

Abstract
Invariant Natural Killer T cells (iNKT cells) have been reported to play a role not only in innate immunity but also to regulate several models of autoimmunity. Furthermore, iNKT cells are necessary for the generation of the prototypic eye-related immune regulatory phenomenon, anterior chamber associated immune deviation (ACAID). Here we explore the role of iNKT cells in regulation of autoimmunity to retina, using a model of experimental autoimmune uveitis (EAU) in mice immunized with a uveitogenic regimen of the retinal antigen (Ag), IRBP. Natural strain-specific variation in iNKT number or induced genetic deficiencies in iNKT did not alter baseline susceptibility to EAU. However, iNKT function seemed to correlate with susceptibility and its pharmacological enhancement in vivo by treatment with iNKT TCR ligands at the time of uveitogenic immunization reproducibly ameliorated disease scores. Use of different iNKT TCR ligands revealed dependence on the elicited cytokine profile. Surprisingly, superior protection against EAU was achieved with α-C-GalCer, which induces a strong IFN-γ but only a weak IL-4 production by iNKT cells, in contrast to the ligands α-GalCer (both IFN-γ and IL-4) and OCH (primarily IL-4). The protective effect of α-C-Gal-Cer was associated with a reduction of adaptive Ag specific IFN-γ and IL-17 production and was negated by systemic neutralization of IFN-γ. These data suggest that pharmacological activation of iNKT cells protects from EAU at least in part by a mechanism involving innate production of IFN-γ and a consequent dampening of the Th1 as well as the Th17 effector responses.
Keywords: NKT cells, autoimmune disease, cytokines
INTRODUCTION
Invariant Natural Killer T cells (iNKT) are considered to represent an innate subset of T cells. iNKT cells have a semi-invariant Vα14-Jα18 TCR repertoire specific for lipid antigens that are presented on the MHC class I-like CD1d molecule. α̂GalCer, a synthetic glycolipid derived from marine sponges, is a well known iNKT TCR ligand (1). Recently, natural ligands of iNKT TCR have been described, such as α-glucuronosylceramide and glycosphingolipids from Sphingomonadaceae (2–4), that resemble cell wall constituents of some Gram-negative bacteria, supporting an innate role of iNKT cells in some infectious diseases. Other studies also revealed regulatory and protective properties in various models of autoimmunity such as experimental autoimmune encephalomyelitis (EAE) and type-1 diabetes (reviewed in 1). Most of these studies utilized synthetic ligands for iNKT cell activation and therapeutic regimen.
Upon ligation of their invariant TCR with α–GalCer, iNKT cells rapidly produce large amounts of cytokines such as IFN-γ and IL-4 (5). Analogs of α-GalCer prepared by total synthesis led to the identification of ligands that induce a cytokine pattern that is more biased toward either a Th1-type (IFN-γ) or a Th2-type (IL-4) response. An example of the former is αC-GalCer, while the latter includes the ligand OCH (6, 7). Some studies demonstrated altered biological effects of these α–GalCer analogs on autoimmunity and cancer that could be ascribed to the cytokine profiles they elicit (6, 7).
Experimental autoimmune uveitis (EAU) induced in animals by immunization with retinal antigens in complete Freund’s adjuvant (CFA) is a model for human autoimmune uveitis, a disease that accounts for about 10–15% of severe visual handicap in the US. EAU is induced by immunization with the same retinal antigens that are recognized by uveitis patients and is dependent on CD4+ Th1 and Th17 effector cells (8, 9)}. EAU in mice is induced with the interphotoreceptor retinoid-binding protein (IRBP) or with its pathogenic fragments emulsified in CFA (8).
The relationship between NKT cells and eye-related immune responses is not well understood. Although iNKT cells have been shown to play an important role in the eye-related regulatory phenomenon known as anterior chamber-associated immune deviation (ACAID) (10, 11), the possible role of iNKT cells in regulation of EAU has not been established. In the present study we examine the role of iNKT cells and the effects of the iNKT cell ligands on EAU. Our data show that although natural strain-specific variations or genetically induced lack of iNKT cells do not seem to affect the threshold of susceptibility to EAU, iNKT function seemed to correlate with susceptibility. Importantly, a pharmacological enhancement of these cells using glycolipid iNKT cell ligands was able to inhibit induction of disease. This appeared to be due at least in part to iNKT-produced IFN-γ and a consequent dampening of the adaptive Th1 and Th17 pathogenic effector responses.
