

Abstract

The syn-selective addition of organozinc compounds to 4α-epoxypyranosides (4α-EPs), generated from methyl β-D-glucoside or aminoglucoside, provides an efficient route to pyranosides with α-L-ido configurations, including L-iduronic acid, neosamine B, and higher monosaccharide derivatives.
Unsaturated pyranosides such as glycals are used extensively in carbohydrate and natural products synthesis.1 One widespread application involves the formation of epoxyglycals by stereoselective addition of dimethyldioxirane (DMDO), which can be employed as glycosyl donors in the synthesis of O- and C-glycosides.2 4-Deoxypentenosides (4-DPs) bear a strong structural homology to glycals, but retain a stereogenic anomeric center. The reactivity profile of 4-DPs is similar to that of glycals but is somewhat more selective with respect to stereocontrol. 4-DPs can be epoxidized by DMDO in a highly facioselective manner,3,4 and 4β-epoxypyranosides (4β-EPs) can undergo anti-selective (SN2) ring opening to generate novel pyranosides with an L-altro configuration.3 4α-epoxypyranosides (4α-EPs), which can also be obtained with high stereoselectivity,4 should be equally valuable as precursors to L-pyranosides. Here we demonstrate the utility of organozinc reagents for syn- selective ring opening of 4α-EPs into pyranosides with an L-ido configuration.
The efficient formation of α-C-glycosides via syn-selective ring openings of α-epoxyglycals has been achieved with mild organometallic reagents having a significant Lewis acid character, namely those derived from aluminum,5,6 boron,5 titanium,7 zirconium,8 or zinc.9 Syn delivery of the nucleophile is assumed to proceed by coordination and simultaneous activation of the epoxide by the metal center. While many of these reagents are likely to be useful for syn-selective addition to 4-EPs,10 organozinc halides (RZnX) are most appealing with respect to atom efficiency and functional group compatibility, and can be generated by several different methods.9,11
4α-EPs 6 and 7 were derived in good yields from β-methyl-D-glucoside 1 and β-methyl-D-glucosaminoside 2 respectively, using an oxidation–decarboxylative elimination sequence.3,10 Phthalimide 4 was transformed into azido derivative 5 by diazo transfer onto the intermediate free amine.12 DMDO oxidations of 4-DPs 3–5 were carried out at −55 °C to produce 4α-EPs 6–8 with 10:1 α:β stereoselectivity and in quantitative yield (Scheme 1).4 The 4α-EPs could be stored at −20 °C for several months without concern for decomposition.
Scheme 1.

