

Abstract
Calmodulin (CaM) binds to a domain near the C-terminus of the plasma-membrane Ca2+-ATPase (PMCA), causing the release of this domain and relief of its autoinhibitory function. We investigated the kinetics of dissociation and binding of Ca2+-CaM with a 28-residue peptide (C28W(1b)) corresponding to the CaM binding domain of isoform 1b of PMCA. CaM was labeled with a fluorescent probe on either the N-terminal domain at residue 34 or on the C-terminal domain at residue 110. Formation of complexes of CaM with C28W(1b) results in a decrease in the fluorescence yield of the fluorophore, allowing the kinetics of dissociation or binding to be detected. Using a maximum entropy method, we determined the minimum number and magnitudes of rate constants required to fit the data. Comparison of the fluorescence changes for CaM labeled on the C-terminal or N-terminal domain suggests sequential and ordered binding of the C-terminal and N-terminal domains of CaM with C28W(1b). For dissociation of C28W(1b) from CaM labeled on the N-terminal domain, we observed three time constants, indicating the presence of two intermediate states in the dissociation pathway. However, for CaM labeled on the C-terminal domain, we observed only two time constants, suggesting that the fluorescence label on the C-terminal domain was not sensitive to one of the kinetic steps. The results were modeled by a kinetic mechanism where an initial complex forms upon binding of the C-terminal domain of CaM to C28W(1b), followed by binding of the N-terminal domain, and then formation of a tight binding complex. Oxidation of methionine residues in CaM resulted in significant perturbations to the binding kinetics. The rate of formation of a tight binding complex was reduced, consistent with the lower effectiveness of oxidized CaM in activating the Ca2+ pump.
Calmodulin is a cellular Ca2+ sensor that interacts with numerous target enzymes in response to changes in intracellular Ca2+ concentrations. The geometries of complexes between CaM and target domains are diverse, as CaM and the target may form not only compact structures, but also more extended bound conformations (1). It is well established that the C-terminal domain of CaM binds Ca2+ with about an order of magnitude higher affinity than the N-terminal domain (2, 3), and it also binds more tightly than the N-terminal domain to some targets (4, 5).
One important CaM target is the plasma membrane Ca2+-ATPase (PMCA), a membrane Ca2+ pump critical for regulation of intracellular Ca2+ levels (6, 7). Several researchers have speculated that an intermediate in the CaM-mediated activation of PMCA corresponds to a bound C-domain of CaM but a free N-domain (4, 5, 8). However, despite being a potentially crucial link in the model of CaM activation of PMCA, it is still uncertain whether this intermediate exists, and if it does, what rate constants govern its appearance and disappearance. A solution structure of CaM bound to C20W, a 20-residue segment of the CaM binding domain of PMCA isoform 4b (5), shows the C-terminal domain of CaM bound to the peptide while the N-terminal domain is free (pdb 1cff (4)). Other experiments show that a peptide with just four more residues (C24W) binds in a final structure where both terminal domains of CaM interact with the target (9, 10). Squier and co-workers showed that CaMC (residues 74-144 of CaM) can activate PMCA, but only upon addition of a high enough concentration of CaMC to allow for two CaMC molecules to bind to each activated PMCA (5). Whereas CaMN (residues 1-74) is also able to fully activate PMCA, a much higher concentration is needed. These results suggest that although the C-terminal domain of CaM may interact more tightly with PMCA, two domains must interact simultaneously with PMCA to fully activate the enzyme.
Despite being the isoform expressed in the most tissues (11), isoform 1 has been rarely studied due to difficulties in expression (12, 13). Recent studies have examined the kinetics of CaM interaction with isoform 4b of PMCA (14, 15). In the current study, kinetic measurements are reported for the complex of CaM with the 28-residue CaM-binding domain of PMCA isoform 1b (denoted C28W(1b)). By comparison of the response for CaM fluorescently labeled in the C-terminal or N-terminal domain, it was possible to determine the order of binding of the C- and N-terminal domains. The goal was to determine the kinetics of C28W(1b) binding under high Ca2+ concentrations. However, experimentally it is useful to probe both the forward and the reserve (dissociation) reactions, which are, of course, governed by the same set of time constants. Interaction of CaM with C28W(1b) was detected by fluorescence quenching of the probe Alexa Fluor 488 (AF488) tethered to CaM via cysteine residues introduced into CaM at either position 34 or 110. The rate of fluorescence change upon binding or dissociation of C28W(1b) by CaM was monitored and compared to kinetic models to obtain information about the kinetics of binding and the existence and nature of intermediate structures. For the complex of CaM with C28W(1b), the dissociation process was best characterized by multiple time constants, demonstrating a dissociation mechanism that includes intermediate species. Slow isomerization steps were also observed, which are likely associated with initial steps toward dissociation of the high-affinity CaM-C28W(1b) complex. An additional rate constant was required to fit the data for CaM labeled at the N-terminal domain relative to the CaM labeled at the C-terminal domain, evidence for an intermediate in the pathway that involves changes experienced uniquely by the N-terminal domain of CaM.
We also investigated the effect of oxidation of methionine residues in CaM on its dissociation from C28W(1b). Methionine residues comprise a large component of the binding clefts of CaM (16). Oxidation of methionine residues to the sulfoxides in CaM, specifically of methionine residues near the C-terminus, is known to greatly decrease the potency of CaM to activate PMCA (17–19). This result has important implications for oxidative stress, calcium homeostasis and aging (18, 20). In addition, oxidation of CaM alters its association with the CaM binding domain of PMCA (21). Thus, using fluorescently labeled oxidized CaM molecules we investigated the effects of CaM oxidation on the kinetics of CaM dissociation from C28W(1b) in order to examine mechanistically how oxidation affects recognition by the PMCA. The observed changes after oxidation suggest a reduced propensity for oxidized CaM to form the tightly binding conformations that may be necessary for full activation of the enzyme.
Experimental Methods
Materials
Escherichia coli cell strain BL21(DE3) and the pET-15b expression vector were purchased from Novagen (Madison, WI). All other restriction and modifying enzymes, reagents, and cells used were purchased from Invitrogen (Carlsbad, CA). Phenyl Sepharose CL-4B resin and Sephedex-G25 (fine grade) were purchased from Amersham Pharmacia Biotech (Uppsala, Sweden). The fluorescent probe conjugation was performed with a maleimide derivative of AF 488 (Molecular Probes) with a C5 linker. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification.
The peptide C28W(1b) from PMCA isoform 1b was synthesized by BioSource International (Hopkinton, MA). Its mass was checked by electrospray ionization mass spectrometry. T34C-CaM and T110C-CaM were expressed, purified, and fluorescently labeled following methods described previously (22). The preparation of T110C-CaM was conducted similarly to that of T34C-CaM. Unlabeled T34C-CaM (used for competition experiments) was purified from E. coli cell paste with a phenyl-sepharose column, eluted by addition of EDTA, and dialyzed twice with a large excess volume of buffer (HEPES, pH 7.4, ~300 μM CaCl2) to return CaM to its high-Ca2+ state.
Fluorescence Labeling and Oxidation
CaM was fluorescently labeled in the N-terminal domain at residue 34 or in the C-terminal domain at residue 110. The labeled proteins are denoted T34C-CaM-AF488 and T110C-CaM-AF488. For conjugation of AF488 C5 maleimide, T34C-CaM or T110C-CaM was diluted to a final concentration of 50–100 μM. A 10-fold molar excess of TCEP was added to prevent intermolecular disulfide bond formation. AF488 C5 maleimide was dissolved in HEPES buffer and added dropwise to a final molar ratio of 12:1 (dye:protein). After stirring at room temperature for 1 hour, the reaction was quenched by the addition of a 5:1 excess of glutathione to dye. The free dye was separated from the protein with the use of a Sephadex G-25 size-exclusion column followed by dialysis in 4 L of buffer (10 mM HEPES-NaOH, pH 7.4, 100 mM KCl, 1.0 mM MgCl2, and 100 μM CaCl2). The labeling percentage was approximately 75%. No attempt was made to separate unlabeled protein, as it does not interfere with the kinetics of peptide release from labeled proteins. Control experiments in our laboratory have shown little or no effect of dye conjugation to CaM in regard to interaction with or activation of target enzymes (refs (22) and Supporting Information in Ref. (23)). For oxidation, CaM-AF488 was incubated with 10 mM H2O2 overnight at room temperature. Following oxidation, extensive dialysis was carried out to remove H2O2. Of the nine methionine residues, some are more solvent exposed than others and are thus oxidized more readily (17). By mass spectrometry, it was shown that the extent of oxidation was nearly uniform: the sample was a mixture of CaM species with 7, 8 or 9 methionine residues oxidized (data not shown).