MATERIALS AND METHODS
Animals
B10.RIII, B10.A, C57BL/6, DBA/2, AKR and BALB/c mice (WT and CD1d-KO) were purchased from The Jackson Laboratory (Bar Harbor, ME). All experiments were approved by the NEI Animal Care and Use Committee. Animal care and use conformed to Institutional guidelines and to the ARVO guidelines on the use of animals in ophthalmic and vision research.
Antigens and Reagents
Bovine IRBP was purified as described (12, 13). Complete Freund’s adjuvant (CFA) was purchased from Difco (Detroit, MI) and was supplemented with additional Mycobacterium tuberculosis H37RA to 2.5 mg/ml. Purified derivative of tuberculin (PPD) was purchased from the Statens Seruminstitut (Copenhagen, Denmark). α-GalCer (KRN7000) was provided by the Kirin Brewery Co, Tokyo, Japan (14). α-C-GalCer was synthesized as described previously (6). OCH was synthesized by Drs. Chi-Huey Wong and Douglas Wu of the Scripps Research Institute, La Jolla, CA. Neutralizing anti-IFN-γ antibodies (clone R4-6A2) were obtained from the Biological Resources Branch, NCI, Frederick MD.
Isolation of lymphoid cells from liver
Livers were perfused in situ through the hepatic portal vein with RT PBS and minced into small pieces PBS with 2%FCS and 0.02% Sodium Azide (PBS/FCS/Az). The tissue was then pressed though a 200 gauge mesh and cells were suspended in cold (4°C) PBS/FCS/Az. Cells were washed twice (500 g for 7 min). The pelleted cells were resuspended in 37.5% isotonic percoll at room temperature (25ml per liver) and spun at 680g for 12 min. The cell pellet made up of lymphocytes and erythrocytes was collected, washed once in PBS/FCS/Az and erythrocytes were lysed with 2mL red cell lysis buffer (Sigma-Aldrich) for 4min. Cells were then recovered by centrifugation through a layer of fetal bovine serum, resuspended in PBS/FCS/Az, filtered through 100 μm mesh, counted and prepared for immunophenotyping by flow cytometry.
Immunization, EAU Induction and EAU Scoring
C57BL/6 and BALB/c mice were immunized with 150 μg emulsified in CFA supplemented with Mycobacterium tuberculosis, strain H37RA from Difco (Detroit, MI) to 2.5 mg/ml. Clinical disease was evaluated by fundus examination in a masked fashion and was scored on a scale from 0 (no inflammation) to 4 (complete destruction of the retina) in half-point increments, as described previously (8). Eyes were harvested for histopathology 21 d after immunization. Disease was scored by an ophthalmic pathologist (C.-C. Chan) in a masked fashion as described previously (8).
α–GalCer and other treatments
Unless otherwise noted, 5 μg of either α-GalCer, α-C-GalCer or OCH were added to and emulsified with the uveitogenic Ag preparation (IRBP/CFA 1:1 v/v). The emulsion was injected subcutaneously, divided into 3 doses (both thighs and base of tail). Systemic IFN-γ neutralization was achieved by treatment with monoclonal anti-IFN-γ Ab, 150 ug/mouse injected i.p. on days −2, 0, 2).
Determination of Immunological Responses
Cytokine productionton to α–GalCer analogs in culture was examined on splenocytes obtained from naive mice. Cell suspensions of 2.5 × 106 cells/ml were incubated with 100ng/ml of the stimulant and supernatants were collected after 48 h. Cytokine production in vivo to α–GalCer analogs was measured in sera collected at the indicated time points after intraperitoneal injection of 5 μg of the analog. For determination of Ag specific cytokine production and proliferation, spleens and lymph nodes draining the site of immunization (inguinal and iliac) were collected on day 21 and were pooled within each group. Proliferation to the indicated doses of antigen was assayed by 3H-Thymidine uptake during the last 16 h of a 72 h culture on triplicate cultures of 0.2 ml, as described (15). Cytokine responses were determined using the Pierce Chemical Co. multiplex SearchLight™ Arrays technology (16 and http://www.endogen.com/services).