Synthesis of 4α-epoxypyranosides a
a Selected abbreviations: DMDO = dimethyldioxirane; DMFDNpA = dimethylformamide dineopentyl acetal; en = ethylenediamine; Phth = phthalimide; TEMPO = tetramethylpiperidine oxide; TfN3 = triflyl azide.
The most reliable syn additions were produced by generating RZnX species via the transmetallation of organolithium or Grignard reagents (see Supporting Information). Systematic examination of reaction conditions revealed that the presence of excess ZnBr2 significantly improved the efficiency of addition (Table 1); adducts were produced in high yields using 2 equiv of RZnBr, whereas stronger Lewis acids such as Zn(OTf)2 or BF3-Et2O were often incompatible with the epoxide. Furthermore, allyltrimethylsilane and other electron-rich olefins tended to react poorly with 4α-EPs in the presence of Lewis acids, in contrast to their efficacy as nucleophiles in C-glycoside synthesis. These studies suggest that ZnBr2 promotes syn addition by forming an active complex with RZnBr (see below), rather than by direct activation of the epoxide. The activation of organometallic reagents by Lewis acids has been well documented over the years.13
Table 1.
Selected conditions for the syn addition of 2-furylzinc halides to 4α-epoxypyranoside 7 in the presence of ZnX2a
 | |||
|---|---|---|---|
| entry | furylZnX, equiv | ZnX2 (equiv) | yield b |
| 1 | 2 | ZnCl2 (2) | 41% |
| 2 | 2 | ZnCl2 (5) | 60% |
| 3 | 4 | ZnCl2 (2) | 64% |
| 4 | 10 | ZnCl2 (2) | 76% |
| 5 | 2 | ZnBr2 (0) | 56% |
| 6 | 2 | ZnBr2 (2) | 67% |
| 7 | 2 | ZnBr2 (5) | 83% |
Preparation of 2-furylzinc halide in THF: (i) Furan, n-BuLi, THF, −78 ºC, 30 min; (ii) ZnX2, THF, 0 ºC, 30 min. Epoxide 7 was added as a CH2Cl2 solution at −78 ºC, warmed to 0 ºC, then stirred for 2 h while warming to rt.
Isolated yields.
The addition of RZnX species to 4α-EPs 6, 7 and 8 proceeded with high selectivity (syn:anti > 20:1) for a variety of sp2- and sp-carbon nucleophiles, producing the corresponding L-idopyranoside derivatives in good to high yields (Table 2). For example, 4α-EP 6 was converted into L-idopyranoside 9a in 84% isolated yield, accompanied by a small amount of anti adduct and a product derived from the minor 4β-EP isomer (<5% combined yield).14 4α-EP 6 also reacted efficiently with activated sp3-carbon nucleophiles such as allylzinc bromide (entry 7) but was less receptive to additions with (Z)-alkenylzinc agents (entries 10, 13). These derivatives could be obtained with higher overall yields by partial hydrogenation of the corresponding alkynyl derivatives.
Table 2. Syn.
additions to 4α-epoxypyranosides 6–8a
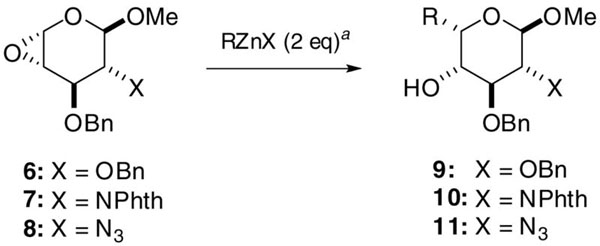 | ||||
|---|---|---|---|---|
| entry | RZnX | prep methodb | product | yieldc |
| 1 |
|
A | 9a | 84% |
| 10a | 83% | |||
| 2 |
|
A | 9b | 74% |
| 10b | 83% | |||
| 3 |

|
9c | 69% | |
| 10c | 82% | |||
| 4 |

|
B | 9d | 80% |
| 10d | 83% | |||
| 5 |
|
B | 9e | 69% |
| 10e | 74% | |||
| 11e | 58% | |||
| 6 |
|
A | 9f | 77% |
| 10f | 74% | |||
| 7 |
|
B | 9g | 73% |
| 8 |
|
C | 9h | 60% |
| 9 |

|
A | 9i | 79% |
| 10 |
|
C | 9j | 37%d |
| (77%)e | ||||
| 11 |
|
C | 9k | 66% |
| 12 |

|
C | 9l | 77% |
| 13 |

|
D |

9m |
23% |
| (73%)f | ||||
See Supporting Information for reaction conditions.
A: (i) RH (4 eq), n-BuLi (2 equiv), THF, −78 °C; (ii) ZnBr2 (7 equiv), THF, 0 °C. B: RMgBr (2 eq), ZnBr2 (7 equiv), 0 °C. C: (i) RBr or RI (2 equiv), t-BuLi (4 equiv), Et2O, −78 °C; (ii) ZnBr2 (7 equiv). D: RI (2 equiv), Zn (activated powder), THF, 45 °C.
Isolated yields.
9f formed as a byproduct (40% yield).
Yield in 2 steps via 9f.
Yield in 4 steps via 9i.
Several of the syn adducts in Table 2 were examined as intermediates for preparing L-idopyranosides with natural or unnatural configurations. In particular, we were interested to develop novel routes to L-iduronic acid, a vital component of heparin and heparan sulfate,15 neosamine B, a 2,6-diaminopyranoside with L-ido configuration found in several aminoglycoside antibiotics,16 and higher monosaccharides with structural analogy to sialic acid and other biologically active congeners.17,18 Syntheses of these sugars typically involve manipulation of acyclic intermediates, either for epimerization of the C5 stereocenter in the cases of L-iduronic acid and neosamine B,19,20 or for chain extension at the reducing end in the case of higher monosaccharides.17,21 In comparison, 4-EPs enable the stereoselective installation of carbon fragments at C5 with retention of the pyranose ring and anomeric configuration.
L-iduronic acid methyl glycoside 12 was prepared in 73% yield from 2-furyl adduct 9a by ozonolysis with Me2S workup (Scheme 2). Neosamine B methyl glycoside 14 was derived in 3 steps from 11e (the C5 vinyl adduct of 4α-EP derivative 8) by ozonolysis, reductive amination, and hydrogenation, and fully characterized as peracetate 15.
Scheme 2.