Sample Conditions
Final buffer conditions were 10 mM HEPES-NaOH, pH 7.4, 100 mM KCl, 1.0 mM MgCl2, and 100 μM CaCl2. The free Ca2+ concentration of the solutions was checked with the Ca2+ indicator dye Rhod-5n calibrated with respect to a calcium calibration kit (Molecular Probes, Eugene, OR). For competition experiments after addition of unlabeled CaM, the free Ca2+ concentration was 150 μM. For binding experiments, the free Ca2+ concentration was 105 μM.
Kinetic Studies
The fluorescence of AF488 was monitored at 517 nm with a Varian fluorimeter with excitation at 490 nm. Excitation slits were set at 1.5 nm, and emission slits were set at 5 nm. For dissociation experiments, CaM-AF488 (70 nM) was incubated with C28W(1b) (3.0 μM) for up to an hour. The fluorescence was monitored while adding a volume of 100 μL of concentrated unlabeled T34C-CaM to yield a final concentration of 25 μM unlabeled T34C-CaM for over a 300 fold molar excess to CaM-AF488. Upon T34C-CaM-AF488 dissociation from C28W(1b), there was a high probability that C28W(1b) would bind to the excess of T34C-CaM and not back to the minority species T34C-CaM-AF488. The fluorescence of T34C-CaM-AF488 was significantly higher in the absence of bound C28W(1b) (see Figure 1), and therefore the overall fluorescence increased as C28W(1b) dissociated. The rise in fluorescence was monitored at 10 Hz. Fluorescence counts were binned before fitting to reduce noise. Control experiments consisting of the addition of 100 μL buffer to the incubated CaM-AF488-C28W(1b) sample were carried out to estimate the mixing time under the stirring conditions. The mixing time was on the order of 0.5 to 1 second. The fluorescence output was recorded over the time scale of 100 ms to 10,000 seconds. No photobleaching was observed in control experiments for up to 1 hour. The dissociation measurement was conducted for the N-terminal mutant, T34C-CaM-AF488, and the C-terminal mutant, T110C-CaM-AF488. Dissociation kinetic experiments were also carried out for T110C-CaMox-AF488 and T34C-CaMox-AF488. For binding studies, a small volume of concentrated C28W(1b) was added to T34C-CaM-AF488 (70 nM) in a stirred 3.0 mL cuvette. The final concentration of C28W(1b) was 3.0 μM.
Figure 1.
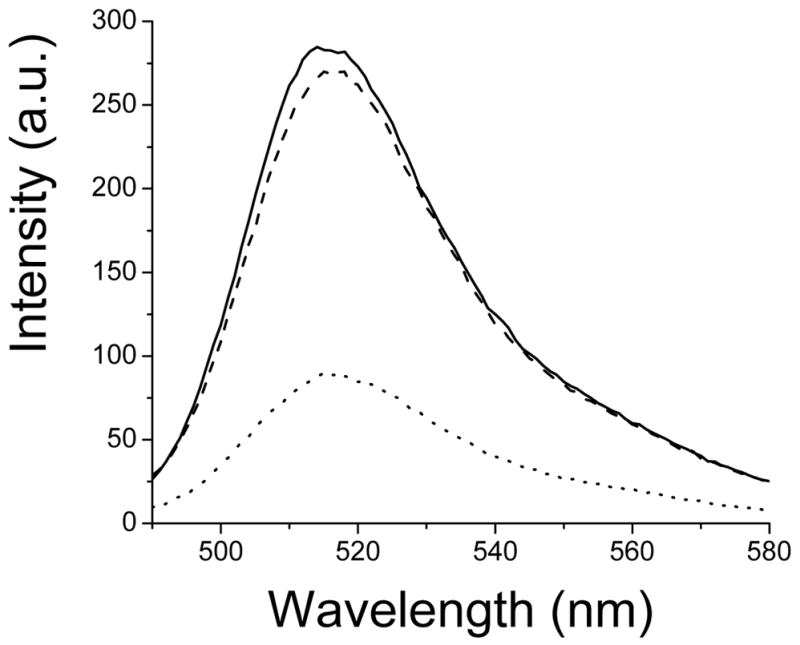
Fluorescence spectra of T34C-CaM-AF488. Top curve (solid): T34C-CaM-AF488; lower curve (dotted): T34C-CaM-AF488 plus C28W(1b) (2.0 μM); middle curve (dashed), T34C-CaM-AF488 showing recovery 1.5 hours after addition of excess, unlabeled CaM (see text).
Maximum Entropy Fitting
The software package MemExp was used to fit all kinetic data (24). MemExp is a hybrid maximum-entropy, least-squares fitting program developed specifically for use in fitting complex kinetic data possessing information on multiple time scales (25). For fitting of the data, the uncertainty in fluorescence counts per period was assumed constant. Both increasing and decreasing contributions to the fluorescence change were allowed, but the data were best fit by maximum-entropy distributions with monotonic changes in fluorescence intensity, decreasing in the case of peptide binding or increasing in the case of peptide dissociation from CaM. In each case, the fit recommended by MemExp was chosen as the best fit. This fit was the last iteration in which the goodness of fit parameter χ2 and the correlation length of the residuals both decreased by more than 1%.
Results
The fluorescence of AF488 conjugated to CaM, either at site 34 or 110, was quenched by 60 to 70% upon C28W(1b) binding. Examples of fluorescence spectra of 250 nM T34C-CaM-AF488 in the presence and absence of 2.0 μM C28W(1b) and after addition of excess unlabeled T34C-CaM are shown in Figure 1. The quenching was likely due to proximity of the fluorescence label and the conserved tryptophan residue in C28W(1b). The results of kinetic measurements are shown in Figure 2. Upon dissociation of C28W(1b) from T34C-CaM-AF488 or T110C-CaM-AF488 in the presence of a large excess of unlabeled T34C-CaM, the fluorescence of the system increased. The free Ca2+ concentration for these solutions was 150 μM, as measured by the Ca2+ indicator dye Rhod-5n.
Figure 2.
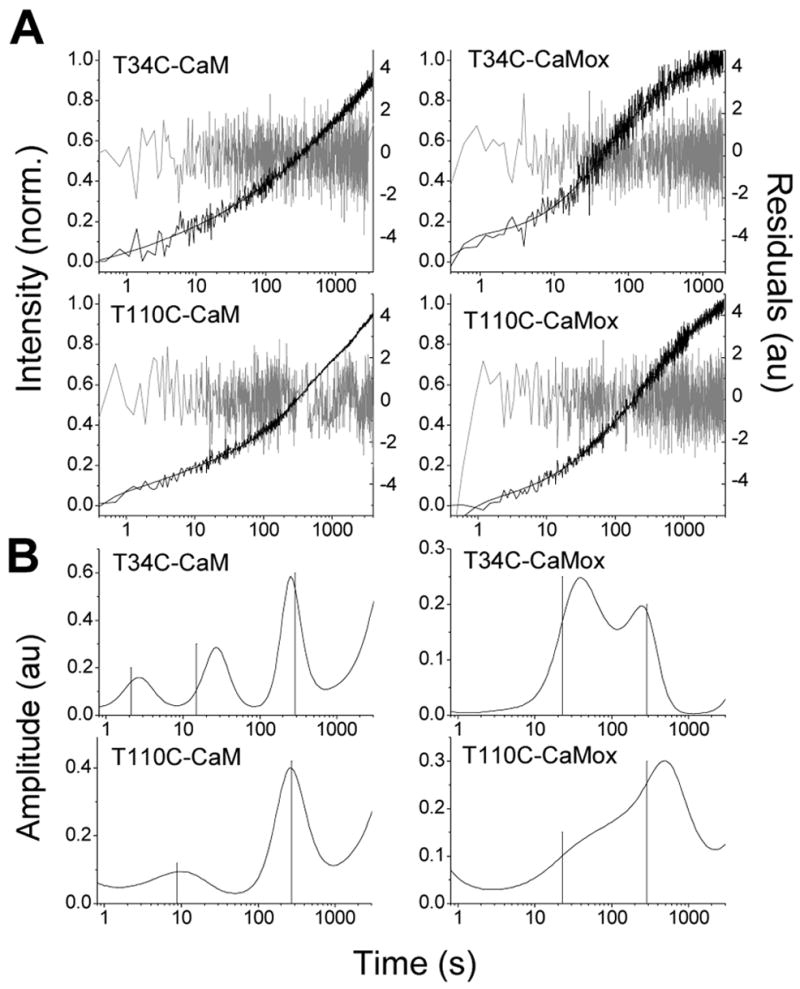
A. Increase in fluorescence of T34C-CaM-AF488, T110C-CaM-AF488, T34C-CaMox-AF488, and T110C-CaMox-AF488 upon dissociation from C28W(1b) plotted with a log time scale. Dissociation was detected by introduction of an excess of unlabeled T34C-CaM. The free Ca2+ concentration was 150 μM in each case. The MEM fits are shown as solid lines through the data and the residuals are plotted in gray. B. MEM fits for CaM dissociation for each of the four CaM-AF488 species. Lines shown are the inverse of the eigenvalues obtained after insertion of the rate constants from the best global fit back into the kinetic rate equations (see eqs S1 and S10).
