11/8H2O) C, H.
7-(3,4-Dimethoxyphenyl)-4-[3-(3,4-dimethoxyphenyl)-acryloyl]-5-hydroxy-hepta-2,4,6-trienoic acid methyl ester (42)
Orange powder; 50% yield; mp 167−168 °C; ESI MS m/z 481.6 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 3.82 (3H, s), 3.93 (12H, s), 5.98 (H, d, J=15.6 Hz), 6.90 (2H, d, J= 8.4 Hz), 6.98 (2H, d, J= 15.6 Hz), 7.07 (2H, d, J= 1.8 Hz), 7. 20 (2H, dd, J= 8.4 Hz, J= 1.8 Hz), 7.76 (2H, d, J= 15.6 Hz), 7.90 (H, d, J= 15.9Hz); Anal. (C27H28O8·1/4H2O) C, H.
7-(3,4-Dimethoxyphenyl)-4-[3-(3,4-dimethoxyphenyl)-acryloyl]-5-hydroxy-hepta-2,4,6-trienoic acid ethylamide (43)
Yellow powder, 16% yield; mp 219−221 °C; ESI MS m/z 516.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 1.32 (3H, t), 3.92 (6H, s), 3.92 (6H, s), 4.00 (2H, quart), 5.85(H, d, J=15.3 Hz), 6.88 (2H, d, J= 8.4 Hz), 6.95 (2H, d, J= 15.6 Hz), 7.06 (2H, d, J= 1.5 Hz), 7.18 (2H, dd, J= 8.4 Hz, J= 1.5 Hz), 7.73 (2H, d, J= 15.6 Hz), 7.82(H, d, J= 15.3Hz); Anal. (C28H31NO7·3/2H2O) C, H.
7-(3,4-Dimethoxyphenyl)-4-[3-(3,4-dimethoxyphenyl)-acryloyl]-5-hydroxy-hepta-2,4,6-trienenitrile (44)
19% yield; mp 204−205 °C; ESI MS m/z 448.3 (M+H)+; 1H NMR (300 MHz, DMSO): δ 3.70 (3H, s), 3.75 (3H, s), 3.79 (3H, s), 3.80 (3H, s), 6.32 (H, d, J=9.6 Hz), 6.99 (2H, dd, J= 8.4 Hz, J= 1.5 Hz), (2H, d, J= 15.6 Hz), (2H, d, J= 1.8 Hz), (2H, dd, J= 8.4 Hz, J= 1.8 Hz), (2H, d, J= 15.6 Hz), (H, d, J= 15.9Hz); Anal. (C26H25NO6·9/4H2O) C, H.
1,7-Bis-(3,4-dimethoxyphenyl)-5-hydroxy-4-(3-hydroxypropenyl)-hepta-1,4,6-trien-3-one (45)
Red powder. 19% yield; mp 178−179 °C; ESI MS m/z 453.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): 3.92 (6H, s), 3.93 (6H, s), 4.40 (2H, d, triplet of doublet, J= 4.5 Hz), 5.30 (0.4 H, s), 5.88 (H, J=15.6 Hz, J= 4.5 Hz), 6.59 (H, d, J= 15.6 Hz), 6.88 (2H, d, J= 8.4 Hz), 6.97 (2H, d, J= 15.6 Hz), 7.06 (2H, d, J= 1.8 Hz), 7.17 (2H, dd, J= 8.4 Hz, J= 1.8 Hz), 7.68 (2H, d, J= 15.6 Hz); Anal. (C26H28O7·3/4H2O) C, H.
3-[1-Hydroxy-3-(4-hydroxy-3-methoxyphenyl)-allylidene]-6-(4-hydroxy-3-methoxyphenyl)-hex-5-ene-2,4-dione (46)
Compound 46 was obtained from vanillin and triacetylmethane by using the same procedure described above for 12 from 5-hydroxymethyl-2-furaldehyde and 2,4-pentanedione. 8% yield; mp 162−164 °C; ESI MS m/z 433.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 2.17 (3H, s), 3.94 (6H, s), 5.80 (H, s), 6.48 (2H, d, J= 15.6 Hz), 6.94 (2H, d, J= 8.1 Hz), 7.05 (2H,d,2H, d, J=1.8 Hz), 7.13 (2H, dd, J= 1.8 Hz, J= 8.1 Hz), 7.59 (2H, d, J= 15.6 Hz). Anal. (C23H22O7) C, H.