Synthesis of L-iduronic acid and neosamine B methyl glycosides
Reactions involving unactivated alkylzinc halides and dialkylzincs did not proceed as smoothly under comparable reaction conditions. For example, phenethylzinc bromide and diphenethylzinc gave low to fair yields of expected L-idopyranoside 16 accompanied by significant amounts of C5 phenethoxy adducts 17 (Scheme 3), an indication of the oxidative sensitivity of alkylzinc species.22 Rigorous removal of oxygen from the reaction mixture did not improve the ratio of C- to O-addition products, implying that alkylzincs could be oxidized by the 4-EPs or an unidentified byproduct of the DMDO reaction (Scheme 1).23
Scheme 3.

Addition of mono- and dialkylzinc species to 4α-epoxypyranoside 6
a Epoxide 6 added as a CH2Cl2 solution at −78 °C to PhCH2CH2ZnBr (4 equiv) and ZnBr2 (3 equiv) in THF, slowly warmed to −30 ºC, then stirred for 12 h at −30 ºC. b Epoxide 6added at −78 °C to (PhCH2CH2)2Zn (4 equiv) in THF, slowly warmed to −30 °C, then stirred for 4 h at 0 °C.
L-Idopyranosides with C5 alkyl substituents can be prepared with considerable structural diversity and in synthetically useful yields via the syn addition of alkenyl groups. We illustrate this with the stereoselective syntheses of several octopyranoside derivatives, all with L-ido configuration but stereochemically divergent at C6 and C7. Compounds 20 and 21 were prepared respectively from trans-allylic ether 9h and cis-allylic ether 18 (via reduction of propargylic ether 9i) by catalytic osmylation, with diastereomeric ratios of 5:1 and 6:1 favoring the desired product (Scheme 4). The configurations of octopyranosides 20 and 21 were tentatively assigned as D-threo-L-ido and L-erythro-L-ido with the stereochemical relationship of C5 and C6 being erythro in each case, according to the empirical rule developed by Kishi and coworkers.24 The stereochemical assignments were confirmed by nuclear Overhauser enhancement (NOE) measurements (Figure 1). The osmylation of α,β-unsaturated lactone 9m, which was expected to proceed with selectivity opposite to that of 18, yielded a single diastereomer which was assigned as D-erythro-L-ido by NOE analysis of diacetate 22. Compound 9m could also be oxidized stereoselectively into epoxide 19 by hypochlorite oxidation25 and converted cleanly by phenylselenol reduction26 into 7-deoxyoctopyranoside 23, whose C6 configuration was assigned to be L-glycero by NOE measurements.
Scheme 4.