Maximum Entropy Fits
The time dependencies of the fluorescence increases in kinetic measurements consisted of multiple phases. In such cases, fitting the data to discrete exponentials requires an a priori assumption of the number of exponential components and is susceptible to the multiple-minima problem (the fitted constants may depend on the initial guesses used in the fit). Such fits may not show conclusively whether an additional discrete time constant is justified, and the addition of an extra component may affect the fitting parameters for other components. The maximum entropy method (MEM) is an excellent alternative for fitting complex data (26, 27). MEM fits data to a distribution of rates in order to minimize χ2 and simultaneously to maximize the smoothness of the fit (28). Additional peaks in the MEM distribution decrease the entropy (smoothness), and so appear only if the corresponding decrease in χ2 is sufficient to offset the decrease in entropy. Thus, MEM does not require an a priori assumption of the number of kinetic constants and is useful in identifying the minimum number of rate constants required to fit the data.
The increase in fluorescence upon the dissociation of T34C-CaM-AF488 and T110C-CaM-AF488 from C28W(1b) are shown in Figure 2, along with the MEM fits, the residuals, and the resulting MEM lifetime distributions. The data are plotted in log time scale, with fluorescence counts normalized from 0 to 1 on the y-axis. The absence of a plateau at long times in the data is exaggerated by the logarithmic time scale and results from a longer phase in the fluorescence recovery (see below). The data and fits for dissociation of C28W(1b) from the oxidized species T34C-CaMox-AF488 and T110C-CaMox-AF488 are also shown in Figure 2. The peaks of the MEM lifetime distributions and their fractional amplitudes are shown in Table 1. Widths in the distributions are an unavoidable result of noise in the experimental data and may also reflect inhomogeneity in the kinetic processes.
Table 1.
Time constants of the MEM fits to kinetic data, with amplitudes in parenthesis.
| Time constants (s) | |||
|---|---|---|---|
| Release kinetics | |||
| T34C-CaM-AF488 | 2.6 (18%) | 26.5 (31%) | 270 (51%) |
| T110C-CaM-AF488 | 8.4 (29%) | 271 (71%) | |
| T34C-CaMox-AF488 | 36 (64%) | 238 (36%) | |
| T110C-CaMox-AF488 | 118 (86%) | 380 (14%) | |
| Binding kinetics | |||
| T34C-CaM-AF488 | 3.2 (20%) | 55 (80%) | |
MEM fits yielded three peaks for dissociation of T34C-CaM-AF488, and two peaks for dissociation of T110C-CaM-AF488 and for the two oxidized CaM species. Peak time constants and amplitudes are listed in Table 1. In all cases, an extremely long time constant on the order of several thousand seconds was also detected with an amplitude estimated to be less than 10% of the total recovery. Quantitative determination of the long time constant or amplitude was experimentally difficult due to photobleaching on this long time scale, even though steps were taken to minimize the influence of photobleaching, including minimizing exposure time and excitation intensity. As this step is over an order of magnitude slower than the other processes, it can be excluded from the kinetic fit of the data with little effect on the modeling. It was therefore omitted to simplify the analysis. The extreme length of this time constant suggests that it is not relevant to the function of CaM. It may result from desorption of protein from the glass walls of the cuvette.
Dissociation of C28W(1b) from T34C-CaM-AF488 showed the fastest peak (2.6 seconds). MemExp also predicted a fast distribution for T110C-CaM-AF488 centered at 8.4 seconds. Corresponding fast release processes were not found for dissociation of C28W(1b) from either of the oxidized species, with the fastest peak for T34C-CaMox-AF488 centered at 36 seconds. The best fit for T110C-CaMox-AF488 did not identify a well resolved peak below 350 seconds, although there was a broad shoulder centered at 118 seconds. There were similarities in the MEM fits among the species that were important for modeling the data (see below). In all cases, a peak time constant was observed in the range of several hundred seconds (270 seconds for T34C-CaM-AF488, 271 seconds for T110C-CaM-AF488, 238 seconds for T34C-CaMox-AF488, and 380 seconds for T110C-CaMox-AF488, see Table 1). In addition the predicted time constants were similar for T34C-CaM-AF488 and T34C-CaMox-AF488, except for the absence of a fast component in the case of T34C-CaMox-AF488. Oxidation also produced a decrease in the resolution of the peaks recovered from the MEM fits, especially for T110C-CaM-AF488, suggesting possible kinetic heterogeneity.
In order to obtain more information for modeling kinetics steps involving the N-terminal domain, the binding of C28W(1b) to T34C-CaM-AF488 was also monitored and the resulting fluorescence decrease was fit by MEM (Figure 3). Two peaks were observed, corresponding to time constants of 3.2 seconds and 54.7 seconds. The relative amplitudes for these peaks were 20% and 80%, respectively (Table 1). These data proved valuable for determining rate constants for intermediate steps in the binding pathway (see below).
Figure 3.

The decrease in fluorescence of T34C-CaM-AF488 upon addition of C28W(1b) is shown on the left, along with the MEM fit (solid line through the data). The residuals are plotted behind in gray. On the right the MEM distribution for C28W(1b) binding to T34C-CaM-AF488 is shown.
Kinetic Modeling
The change in fluorescence intensity upon peptide release from fluorescently labeled CaM contains information about the mechanism of target binding and release (29). The MEM fit for the rise in fluorescence accompanying dissociation of C28W(1b) from T34C-CaM-AF488 had three peaks (excluding the longest component) suggesting a model with two intermediate species. One such model was proposed previously by Caride and coworkers for the kinetics of CaM dissociation from the CaM binding domain (C28W) of isoform 4b of PMCA (14, 15) and is shown in Scheme 1: where B, I* and UB represent ‘bound’, ‘intermediate’ and ‘unbound’ species, respectively, and IN is an intermediate possibly related to the release of the N-domain of CaM from C28W(1b) (see Discussion). Under the experimental conditions used (large molar excess of unlabeled T34C-CaM relative to T34C-CaM-AF488), the formation of UB can be considered irreversible due to trapping by the non-fluorescent CaM(wt)-C28W(1b) complex. The kinetic rate equations for this model can be solved by standard methods as described in the Supporting Information, yielding an expression for the predicted observable time constants in terms of the rates constants in Scheme 1 (eq S2 in Supporting Information).
Scheme 1.

Upon addition of unlabeled T34C-CaM to the complex of C28W(1b) with T110C-CaM-AF488, the MEM fit indicated two kinetic processes and was best fit by MEM to two lifetime distributions centered at 8.4 seconds and 271 seconds with relative amplitudes of 29% and 71%, respectively (Table 1, Figure 2). Therefore, these results can be described by a mechanism for dissociation that includes a single intermediate step. Such a mechanism is shown below in Scheme 2, which has been proposed previously by Török and Trentham for dissociation of peptides derived from smooth-muscle myosin light-chain kinase (29). The kinetic rate equations for this model were solved by the same method used for Scheme 1, yielding expressions for the predicted observable rates (eq S11 in Supporting Information).