Flow cytometry
iNKT cells were enumerated by flow cytometry after exclusion of dead cells by DNA staining with 7-Amino-Actinomycin D (7-AAD). Cells were reacted with PE-labeled CD1d/α–GalCer tetramers, APC-labeled β-TCR and FITC-labeled anti-CD4 antibodies. β-TCR positive cells were gated and the percentage of α-GalCer positive cells (either CD4+ or−) was determined by counting of 1 × 105 viable β-TCR positive cells. Multiplication of percentages by absolute numbers of lymphocytes that were isolated from each organ (beta counter) resulted in the total iNKT numbers shown in figure 1.
Fig. 1. Total iNKT cell numbers and percentages of CD4+ iNKT cells seem unrelated to susceptibility to EAU.

Six different mouse strains (six mice per strain, 8 weeks of age) were immunized with 150 μg IRBP in Complete Freund’s Adjuvant and an additional i.p. injection of 0.3 μg Pertussis Toxin (PTx). B10.RIII mice were immunized with 10 μg IRBP without additional PTx. (a): Schematic representation of typical strain-specific disease scores and representative histopathology (inset), based on established data. Lymphocytes from naïve livers, thymuses and spleens were isolated on a density gradient and iNKT cell numbers were determined by flow cytometry using labeled CD1d/α–GalCer tetramers, anti-CD4 and anti-β-TCR antibodies as described in Materials &Methods. (b): Average of isolated iNKT cell numbers in thymus, LN and spleen, expressed as a total cell number collected from each mouse. (c): Average of isolated iNKT cell numbers in thymus, LN and spleen, expressed as a percentage of the total cell number collected from each mouse. (d): Proportion of CD4+ iNKT cells expressed as a percentage of total iNKT cells. The data are pooled from 2 identical experiments of 3 mice each (total 6 individual animals per point).
Statistical Analysis and data presentation
All experiments were performed at least twice and results were highly reproducible. Figures show data from representative or from pooled experiments, as specified. EAU severity is represented by fundoscopy scores determined on day 19–21. All fundoscopy scores were confirmed by histopathology. Where appropriate, statistical analysis of EAU severity was performed using the Snedecor and Cochran z test for linear trend in proportions (17). This is a nonparametric, frequency-based test that takes into account both disease severity and incidence. Probability values of < 0.05 were considered to be significant. Values determined to be significantly different from controls are marked with an asterisk in the figures.
RESULTS
iNKT cytokine profile, but not their number, may correlate with susceptibility to EAU
EAU is a disease model where susceptibility varies considerably among different mouse strains. The strain with the highest known susceptibility is B10.RIII. B10.A is another susceptible strain that can develop high disease scores, but unlike the B10.RIII strain it requires administration of pertussis toxin at the time of immunization in order to develop EAU, as do all other susceptible strains. C57BL/6 and DBA/2 mice have mild to moderate susceptibility and AKR as well as BALB/c mice are resistant to disease. We asked the question whether susceptibility to disease in a series of EAU-characterized mouse strains correlated with their numbers of iNKT cells. Figure 1a shows a schematic representation of typical EAU scores for six mouse strains, summarizing previously reported findings (18–20). Representative pictures of severe mild and no disease are shown in the inset.
Most of the iNKT cells in the body are concentrated in the thymus, liver and spleen. We enumerated invariant TCR-bearing NKT cells in these 3 organs in the different mouse strains using flow cytometry, by binding of PE-labeled CD1d/α–GalCer-Tetramers. Although there were strain-specific differences in iNKT cell numbers and variations in their content in the different organs, there was no correlation with strain-specific differences in disease susceptibility (Fig. 1b and 1c). In addition, a low iNKT number in one organ (e.g. thymus B10.A, spleen AKR) tended to be counterbalanced by a higher number in another organ (e.g. liver of B10.A and AKR) in some strains. Consequently, the total number of iNKT cells was often similar between strains with different EAU susceptibilities (Fig. 1b).