Stereoselective synthesis of octopyranosides with L-ido configuration a
a Selected abbreviations: BAIB = bisacetoxyiodobenzene; CSA = camphorsulfonic acid; MP = methoxypropene; NMO = morpholine N-oxide; TBS = t-butyldimethylsilyl; TEMPO = tetramethylpiperidinoxy radical.
Figure 1.
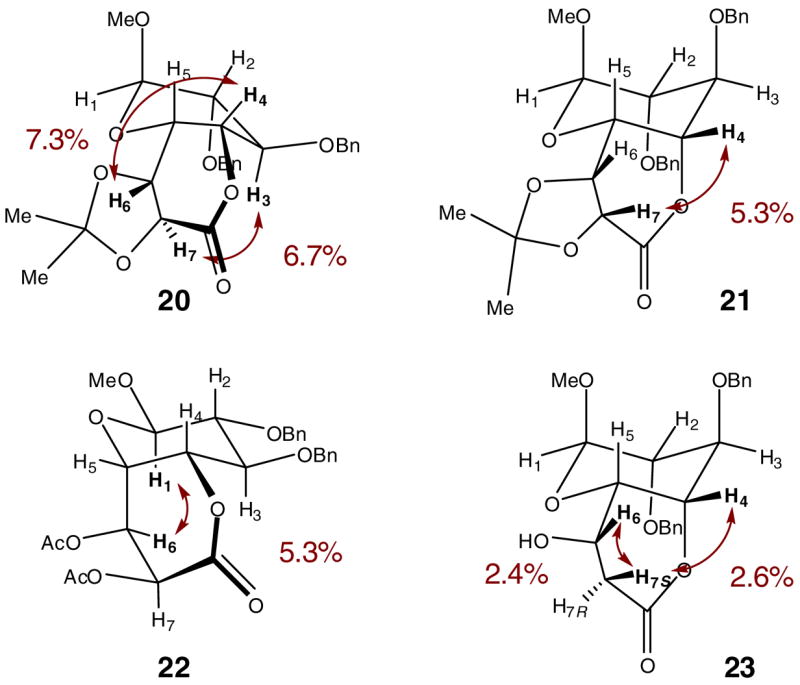
Stereochemical assignments of bicyclic lactones 20–23. Conformations are drawn to emphasize NOE interactions.
In conclusion, ZnBr2-mediated syn additions to 4α-epoxypyranosides provides a general and efficient route to natural and unnatural L-idopyranosides.
Supplementary Material
Experimental procedures and spectroscopic characterization of compounds 9a–23 (PDF). This material is available free of charge via the Internet.
Acknowledgments
This work was supported by the National Institutes of Health (GM-06982). The authors also gratefully acknowledge support from the Purdue Cancer Center and the Purdue interdepartmental NMR facility, and Mr. Zhihong Huang (Purdue Univ.) for helpful discussions on organozinc chemistry.
References
- 1.Ferrier RJ, Hoberg JO. In: Adv Carbohydr Chem Biochem. Horton D, editor. Vol. 58. Academic Press; New York: 2003. pp. 55–119. [DOI] [PubMed] [Google Scholar]
- 2.(a) Halcomb RL, Danishefsky SJ. J Am Chem Soc. 1989;111:6661–6666. [Google Scholar]; (b) Cheshev P, Marra A, Dondoni A. Carbohydr Res. 2006;341:2714–2716. doi: 10.1016/j.carres.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Boulineau FP, Wei A. Org Lett. 2002;4:2281–2283. doi: 10.1021/ol026175q. [DOI] [PubMed] [Google Scholar]
- 4.Cheng G, Boulineau FP, Liew ST, Shi Q, Wenthold PG, Wei A. Org Lett. 2006;9:4545–4548. doi: 10.1021/ol0617401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Rainier JD, Cox JM. Org Lett. 2000;2:2707–2709. doi: 10.1021/ol006286u. [DOI] [PubMed] [Google Scholar]; (b) Allwein SP, Cox JM, Howard BE, Johnson WB, Rainier JD. Tetrahedron. 2002;58:1997–2009. [Google Scholar]
- 6.Bailey JM, Craig D, Gallagher PT. Synlett. 1999:132–134. [Google Scholar]
- 7.Parrish JD, Little RD. Org Lett. 2002;4:1439–1442. doi: 10.1021/ol025575a. [DOI] [PubMed] [Google Scholar]
- 8.Wipf P, Pierce JG, Zhuang N. Org Lett. 2005;7:483–485. doi: 10.1021/ol0475414. [DOI] [PubMed] [Google Scholar]
- 9.(a) Leeuwenburgh MA, Timmers CM, van der Marel GA, van Boom JH, Mallet JM, Sinaÿ PG. Tetrahedron Lett. 1997;38:6251–6254. [Google Scholar]; (b) Steinhuebel DP, Fleming JJ, Du Bois J. Org Lett. 2002;4:293–295. doi: 10.1021/ol010273e. [DOI] [PubMed] [Google Scholar]; (c) Xue S, Han KZ, He L, Guo QX. Synlett. 2003:870–872. [Google Scholar]