Scheme 2.

We assumed that the predicted time constants for Scheme 1 or 2 correspond to the experimentally measured time constants that are represented by peaks in the MEM distributions.
Therefore, we varied the values of the rate constants in Scheme 1 (k3, k−3, k2, k−2, and k−1) or Scheme 2 (k3, k−3, k−c, and k−1) to obtain the best fit of the time constants predicted by the model to the observed MEM peaks. The determination of these rate constants allowed for calculation of the concentrations of the reactants, products, and intermediate species as a function of time (see Supporting Information, eq S4). As a function of time, the rise in fluorescence F(t) for both T34C-CaM-AF488 and T110C-CaM-AF488 is given by:
| (1) |
where FX is the relative fluorescence efficiency in state X.
In determining fits to the rate constants, we discuss first the determination of rate constants for Scheme 2. Then, results from fits to Scheme 2 are used as constraints to determine the rate constants for Scheme 1. Excluding the longest component (thousands of seconds), it is convenient to fit the data for T110C-CaM-AF488 with the model proposed by Török and Trentham (29) (Scheme 2). With the rates and relative amplitudes (Table 1) obtained from the MEM fit for dissociation of C28W(1b) from T110C-CaM-AF488, the rate constants k−3, k3, and k−c were obtained (Table 2). The relative amplitudes obtained for the time constants from the MEM fit depend on the relative kinetic amplitudes from each kinetic step (or combination of steps) scaled by the relative fluorescence intensities for each species (FB, FI*, and FUB). Initially, relative values of 1.0, 0.8, and 0.7 were chosen for FUB, FI*, and FB, respectively. The rate constants changed only slightly with changes in the values of FUB, FI*, and FB, provided that the fluorescence intensity of UB was higher than B, which must be true based on the steady-state results (Figure 1).
Table 2.
Rate constants obtained from modeling with eq 1 (see Supporting Information for further details).
| Rate constants (s−1) | ||||||
|---|---|---|---|---|---|---|
| k−3 | k3 | k−2 | k2 | k−1 | k−c | |
| Native CaM | 0.0039 | 0.0088 | 0.14 | 0.20 | 0.21 | 0.11 |
| k−3ox | k3ox | k−cox | ||||
| Oxidized CaM | 0.0039 | 0.0037 | 0.039 | |||
The MEM fit for dissociation of C28W(1b) from T34C-CaM-AF488 showed three time constants (see Fig. 2). Differences in MEM distributions for T34C-CaM-AF488 and T110C-CaM-AF488 were assumed to be due to differences in the sensitivity of the fluorescence intensity of the two domains to each of the kinetic steps. Thus, although Scheme 1 describes dissociation of C28W(1b) from both the N-terminal- and C-terminal-labeled mutants of CaM, the relative fluorescence yields of the species B, I*, and IN are not the same for T34C-CaM-AF488 and T110C-CaM-AF488. Based on the additional peak in the MEM distribution for T34C-CaM-AF488 relative to T110C-CaM-AF488, it is apparent that two of the T110C-CaM-AF488 species have similar fluorescence intensities so that their interconversion was not resolved. We therefore expected that the ca. 8-s time constant reported by the MEM fit of T110C-CaM-AF488 corresponds to the steps with time constants of 2.6 s and 26 s detected for T34C-CaM-AF488, and the individual steps are not resolved by the MEM fit for T110C-CaM-AF488 because the CaM labeled on the C-terminal domain is not sensitive to the formation of the IN intermediate Therefore in the fit of the fluorescence data to eq 1 the constraint FI* = FIn was set for T110C-CaM-AF488 but not necessarily for T34C-CaM-AF488. With this restriction, the amplitudes for each kinetic rate were varied along with the fluorescence intensity of each species to fit the fluorescence data (see Supporting Information, eqs S7 and S9).
The equations obtained by solving the kinetic rate equations for Scheme 1 were insufficient to determine all of the rate constants, as there are five rate constants in Scheme 1 but only three equations (eq S2, for each nonzero eigenvalue ρ). Additional constraints were therefore necessary to fit the data. First, k−3 and k3 were obtained from fits of the data for T110C-CaM-AF488 to Scheme 2 (see above). This is reasonable because the same slow time constant (~270 s) was obtained for both T34C-CaM-AF488 and T110C-CaM-AF488. Given these assumptions, multiple sets of values for k−2, k2, and k−1 still fit the experimental data. Further constraints on the values of the rate constants were therefore needed. These were obtained from the binding kinetics measured by the addition of concentrated C28W(1b) to T34C-CaM-AF488 shown in Figure 3.
For analysis of the binding experiment, it was necessary to include the binding step from UB to IN. Scheme 1 was therefore modified to include k1: The system of rate equations for Scheme 3 (see eq S13 in the Supporting Information) yielded further conditions for the kinetic rate constants. The fluorescence experiment reported here lacked the time resolution to directly measure k1, which has been reported for various enzymes to be on the order of 108 M−1s−1 (14, 15, 30). Caride and co-workers recently analyzed CaM binding to the CaM binding domain of isoform 4b of PMCA (14, 15). It is widely believed that the initial encounter between CaM and its targets involves the C-terminal domain of CaM interacting with the N-terminal region of the target domain (4, 5, 14, 31). The 18 N-terminal residues of C28W from isoforms 1b and 4b are identical. Therefore we assumed that the rate constants for the initial encounter between CaM and these two isoforms are similar. The observed differences in the overall kinetics of CaM association with the 1b and 4b isoforms of C28W are therefore likely due to differences in the contacts between CaM and the C-terminal regions of the C28W isoforms, which show some sequence variability (see below). Therefore, in order to model the data, we used the value for k1 reported for CaM binding to isoform 4b of C28W (4.6 × 108 M−1s−1 (14)).
Scheme 3.

With k3 and k−3 fixed as described above, the remaining rates constants were varied to find the best fit of the predicted rates in both Schemes 1 and 3 to the peaks found by MEM analysis. The rate constants that produced the best fit are listed in Table 2. From the best fit, the concentration of each species as a function of time is shown in Figure 4, and the relative fluorescence intensities of each species are shown in Table 3. As a check, the rate constants were substituted back into the kinetic rate equations (see Supporting Information, eqs S2 and S11), yielding time constants of 2.1 s, 15 s, and 291 s for T34C-CaM-AF488 in Scheme 1 (eq S2) and 8.8 s and 276 s for T110C in Scheme 2 (eq S11). The values for T34C-CaMox-AF488 and T110C-CaMox-AF488 were 23 s and 291 s. These values are shown as vertical lines in Fig. 2. These time constants are consistent with the experimental dissociation time constants (Table 1). Sources of possible error include the use of a single time constant for a process that MEM fits with a distribution and the assumption that k1 for isoform 4b (14) is the same as k1 for isoform 1b (see Scheme 3).
Figure 4.
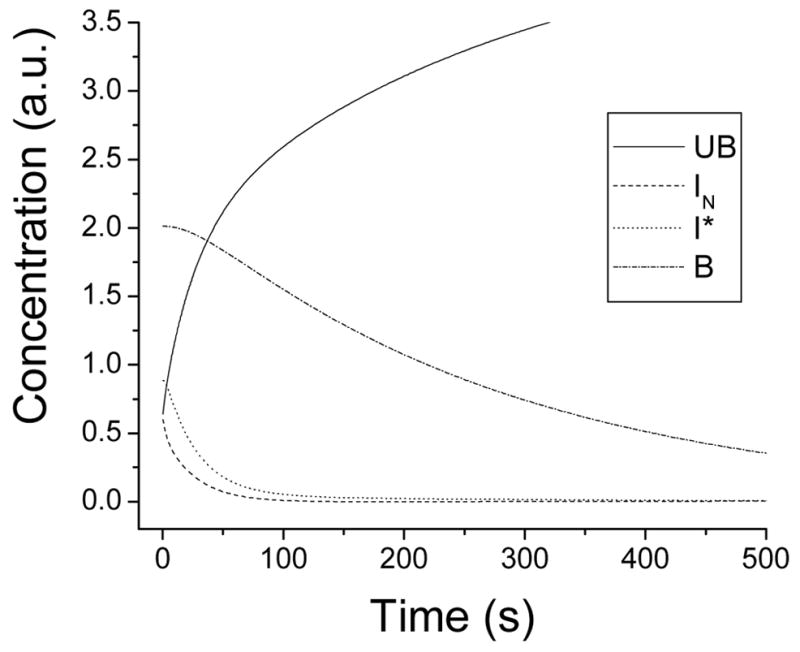
Modeled concentration of each species as a function of time for dissociation of C28W(1b) from CaM-AF488.