4-Fluoro-7-[3-methoxy-4-(tetrahydropyran-2-yloxy)-phenyl]-4-{3-[3-methoxy-4-(tetrahydropyran-2-yloxy)-phenyl]-acryloyl}-5-oxo-hept-6-enoic acid ethyl ester (47)
13% yield; mp 59− 60 °C; EIMS m/z 677.3 (M+Na)+, 655.3 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 1.25 (3H, t), 1.5−2.1 (12H, m), 2.45(2H, m), 2.65 (2H, m), 3.62 (4H, t), 3.90 (6H, s), 4.13 (2H, q), 5.49 (2H, t), 7.06 (2H, dd, J=3 Hz, J=15.6 Hz), 7.12−7.15 (6H, m), 7.75 (2H, d, J=15.6 Hz); Anal. (C36H44FO10) C, H.
4-Fluoro-7-(4-hydroxy-3-methoxyphenyl)-4-[3-(4-hydroxy-3-methoxyphenyl)-acryloyl]-5-oxo-hept-6-enoic acid ethyl ester (48)
97% yield; mp 63− 63.5 °C; EIMS m/z 509.3 (M+Na)+, 487.3 (M+H)+; 1H NMR (300 MHz, CDCl3): δ 1.25 (3H, t), 2.45(2H, m), 2.65 (2H, m), 3.95 (6H, s), 6.93 (2H, d, J=7.8 Hz), 7.06 (2H, dd, J= 3 Hz, J=15.6 Hz), 7.09(2H, d, J= 1.8 Hz), 7.16 (2H, dd, J=1.8 Hz, J=7.8 Hz), 7.75 (2H, d, J=15.6 Hz); Anal. (C26H27FO8·3/4H2O) C, H.
1,7-Bis-(3,4-dimethoxyphenyl)-4-methyl-hepta-1,6-diene-3,5-dione (49)
Compound 49 was prepared from 3-methyl-2,4-pentanedione and 3,4-dimethoxybenzaldehyde by using the same procedure described above for 12 from 2,4-pentanedione and 5-hydroxymethyl-2-furaldehyde. 51% yield; mp 129−130 °C (lit.28 142−145 °C); ESI MS m/z 433.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 2.19 (3H, s), 3.91 (3H, s), 3.93 (3H, s), 3.95 (3H, s), 6.89 (2H, d, J=8.1 Hz), 6.99 (2H, d, J=15.3 Hz), 7.10 (2H, d, J=1.8 Hz), 7.19(2H, dd, J= 8.1Hz, J=1.8 Hz), 7.70 (2H, d, J=15.6 Hz).
1,7-Bis-(3,4-dimethoxyphenyl)-4-fluoro-4-methyl-hepta-1,6-diene-3,5-dione (50)
Yellow powder. 48% yield; mp 44−45 °C; ESI MS m/z 451.2 (M+Na)+; 1H NMR (300 MHz, CDCl3): δ 1.97 (3H, s), 3.90 (6H, s), 3.95 (6H, s), 6.83 (2H, d, J=7.8 Hz), 6.87 (2H, dd, J=3 Hz,J=15.6 Hz), 6.90(2H, d, J= 1.8 Hz), 6.95 (2H, dd, J=1.8 Hz, J=7.8 Hz), 7.75 (2H, d, J=15.6 Hz); Anal. (C24H25FO6·5/2H2O) C, H.
Cytotoxicity bioassay in human prostate cancer cell lines LNCaP and PC-3
The in vitro cytotoxicity bioassay was performed according to the procedures described in Rubinstein et al.29 Drug stock solutions were prepared in DMSO, and the final solvent concentration was not greater than 1% DMSO (v/v), a concentration without effect on cell replication. The human prostate cancer cells were exposed to doses of the compounds for 2 days and variation between replicate experiments was ≤5%. The IC50 values were determined from dose-response graphs.
Antiandrogenic bioassay in human prostate cancer cells
Human prostate cancer LNCaP and PC-3 cells were maintained in RPMI medium and Dulbecco's minimum essential medium (DMEM), respectively. Both media were supplemented with penicillin (25 units/mL), streptomycin (25 μg/mL), and 10% fetal calf serum. For the androgen receptor transactivation assay, an androgen-dependent reporter gene transcription test was employed as the primary screening for potential antiandrogen identification. This assay was first performed in LNCaP cells, which express a clinically relevant mutant AR. Once anti-androgenic activity was detected in the LNCaP AR transactivation assay, compounds were re-examined for their potential activity against wild type AR. Wild type AR transactivation assay was performed in PC-3 host cells, which lack an endogenous, functional AR. The method and conditions of cell and gene transfection have been described previously. In brief, cells were plated in 24-well tissue culture dishes for 24 (PC-3 cells) or 48 (LNCaP cells) h prior to transfection. Subsequently, LNCaP cells were transfected with a reporter gene, MMTV-luciferase, which contains MMTV-LTR promoter and androgen receptor binding element, and PRL-SV40, which served as an internal control for transfection efficiency. PC-3 cells were transfected with a wild type AR expression plasmid, pSG5AR, in addition to the above-mentioned MMTV-luciferase reporter gene and PRL-SV40 internal control. SuperFect (Qiagen, Chatsworth, CA) was employed as the transfection reagent following manufacturer's recommendations. At the end of a five-hour transfection, the medium was changed to DMEM or RPMI supplemented with 10% charcoal dextran-stripped, i.e., androgen-depleted, serum. After 24 h, the cells were treated with 1 nM of DHT and/or test compounds at the designated concentration for another 24 h. The cells were harvested for luciferase activity assay using Dual Luciferase Assay System (Promega, Madison, WI). The derived data were expressed as relative luciferase activity normalized to the internal luciferase control. Cells cultured in medium containing DHT (androgen), as a positive control, induced a marked reporter gene expression. Test compounds capable of significantly suppressing this DHT-induced reporter gene expression were identified as potential antiandrogens.