- 10.Boulineau FP, Wei A. J Org Chem. 2004;69:3391–3399. doi: 10.1021/jo035789l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Knochel P, Almena JJ, Jones P. Tetrahedron. 1998;54:8275–8319. [Google Scholar]; (b) Ikegami R, Koresawa A, Shibata T, Takagi K. J Org Chem. 2003;68:2195–2199. doi: 10.1021/jo026746s. [DOI] [PubMed] [Google Scholar]; (c) Huo S. Org Lett. 2003;5:423–425. doi: 10.1021/ol0272693. [DOI] [PubMed] [Google Scholar]; (d) Kneisel FF, Dochnahl M, Knochel P. Angew Chem Int Ed. 2004;43:1017–1021. doi: 10.1002/anie.200353316. [DOI] [PubMed] [Google Scholar]; (e) Huang Z, Negishi E. Org Lett. 2006;8:3675–3678. doi: 10.1021/ol061202o. [DOI] [PubMed] [Google Scholar]
- 12.Alper PB, Hung SC, Wong CH. Tetrahedron Lett. 1996;37:6029–6033. [Google Scholar]
- 13.(a) Negishi E-i. Pur Appl Chem. 1981;53:2333–2356. [Google Scholar]; (b) Olah GA. Angew Chem Int Ed. 1993;32:767–922. [Google Scholar]; (c) Negishi E-i. Bull Chem Soc Japan. 2007;80:233–257. [Google Scholar]
- 14.Addition to the 4β-EP isomer is presumed also to proceed with high syn selectivity.
- 15.(a) Conrad HE. Heparin-Binding Proteins. Academic Press; San Diego: 1998. [Google Scholar]; (b) Linhardt RJ. J Med Chem. 2003;46:2551–2564. doi: 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- 16.Umezawa S. Adv Carbohydr Chem Biochem. 1974;30:111–182. doi: 10.1016/s0065-2318(08)60264-4. [DOI] [PubMed] [Google Scholar]
- 17.Danishefsky SJ, DeNinno MP. Angew Chem Int Ed. 1987;26:15–23. [Google Scholar]
- 18.(a) Unger FM. In: Adv Carbohydr Chem Biochem. Horton D, editor. Vol. 38. Academic Press; New York: 1982. pp. 323–388. [Google Scholar]; (b) Stenutz R, Weintraub A, Widmalm G. FEMS Microbiol Rev. 2006;30:382–403. doi: 10.1111/j.1574-6976.2006.00016.x. [DOI] [PubMed] [Google Scholar]
- 19.(a) Blanc-Muesser M, Defaye J. Synthesis. 1977:568–569. [Google Scholar]; (b) Jacquinet JC, Petitou M, Duchaussoy P, Lederman I, Choay J, Torri G, Sinaÿ P. Carbohydr Res. 1984;130:221–241. doi: 10.1016/s0008-6215(00)90633-5. [DOI] [PubMed] [Google Scholar]; (c) Ojeda R, de Paz JL, Martín-Lomas M, Lassaletta JM. Synlett. 1999;8:1316–1318. [Google Scholar]; (d) Takahashi H, Hitomi Y, Iwai Y, Ikegami S. J Am Chem Soc. 2000;122:2995–3000. [Google Scholar]
- 20.Usui T, Takagi Y, Tsuchiya T, Umezawa S. Carbohydr Res. 1984;130:165–177. [Google Scholar]
- 21.(a) Györgydeák Z, Pelyvás I. Monosaccharide Sugars: Chemical synthesis by chain elongation, degradation, and epimerization. Academic Press; San Diego: 1998. [Google Scholar]; (b) Krülle T, Holst O, Brade H, Schmidt RR. Carbohydr Res. 1993;247:145–158. doi: 10.1016/0008-6215(94)84248-5. [DOI] [PubMed] [Google Scholar]
- 22.Katritzky AR, Luo Z. Heterocycles. 2001;55:1467–1474. [Google Scholar]
- 23.In some instances the deoxygenated reactant (4-DP 3) could be recovered in low yield.
- 24.Cha JK, Christ WJ, Kishi Y. Tetrahedron. 1984;40:2247–55. [Google Scholar]
- 25.Zhu L, Kedenburg PJ, Xian M, Wang GP. Tetrahedron Lett. 2005;46:811–813. [Google Scholar]
- 26.Ding F, Jennings PM. Org Lett. 2005;7:2321–2324. doi: 10.1021/ol0504897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectroscopic characterization of compounds 9a–23 (PDF). This material is available free of charge via the Internet.