Table 3.
Relative fluorescence of each species from a fit of the data to eq 1. For oxidized CaM, the fluorescence of each species was found from the iterative fit of T34C-CaMox-AF488 and T110C-CaMox-AF488 to eqs (A11) and (A12).
| F (B) | F (I*) | F (IN) | F (UB) | |
|---|---|---|---|---|
| T34C-CaM-AF488 | 0.69 | 0.57 | 0.96 | 1.0 |
| T110C-CaM-AF488 | 0.59 | 0.79 | 0.79 | 1.0 |
| F (Box) | F (I*ox) | F (UBox) | ||
| T34C-CaMox-AF488 | 0.65 | 0.68 | 1.0 | |
| T110C-CaMox-AF488 | 0.72 | 0.93 | 1.0 | |
Oxidized CaM
With only two experimental rate constants, an analysis based on Scheme 2 was sufficient for the fit of the dissociation data for oxidized CaM. The rates kc, k−3, and k3 are here denoted k−cox, k−3ox, and k3ox. The MEM distributions for T34C-CaMox-AF488 and T110C-CaMox-AF488 (Figure 2) did not show a peak on the time scale of 1 to 20 seconds, suggesting that the fluorescence of AF488 at either domain is insensitive to any rapid interchange among intermediate species, or that such interchange does not occur. Further, as opposed to the unoxidized species, for oxidized CaM the slow process (time scale of hundreds of seconds) is lower in amplitude than the faster process (time scale of tens of seconds for T34C-CaMox-AF488 or ~100 seconds for T110C-CaMox-AF488). For T110C-CaMox-AF488 and T34C-CaMox-AF488, the slower components had amplitudes of 14% and 36%, respectively. For the unoxidized species, the corresponding amplitudes were 71% and 51% (Table 1).
The experimental time constants for CaMox were fit following the methods used for T110C-CaM-AF488 to yield values for the rate constants k3ox, k−3ox, and k−cox. For the initial analysis, FBox was set to 1.5 and FI*ox and FUBox were set to 1.0. This analysis yielded values for k−3ox for both T110C-CaMox-AF488 and T34C-CaMox-AF488 that were similar to the value of k−3 for unoxidized CaM. This rate constant was therefore fixed to the value for unoxidized CaM. The same rate constants were used for T110C-CaMox-AF488 and T34C-CaMox-AF488, and differences in the MEM distributions were assumed to be due to differences in the relative fluorescence intensities of the intermediates for the N-terminal and C-terminal labeled proteins. With the amplitudes and experimental time constants from the MEM distributions, rate constants k3ox and k−cox were varied along with the fluorescence intensities of FBox, FI*ox, and FUBox to generate a global fit to the dissociation data for both T110C-CaMox-AF488 and T34C-CaMox-AF488. The results of an iterative fit yielded the rate constants given in Table 2 and the relative fluorescence intensities of each species shown in Table 3. The value of k3ox for the final fit is lower for the oxidized proteins than k3 for unoxidized CaM as expected based on the lower relative amplitude of the slower MEM peak for the oxidized proteins.
Discussion
Binding Model
A number of studies have examined the kinetics of CaM binding and dissociation from targets such as myosin light chain kinase, CaM-dependent protein kinase II, and isoform 4b of PMCA (14, 15, 32, 33). In each of these studies, a multi-exponential time response was observed, suggesting mechanisms with at least one intermediate. To our knowledge, this is the first study that systematically monitored dissociation kinetics with CaM labeled on both the N-terminal and C-terminal domains. Perhaps the most interesting finding is the necessity for an additional time constant to fit the data for dissociation of C28W(1b) from T34C-CaM-AF488 relative to T110C-CaM-AF488. This result suggests, as has been proposed (4, 5, 14, 31), that an intermediate step in the interaction of CaM with PMCA involves release of the N-terminal domain of CaM. T34C-CaM-AF488 would be expected to be sensitive to such a step, while T110C-CaM-AF488 may not be.
Based on the number of peaks in the MEM distributions, the model for CaM dissociation from C28W(1b) must include two intermediate states. A similar model was employed previously for CaM binding to C28W from isoform 4b of PMCA (15). Two of the intermediates must have similar fluorescence characteristics for CaM labeled on the C-terminal domain, because only one intermediate was detected for dissociation of T34C-CaM-AF647. A model that satisfies all of these conditions, shown in Figure 5, is based on Scheme 1 and on a model proposed previously for interaction of CaM with the binding domain of myosin light-chain kinase (34). NMR results suggest that the formation of a tightly bound complex involves tight ionic contacts between the target helix and CaM (34, 35). The model consists of a bound complex B between CaM and C28W(1b) where the domains of CaM interact tightly with a fully helical C28W(1b) peptide.
Figure 5.

Model for CaM dissociation from C28W(1b). B represents the bound CaM-C28W(1b) complex with a helical CaM binding domain. In the intermediate I*, the C28W(1b) binding domain is not fully helical. In IN the N-terminal domain of CaM is dissociated from the peptide. UB represents the fully unbound state.
The first step in the dissociation pathway involves structural rearrangements from the compact, collapsed complex where the C28W(1b) peptide is predominantly helical to a less compact and more open form (34, 35). The C28W(1b) helix is destabilized and partial unwinding of the helix occurs during this step. The IN state is then formed as the C28W(1b) peptide unwinds to form a random coil structure and the N-terminal domain of CaM releases. Release of the C-terminal domain of CaM completes the dissociation process. The experiments presented here were conducted in the presence of 100 μM free Ca2+, a concentration that is high enough to saturate Ca2+ binding in both domains of CaM. Therefore, it is unlikely that any of the observed intermediates in the current study is due to release of a Ca2+-free N-terminal domain. Rather, it is likely that the ordered release of the N- and C-terminal domains occurs with two Ca2+ ions still bound in each domain.
Within the framework of this mechanism, the results in Table 3 reflect the structural changes that accompany rearrangement of the compact complex B to the intermediate I*. The distance between the tryptophan in C28W(1b) (Trp 8, responsible for the fluorescence quenching in the complex) and the fluorescence probe is increased substantially upon conversion of B to I* when the probe is in the C-terminal domain, as indicated by the relatively large fluorescence increase. The side chain of this tryptophan residue is presumably bound in the hydrophobic cleft of the C-terminal domain of CaM (4). Therefore, upon conversion of B to I*, both orientational changes of the peptide in the C-terminal binding cleft and concomitant structural changes in the C-terminal domain promote a significant distance change between the C-terminal fluorescent probe and the tryptophan in the C28W(1b) peptide. Conversely, the distance between the tryptophan in C28W(1b) and the fluorescence probe is decreased when the probe is in the N-terminal domain (T34C-CaM-AF488), as is demonstrated by the moderate decrease in fluorescence when B is converted to I*. This change is smaller than that observed when the probe is in the C-terminal domain, and reflects a repositioning of the N-terminal domain relative to the tryptophan in the C28W(1b) peptide perhaps due to unwinding and bending of the C28W(1b) helix. Conversion to IN is accompanied by a large change in fluorescence of the complex when the probe is in the N-terminal domain, consistent with dissociation of the N-terminal domain of CaM from the peptide, but no fluorescence change when the probe is in the C-terminal domain, indicating little change in the interaction of the C-terminal domain and the C28W(1b) peptide.