Supplementary Material
Figure 1.

Structures of curcumin (1), DMC(2), ECECur(3) and 4
Acknowledgement
This work was supported by National Cancer Institute Grant CA-17625 awarded to K. H. Lee.
Footnotes
Antitumor agents 250. For prior paper in the series, see ref 1.
Supporting Information Available: Results of the elemental analysis of 4, 8−15, 17, 19−21, 23, 25, 26, 29−31, 35, 36, 38−48, and 50 are reported.
References
- 1.Tatsuzaki J, Bastow KF, Nakagawa-Goto K, Nakamura S, Itokawa H, Lee KH. Antitumor Agents 249. Synthesis and evaluation of dehydrozingerone analogues as cytotoxic agents. Cancer Lett. submitted. [Google Scholar]
- 2.Jemal A, Samuels A, A. G, Ward E, Thun M. Cancer statistics, 2003. CA Cancer J. Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 3.Ross RK, Pike MC, Coetzee GA, Reichards MC, Yu MC, Feigelson H, Stanczyk FZ, Kolonel LN, Henderson BE. Androgen metabolism and prostate cancer: establishing a model of genetic susceptibility. Cancer Res. 1998;58:4497–4504. [PubMed] [Google Scholar]
- 4.Goldenberg SL, Bruchovsky N. Use of cyproterone acetate in prostate cancer. Urol. Clin. North. Am. 1991;18:111–112. [PubMed] [Google Scholar]
- 5.de Voogt HJ. The position of cyproterone acetate (CPA), a steroid anti-androgen, in the treatment of prostate cancer. Prostate. 1992;(Supply 4):91–95. doi: 10.1002/pros.2990210514. [DOI] [PubMed] [Google Scholar]
- 6.de Voogt HJ, Smith PH, Pavone-Macaluso M, de Pauw M, Suciu S. Cardiovascular side effects of diethylstilbestrol, cyproterone acetate, medroxyprogesterone acetate and estraumustine phosphate used for the treatment of advanced prostate cancer: Results from European Organization for Research on Treatment of Cancer Trials 30761 and 30762. J. Urology. 1986;135:303–307. doi: 10.1016/s0022-5347(17)45620-5. [DOI] [PubMed] [Google Scholar]
- 7.Kelly WK, Scher HI. Prostate specific antigen decline after antiandrogen withdrawal: The flutamide withdrawal syndrome. J. Urology. 1993;149:607–609. doi: 10.1016/s0022-5347(17)36163-3. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: relation to antiandrogen withdrawal syndrome. Prostate. 1996;29:153–158. doi: 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sc. U.S.A. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsing AW, Tsao L, Devesa SS. International trends and patterns of prostate cancer incidence and mortality. Intl. J. Cancer. 2000;85:60–67. doi: 10.1002/(sici)1097-0215(20000101)85:1<60::aid-ijc11>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 11.Ruby AJ, Kuttan G, Babu KD, Rajasekharan KN, Kuttan R. Antitumor and antioxidant activity of natural curcuminoids. Cancer Lett. 1995;94:79–83. doi: 10.1016/0304-3835(95)03827-j. [DOI] [PubMed] [Google Scholar]
- 12.Huang M-T. Antioxidant and antitumorigenic properties of curcumin. Food Factors for Cancer Prevention, [International Conference on Food Factors: Chemistry and Cancer Prevention], Hamamatsu, Japan, Dec., 1995. 1997:249–252. [Google Scholar]
- 13.Jordan WC, Drew CR. Curcumin--a natural herb with anti-HIV activity. J. Natl. Med. Assoc. 1996;88:333. [PMC free article] [PubMed] [Google Scholar]
- 14.Kawamori T, Lubet R, Steele VE, Kelloff GJ, Kaskey RB. Chemopreventive effect of curcumin, a naturally occuring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res. 1999;59:597–601. [PubMed] [Google Scholar]
- 15.Aggarwal BB, Kumar AP, Bharti AC. Anticancer potential of curcumin preclinical and clinical studies. Anticancer Res. 2003;23:363–398. [PubMed] [Google Scholar]