Relationship of Model to Previous work
The model in Figure 5 is consistent with other models presented in the literature. Török and coworkers measured the binding and dissociation kinetics for CaM with peptides from the CaM binding domains of myosin light chain kinase (29) and CaM-dependent protein kinase II (33), with CaM labeled in the central linker region or with a donor-acceptor pair. The kinetics could be modeled by a two-step mechanism with a single intermediate state. Caride and co-workers (for PMCA isoform 4b) and Persechini and coworkers (for myosin light chain kinase) suggested a CaM-peptide binding pathway where the C-domain of CaM binds first, then the N-domain, followed by further collapse of CaM around the target to form the final complex (14, 31). The model presented here is also consistent with one proposed by Wand and co-workers based on NMR data of CaM binding to the CaM-binding domain of smooth-muscle myosin light chain kinase, which predicts a binding intermediate consisting of CaM complexed with a partially helical target CaM binding domain and a fully bound complex with a helical target binding domain (34, 35).
Relationship to Isoform 4b
There are four isoforms of PMCA, and changes of only a few residues in the CaM binding domain of some isoforms can greatly affect the interaction between PMCA and CaM (6). Despite being the most common isoform, isoform 1 has been rarely studied due to difficulties in expression (12, 13). A detailed expression protocol has been developed and published recently (36). The characterization of CaM binding to the peptide representing the CaM binding domain of this isoform may be a useful precursor to studies with the intact enzyme.
Recently, two studies have examined the kinetics of CaM binding to isoform 4b of PMCA and the CaM-binding domain of this isoform (14, 15). The results of those studies suggest a mechanism for CaM binding to isoform 4b whereby the C-terminal domain of CaM binds first, followed by binding of the N-terminal domain and subsequent collapse of CaM around the target. Thus, the results reported here are consistent with the kinetics of CaM binding to C28W from isoform 4b of PMCA. However, the rates reported here for binding to isoform 1b are much slower than those reported for CaM binding to C28W from isoform 4b.
The fact that amino-acid sequence differences between the CaM binding regions of these two isoforms are limited to the C-terminal segment (see below) suggests that these C-terminal amino acids control the rates of interaction with CaM. Sequences of the CaM binding domains for different isoforms of PMCA are shown in Figure 6. The first 18 N-terminal residues of the CaM binding domains of these isoforms are identical. However, differences in the ten C-terminal residues of these isoforms are sufficient to dramatically alter their CaM binding affinities (36, 37). For instance, a recent study reported dissociation constants of CaM binding to C28W peptides from isoforms 3f and 2b that were nearly two orders of magnitude apart (38), and these differences must originate in the sequence differences in the C-terminal portions of the respective CaM binding regions. Thus, the differences in the kinetics reported for isoforms 1b and 4b must be due to the amino-acid sequence differences (only three of them, shown in lower case in Figure 6) in the C-terminal 10 residues of the CaM binding domains of isoforms 1b and 4b.
Figure 6.

Sequences of the CaM binding domains for different isoforms of PMCA (37,38). The first 18 residues (underlined) are identical for all isoforms. Differences between isoforms 1b and 4b are shown in lower case.
Hydrophobic interactions are thought to be the dominant force in target recognition and binding by CaM (1, 16). However, it is unlikely that the changes in hydrophobicity can account for the changes in binding kinetics between the peptides for isoforms 1b and 4b. Each of the residues involved in these differences have similar hydrophobicities. (Kyte and Doolittle hydrophobicity parameters are −4.5, −3.9, −3.5, and −3.2 for arginine, lysine, asparagine, and histidine, respectively (39).) Charge-charge interactions may contribute to the differences observed for the two isoform of PMCA. Isoform 1b has an asparagine at residue 23 and an arginine at residue 26. At neutral pH, the asparagine would be neutral while the arginine would have a positive charge. In isoform 4b, these charges are switched: the lysine at reside 23 would have a positive charge while the histidine at residue 26 would be neutral. Altered interactions between negatively charged CaM residues and positively charged residues in C28W(1b) may thus explain the differences between the affinity of CaM with isoform 1b observed here relative to CaM and isoform 4b. This notion is consistent with the idea that these electrostatic contributions are important for the specificity of interactions (34, 35). It should also be noted that the helix propensity of asparagine is significantly lower than that of arginine, lysine or histidine, and a decreased helicity in the peptide domain could contribute to decreased rates of association if the N-terminal domain of CaM interacts preferentially with a helical segment.
Relation to PMCA Activation
The peptide C28W(1b), which consists of the CaM binding domain of PMCA, is located in the autoinhibitory region of the enzyme. This domain of PMCA blocks activity of the enzyme in the absence of CaM (40) but is released or dissociated when CaM binds, thus activating the enzyme. The formation of a helix in the CaM-binding domain of PMCA is believed to be necessary for the release of the autoinhibitory domain. The C-terminal domain of CaM is thought to bind more tightly to target binding domains than the N-terminal domain, even when both domains are Ca2+-loaded (5). Binding of both the C- and N-terminal domains of CaM to the CaM-binding domain is required for activation of the enzyme (5). The formation of the fully bound complex B in C28W(1b) may thus be related to the induction of conformational changes in PMCA necessary for release of the autoinhibitory domain. Nevertheless, as demonstrated by Caride and co-workers (14), the kinetics of CaM binding to PMCA is more complex than binding to C28W because of participation of the rest of the enzyme and the possibility of CaM binding to both the closed and open conformations of the autoinhibitory domain.
Changes upon Oxidation
The oxidation of methionine residues has been shown to reduce the potency of CaM to maximally activate PMCA (17–19). Interestingly, this loss in activity is not due to a lack of binding of CaMox to PMCA, but rather to a reduced activity of PMCA-CaMox complexes (19, 41). The reduced productivity of CaMox binding has been linked to a reduced tendency for CaMox to generate structural changes in the CaM binding domain of PMCA that are necessary to induce dissociation of the autoinhibitory domain from the catalytic region of the enzyme to relieve autoinhibition (21, 41, 42). A recent study of structural changes in CaMox binding to C28W(1b) suggests that these functional changes result from a reduced helical propensity upon oxidation of Met144 and Met145 in the C-terminal helix of CaM (21).
In this study, we found that the kinetics of dissociation of C28W(1b) are perturbed by oxidation of the methionine residues in CaM in the following ways. First, the fast dissociation time observed with CaM (2 to 10 s) was not observed in CaMox. Second, the long dissociation time constant (36 s to 118 s) had a higher relative amplitude for CaMox than the corresponding time constants for unoxidized CaM. This result corresponds to a lower fraction of tight binding orientations for oxidized CaM relative to unoxidized. The resulting shift in the equilibrium away from a fully helical binding domain is consistent with the reduced tendency for release of the autoinhibitory domain in PMCA bound to CaMox (42). As a result, the net dissociation time for C28W(1b) is faster from CaMox than from unoxidized CaM.
The dissociation kinetics for CaMox suggest that a mechanistic model based on Scheme 2 can be applied to the dissociation kinetics for both T34C-CaMox-AF488 and T110C-CaMox-AF488. The absence of a third time constant for T34C-CaMox-AF488 in contrast to T34C-CaM-AF488 suggests that the final step in the dissociation of C28W(1b) from CaMox does not result in different fluorescence signals when for the fluorescent probe is in the C- or N-terminal domains of CaMox. Thus, a distinct intermediate with one domain dissociated (IN in Scheme 1) was not detected. This suggests either that dissociation of one domain is rapidly followed by dissociation of the other so that the step IN → UB (Scheme 1) is not resolved, as suggested by the absence of a time constant of less than 10 s for CaMox, or that binding of the N-terminal and C-terminal domains of CaMox may no longer be ordered and sequential, as it is for unoxidized CaM.
The rate constant k3ox for formation of the final complex (species B) of oxidized CaM and C28W(1b) is slower for oxidized CaM than it is for native CaM (Table 2), significantly altering the equilibrium for interconversion of B and I* relative to native CaM. The reduced rate of formation of species B with CaMox results in a predicted drop in the equilibrium constant for this step in Scheme 2 from 2.3 to 0.95. The resulting shift in the equilibrium away from a fully helical binding domain is consistent with the reduced tendency for release of the autoinhibitory domain in PMCA. The reduction of this rate may shift the equilibrium sufficiently to reduce the likelihood for oxidized CaM to induce the necessary structural changes in the target that ultimately promote dissociation of the autoinhibitory domain. This result is consistent with the observation that for species B, the N- and C-terminal domains of CaM are in closer proximity in the CaMox-C28W(1b) complex relative to the CaM-C28W(1b) complex (41). As described above, we speculate that elongation of C28W(1b) as it becomes helical in the final binding step for unoxidized CaM is responsible for the increase in fluorescence of T34C-CaM-AF488 for species B relative to I* (Table 3). A lack of full helix formation would preclude this result. As expected for oxidized CaM, the fluorescence yield of B was not increased relative to I* for T34C-CaMox-AF488 (Table 3), consistent with a final bound complex with oxidized CaM where C28W(1b) is not fully helical.
Conclusions
The fluorescence of AF488 conjugated to CaM is sensitive to the binding and dissociation of C28W(1b), the CaM binding domain of PMCA. Two CaM mutants, one with AF488 conjugated to residue 34 in the N-domain and the other to residue 110 in the C-domain were used to elucidate the nature of different species in the dissociation pathway. The results indicate that binding of C28W(1b) involves an intermediate consisting of a bound C-domain with a free N-domain, followed by N-domain binding with partial helix formation of the target, and finally a slight increase in distance between lobes of CaM as the helix fully forms. Previous measurements with CaM labeled at Lys 75 (15) also favored a model where CaM binds to C28W from isoform 4b of PMCA in three reversible steps with two intermediates. The present results for isoform 1b are consistent with that kinetic model, but with rates constants that are lower by at least an order of magnitude. Furthermore, by use of CaM with fluorescence labels in either the N- or C-terminal domains, the present results also reveal the different roles of the N- and C-terminal domains in the individual kinetic steps. Analysis of the dissociation kinetics for oxidized CaM suggests that CaMox is less likely to form a tight binding complex due to a decrease in the rate of formation of this complex. These results support the idea that induction of helical structure in the CaM binding domain of PMCA is an obligatory step for relief of autoinhibition and enzyme activation.
Supplementary Material
Supporting information includes the kinetic rate equations and solutions required for analysis of the kinetic data. This material is available free of charge via the internet at http://pubs.acs.org
Acknowledgments
We thank Professor Richard Schowen for insightful discussions regarding binding kinetics. This research was supported by NIH grant R01 GM58715. B.D.S. acknowledges support from the Dynamic Aspects of Chemical Biology NIH training grant (NIH 5 T32 GM08545-09).
Abbreviations
- C28W(1b)
28-residue CaM binding domain from isoform 1b of the plasma membrane Ca2+-ATPase
- CaM
calmodulin
- CaMox
oxidized calmodulin
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(4-butanesulfonic acid)
- PMCA
plasma-membrane Ca2+-ATPase
- T34C-CaM-AF488
calmodulin T34C mutant fluorescently labeled at Cys34 with Alexa Fluor 488
- T110C-CaM-AF488
calmodulin T110C mutant fluorescently labeled at Cys110 with Alexa Fluor 488
References
- 1.Vetter SW, Leclerc E. Novel Aspects of Calmodulin Target Recognition and Activation. Eur J Biochem. 2003;270:404–414. doi: 10.1046/j.1432-1033.2003.03414.x. [DOI] [PubMed] [Google Scholar]
- 2.Bayley P, Ahlstrom P, Martin SR, Forsen S. The Kinetics of Calcium Binding to Calmodulin: Quin 2 and ANS Stopped-Flow Fluorescence Studies. Biochem Biophys Res Commun. 1984;120:185–191. doi: 10.1016/0006-291x(84)91431-1. [DOI] [PubMed] [Google Scholar]
- 3.VanScyoc WS, Sorensen BR, Rusinova E, Laws WR, Ross JBA, Shea MA. Calcium Binding to Calmodulin Mutants Monitored by Domain-Specific Intrinsic Phenylalanine and Tyrosine Fluorescence. Biophys J. 2002;83:2767–2780. doi: 10.1016/S0006-3495(02)75286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elshorst B, Hennig M, Foersterling H, Diener A, Maurer M, Schulte P, Schwalbe H, Griesinger C, Krebs J, Schmid H, Vorherr T, Carafoli E. NMR Solution Structure of a Complex of Calmodulin with a Binding Peptide of the Calcium Pump. Biochemistry. 1999;38:12320–12332. doi: 10.1021/bi9908235. [DOI] [PubMed] [Google Scholar]
- 5.Sun H, Squier TC. Ordered and Cooperative Binding of Opposing Globular Domains of Calmodulin to the Plasma Membrane Ca-ATPase. J Biol Chem. 2000;275:1731–1738. doi: 10.1074/jbc.275.3.1731. [DOI] [PubMed] [Google Scholar]
- 6.Penniston JT, Enyedi A. Modulation of the Plasma Membrane Ca2+ Pump. J Membr Biol. 1998;165:101–109. doi: 10.1007/s002329900424. [DOI] [PubMed] [Google Scholar]
- 7.Carafoli E, Brini M. Calcium Pumps: Structural Basis for and Mechanism of Calcium Transmembrane Transport. Curr Opin Chem Biol. 2000;4:152–161. doi: 10.1016/s1367-5931(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 8.Gao J, Yin DH, Yao Y, Sun H, Qin Z, Schöneich C, Williams TD, Squier TC. Loss of Conformational Stability in Calmodulin Upon Methionine Oxidation. Biophys J. 1998;74:1115–1134. doi: 10.1016/S0006-3495(98)77830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kataoka M, Head JF, Vorherr T, Krebs J, Carafoli E. Small-Angle X-Ray Scattering Study of Calmodulin Bound to Two Peptides Corresponding to Parts of the Calmodulin-Binding Domain of the Plasma Membrane Ca2+ Pump. Biochemistry. 1991;30:6247–6251. doi: 10.1021/bi00239a024. [DOI] [PubMed] [Google Scholar]
- 10.Chapman ER, Alexander K, Vorherr T, Carafoli E, Storm DR. Fluorecence Energy Transfer Analysis of Calmodulin-Peptide Complexes. Biochemistry. 1992;31:12819–12825. doi: 10.1021/bi00166a016. [DOI] [PubMed] [Google Scholar]
- 11.Stauffer TP, Hilfiker H, Carafoli E, Strehler EE. Quantitative Analysis of Alternative Splicing Options of Human Plasma Membrane Calcium Pump Genes. J Biol Chem. 1993;268:25993–26003. [PubMed] [Google Scholar]
- 12.Adamo HP, Verma AK, Sanders MA, Heim R, Salisbury JL, Wieben ED, Penniston JT. Overexpression of the Erythrocyte Plasma Membrane Ca2+ Pump in Cos-1 Cells. Biochem J. 1992;285:791–797. doi: 10.1042/bj2850791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu BF, Xu X, Fridman R, Muallem S, Kuo TH. Consequences of Functional Expression of the Plasma Membrane Ca2+ Pump Isoform 1a. J Biol Chem. 1996;271:5536–5544. doi: 10.1074/jbc.271.10.5536. [DOI] [PubMed] [Google Scholar]
- 14.Penheiter AR, Bajzer Z, Filoteo AG, Thorogate R, Török K, Caride AJ. A Model for the Activation of Plasma Membrane Calcium Pump Isoform 4b by Calmodulin. Biochemistry. 2003;42:12115–12124. doi: 10.1021/bi027098+. [DOI] [PubMed] [Google Scholar]
- 15.Penheiter AR, Filoteo AG, Penniston JT, Caride AJ. Kinetic Analysis of the Calmodulin-Binding Region of the Plasma Membrane Calcium Pump Isoform 4b. Biochemistry. 2005;44:2009–2020. doi: 10.1021/bi0488552. [DOI] [PubMed] [Google Scholar]
- 16.Crivici A, Ikura M. Molecular and Structural Basis of Target Recognition by Calmodulin. Annu Rev Biophys Biomol Struct. 1995;24:85–116. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- 17.Yao Y, Yin D, Jas GS, Kuczera K, Williams TD, Schöneich C, Squier TC. Oxidative Modification of a Carboxyl-Terminal Methionine in Calmodulin by Hydrogen Peroxide Inhibits Calmodulin-Dependent Activation of the Plasma Membrane Ca-ATPase. Biochemistry. 1996;35:2767–2787. doi: 10.1021/bi951712i. [DOI] [PubMed] [Google Scholar]