- 16.Ohtsu H, Itokawa H, Xiao Z, Su C-Y, Shih CCY, Chiang T, Chang E, Lee Y, Chiu S-Y, Chang C, Lee K-H. Antitumor agents 222. Synthesis and anti-androgen activity of new diarylheptanoids. Bioorg. Med. Chem. 2003;11:5083–5090. doi: 10.1016/j.bmc.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 17.Ohtsu H, Xiao Z, Ishida J, Nagai M, Wang H-K, Itokawa H, Su C-Y, Shih C, Chiang T, Chang E, Lee Y, Tsai M-Y, Chang C, Lee K-H. Antitumor agents. 217.Curcumin analogues as novel androgen receptor antagonists with potential as anti-prostate cancer agents. J. Med. Chem. 2002;45:5037–5042. doi: 10.1021/jm020200g. [DOI] [PubMed] [Google Scholar]
- 18.Lin L, Shi Q, Su C-Y, Shih CCY, Lee K-H. Antitumor agents 247. New 4-ethoxycarbonylethyl curcumin analogs as potential antiandrogenic agents. Bioorg. Med. Chem. 2005 doi: 10.1016/j.bmc.2005.11.034. in press. [DOI] [PubMed] [Google Scholar]
- 19.Masuda T, Matsumura H, Oyama Y, Takeda Y, Jitoe A, Kida A, Hidaka K. Synthesis of (±)-cassumunins A and B, new curcuminoid antioxidants having protective activity of the living Cell against oxidative damage. J. Nat. Prod. 1998;61:609–613. doi: 10.1021/np970555g. [DOI] [PubMed] [Google Scholar]
- 20.Pedersen U, Rasmussen PB, Lawesson S-O. Synthesis of naturally occuring curcuminoids and related compounds. Liebigs Ann. Chem. 1985:1557–1569. [Google Scholar]
- 21.Baranovsky A, Schmitt B, Fowler DJ, Schneider B. Synthesis of aew biosynthetically important diarylheptanoids and their oxa- and fluoro- analogues by three different strategies. Syn. Comm. 2003;33:1019–1045. [Google Scholar]
- 22.Bowser JR, Williams PJ, Kurz K. Cleavage of silicon-nitrogen bonds by acid chlorides: an unusual synthetic route to amides. J. Org. Chem. 1983;48:4111–4113. [Google Scholar]
- 23.Lal GS. Site-selective fluorination of organic compounds using 1-alkyl-4-fluoro-1,4-diazabicyclo[2,2,2]octane salts (Selectfluor Reagents). J. Org. Chem. 1993;58:2791–2796. [Google Scholar]
- 24.Belliotti TR, Connor DT, Flynn DL, Kostlan CR, Nies DE. Eur. Pat. Appl. Warner-Lambert Co., USA; USA: 1987. Preparation of novel styrylpyrazoles, styrylisoxazoles, and analogs as 5-lipoxygenase inhibitors. p. 58. [Google Scholar]
- 25.Weber WM, Hunsaker LA, Abcouwer SF, Deck LM, Vander Jagt DL. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005;13:3811–3820. doi: 10.1016/j.bmc.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 26.Chowdhury H, Walia S, Saxena VS. Isolation, characterization and insect growth inhibitory activity of major turmeric constituents and their derivatives against Schistocerca gregaria (Forsk) and Dysdercus koenigii (Walk). Pest Management Science. 2000;56:1086–1092. [Google Scholar]
- 27.Youssef KM, El-Sherbeny MA, El-Shafie FS, Farag HA, Al-Deeb OA, Awadalla SAA. Synthesis of curcumin analogues as potential antioxidant, cancer chemopreventive agents. Archiv der Pharmazie. 2004;337:42–54. doi: 10.1002/ardp.200300763. [DOI] [PubMed] [Google Scholar]
- 28.Ishida J, Ohtsu H, Tachibana Y, Nakanishi Y, Bastow KF, Nagai M, Wang H-K, Itokawa H, Lee K-H. Antitumor agents. Part 214: Synthesis and evaluation of curcumin analogues as cytotoxic agents. Bioorg. Med. Chem. 2002;10:3481–3487. doi: 10.1016/s0968-0896(02)00249-3. [DOI] [PubMed] [Google Scholar]
- 29.Rubinstein LV, Shoemaker RH, Paull KD, Simo RM, Tosini S, Skehan P, Scudiero PA, Monks A, Boyd MR. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990;82:1113. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.