- 18.Gao J, Yao Y, Williams TD, Squier TC. Progressive Decline in the Ability of Calmodulin Isolated from Aged Brain to Activate the Plasma Membrane Ca-ATPase. Biochemistry. 1998;37:9536–9548. doi: 10.1021/bi9803877. [DOI] [PubMed] [Google Scholar]
- 19.Bartlett RK, Bieber Urbauer RJ, Anbanandam A, Smallwood HS, Urbauer JL, Squier TC. Oxidation of Met144 and Met145 in Calmodulin Blocks Calmodulin Dependent Activation of the Plasma Membrane Ca-ATPase. Biochemistry. 2003;42:3231–3238. doi: 10.1021/bi026956z. [DOI] [PubMed] [Google Scholar]
- 20.Squier TC, Bigelow DJ. Protein Oxidation and Age-Dependent Alterations in Calcium Homeostasis. Front Biosci. 2000;5:d504–526. doi: 10.2741/squier. [DOI] [PubMed] [Google Scholar]
- 21.Anbanandam A, Bieber Urbauer RJ, Bartlett RK, Smallwood HS, Squier TC, Urbauer JL. Mediating Molecular Recognition by Methionine Oxidation: Conformational Switching by Oxidation of Methionine in the Carboxyl-Terminal Domain of Calmodulin. Biochemistry. 2005;44:9486–9496. doi: 10.1021/bi0504963. [DOI] [PubMed] [Google Scholar]
- 22.Allen MW, Urbauer RJB, Zaidi A, Williams TD, Urbauer JL, Johnson CK. Fluorescence Labeling, Purification and Immobilization of a Double Cysteine Mutant Calmodulin Fusion Protein for Single-Molecule Experiments. Anal Biochem. 2004;325:273–284. doi: 10.1016/j.ab.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 23.Slaughter BD, Unruh JR, Price ES, Huynh JL, BieberUrbauer RJ, Johnson CK. Sampling Unfolding Intermediates in Calmodulin by Single-Molecule Spectroscopy. J Am Chem Soc. 2005;127:12107–12114. doi: 10.1021/ja0526315. [DOI] [PubMed] [Google Scholar]
- 24.Steinbach PJ, Ionescu R, Matthews CR. Analysis of Kinetics Using a Hybrid Maximum-Entropy/Nonlinear-Least-Squares Method: Application to Protein Folding. Biophys J. 2002;82:2244–2255. doi: 10.1016/S0006-3495(02)75570-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornwell TJ, Evans KF. A Simple Maximum Entropy Deconvolution Algorithm. Astron Astrophys. 1985;143:77–83. [Google Scholar]
- 26.Swaminathan R, Periasamy N. Analysis of Fluorescence Decay by the Maximum Entropy Method: Influence of Noise and Analysis Parameters on the Width of the Distribution of Lifetimes. Proc Indian Acad Sci Chem Sci. 1996;108:39–49. [Google Scholar]
- 27.Steinbach PJ. Two-Dimensional Distributions of Activation Enthalpy and Entropy from Kinetics by the Maximum Entropy Method. Biophys J. 1996;70:1521–1528. doi: 10.1016/S0006-3495(96)79714-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brochon JC. Maximum Entropy Method of Data Analysis in Time-Resolved Spectroscopy. Methods Enzymol. 1994;240:262–311. doi: 10.1016/s0076-6879(94)40052-0. [DOI] [PubMed] [Google Scholar]
- 29.Török K, Trentham DR. Mechanism of 2-Chloro-(E-Amino-Lys75)-[6-[4-(N,N-Diethylamino)Phenyl]-1,3,5-Triazin-4-Yl]Calmodulin Interactions with Smooth Muscle Myosin Light Chain Kinase and Derived Peptides. Biochemistry. 1994;33:12807–12820. doi: 10.1021/bi00209a012. [DOI] [PubMed] [Google Scholar]
- 30.Török K, Cowley DJ, Brandmeier BD, Howell S, Aitken A, Trentham DR. Inhibition of Calmodulin-Activated Smooth-Muscle Myosin Light-Chain Kinase by Calmodulin-Binding Peptides and Fluorescent (Phosphodiesterase-Activating) Calmodulin Derivatives. Biochemistry. 1998;37:6188–6198. doi: 10.1021/bi972773e. [DOI] [PubMed] [Google Scholar]
- 31.Persechini A, Yano K, Stemmer PM. Ca2+ Binding and Energy Coupling in the Calmodulin-Myosin Light Chain Kinase Complex. J Biol Chem. 2000;275:4199–4204. doi: 10.1074/jbc.275.6.4199. [DOI] [PubMed] [Google Scholar]
- 32.Brown SE, Martin SR, Bayley PM. Kinetic Control of the Dissociation Pathway of Calmodulin-Peptide Complexes. J Biol Chem. 1997;272:3389–3397. doi: 10.1074/jbc.272.6.3389. [DOI] [PubMed] [Google Scholar]
- 33.Török K, Tzortzopoulos A, Grabarek Z, Best SL, Thorogate R. Dual Effect of ATP in the Activation Mechanism of Brain Ca2+ Calmodulin-Dependent Protein Kinase II by Ca2+ Calmodulin. Biochemistry. 2001;40:14878–14890. doi: 10.1021/bi010920+. [DOI] [PubMed] [Google Scholar]
- 34.Ehrhardt MR, Urbauer JL, Wand AJ. The Energetics and Dynamics of Molecular Recognition by Calmodulin. Biochemistry. 1995;34:2731–2738. doi: 10.1021/bi00009a001. [DOI] [PubMed] [Google Scholar]
- 35.Kranz JK, Flynn PF, Fuentes EJ, Wand AJ. Dissection of the Pathway of Molecular Recognition by Calmodulin. Biochemistry. 2002;41:2599–2608. doi: 10.1021/bi011818f. [DOI] [PubMed] [Google Scholar]
- 36.Brini M, Coletto L, Pierobon N, Kraev N, Guerini D, Carafoli E. A Comparative Functional Analysis of Plasma Membrane Ca2+ Pump Isoforms in Intact Cells. J Biol Chem. 2003;278:24500–24508. doi: 10.1074/jbc.M300784200. [DOI] [PubMed] [Google Scholar]
- 37.Enyedi A, Filoteo AG, Gardos G, Penniston JT. Calmodulin-Binding Domains from Isozymes of the Plasma Membrane Ca2+ Pump Have Different Regulatory Properties. J Biol Chem. 1991;266:8952–8956. [PubMed] [Google Scholar]
- 38.Filoteo AG, Enyedi A, Verma AK, Elwess NL, Penniston JT. Plasma Membrane Ca2+ Pump Isoform 3f Is Weakly Stimulated by Calmodulin. J Biol Chem. 2000;275:4323–4328. doi: 10.1074/jbc.275.6.4323. [DOI] [PubMed] [Google Scholar]
- 39.Kyte J, Doolittle RF. A Simple Method for Displaying the Hydropathic Character of a Protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 40.Enyedi A, Vorherr T, James P, McCormick DJ, Filoteo AG, Carafoli E, Penniston JT. The Calmodulin Binding Domain of the Plasma Membrane Ca2+ Pump Interacts Both with Calmodulin and with Another Part of the Pump. J Biol Chem. 1989;264:12313–12321. [PubMed] [Google Scholar]
- 41.Gao J, Yao Y, Squier TC. Oxidatively Modified Calmodulin Binds to the Plasma Membrane Ca-ATPase in a Nonproductive and Conformationally Disordered Complex. Biophys J. 2001;80:1791–1801. doi: 10.1016/S0006-3495(01)76149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osborn KD, Bartlett RK, Mandal A, Zaidi A, Urbauer RJB, Urbauer JL, Galeva N, Williams TD, Johnson CK. Single-Molecule Dynamics Reveal an Altered Conformation for the Autoinhibitory Domain of Plasma-Membrane Ca2+-ATPase Bound to Oxidatively Modified Calmodulin. Biochemistry. 2004;43:12937–12944. doi: 10.1021/bi048806p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information includes the kinetic rate equations and solutions required for analysis of the kinetic data. This material is available free of charge via the internet at http://pubs.acs.org