We next examined the percent of CD4+ iNKT cells out of the total iNKT cells, as this subset plays a role in the eye-specific regulatory phenomenon known as ACAID (11, 21). CD4+ iNKT cells were enumerated by double staining for CD4 and for the invariant TCR using CD1d/α–GalCer tetramers. The data revealed that the differences in CD4+ iNKT cells between the strains were even less pronounced than total iNKT numbers and did not correlate with disease susceptibility (Fig. 1d).
Lastly, we selected 3 strains that were either resistant, moderately susceptible or highly susceptible to EAU (Balb/c, C57Bl/6 and B10RIII, respectively) and tested their iNKT cells in vitro using two different ligands: α-GalCer and OCH, each known to elicit differing cytokine profiles (4, 22). Single cell suspensions were made from whole spleens and were stimulated with the indicated ligands for 48 hours. Analysis of culture supernatants revealed that iNKT cells in resistant Balb/c mice produce significantly higher levels of IFN-γ, IL-4 and IL-2, irrespective of the ligand used for stimulation (Figure 2). Interestingly, the opposite was the case with IL-17, with the highest levels detected in supernatant of B10RIII splenocytes. iNKT cells have recently been identified as a source of innate IL-17 (23), although the significance of this response for autoimmune disease is yet to be defined. No inter-strain differences were found in other cytokines examined such as IL-10, IL-13 and IL-5 (not shown).
Fig. 2. Functional differences are present in iNKT cells from susceptible, compared with resistant strains.

Splenocytes from Balb/c, C57Bl/6 and B10RIII mice were isolated and cultured in HL-1 media containing 1% normal mouse serum and were left either unstimulated, or were treated with 100ng/ml of either α-GalCer or OCH, as indicated in the figure. Cell supernatants were collected after 48 hours and cytokines were analyzed by Pierce Searchlight Technology. Data are representative of 2 experiments with 3 individual mice in each group. Supernatants were pooled prior to analysis.
Genetic lack of iNKT cells does not enhance susceptibility to EAU
If iNKT cells had a role in raising the threshold of susceptibility to EAU, we would expect that iNKT deficiency would result in more severe disease. However, mice deficient in CD1d (lacking CD1d-dependent NKT cells) (24) did not show enhanced EAU susceptibility compared to their WT counterparts (Fig. 3). The time of onset as well as the course of disease as determined by periodic fundus examinations were also not affected (data not shown). Mice deficient in CD1d on the resistant BALB/c background remained resistant (Fig. 3). These data are consistent with observations made by others in the EAE model (25–27).
Fig. 3. Lack of iNKT cells does not seem to alter the disease course or the susceptibility to EAU.
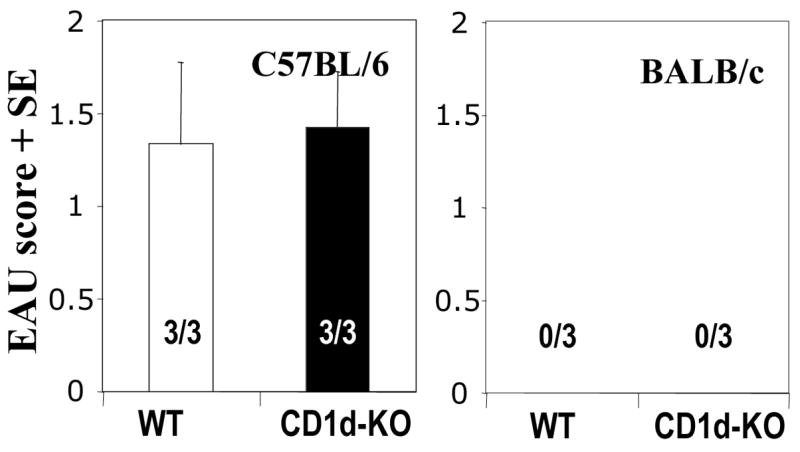
An EAU susceptible (C57BL/6) and resistant strain (BALB/c) were immunized as described in Fig. 1a. Likewise iNKT deficient strains on these genetic backgrounds were immunized and EAU scores were compared with the WT mice. (a) EAU scores of C57BL/6 WT and Jα18-KO mice. (b) EAU scores of C57BL/6 WT and CD1d-KO mice. (c) EAU scores of BALB/c WT and CD1d-KO mice. Representative experiment of 2 showing the same pattern (data were not pooled due to inter-experiment variation in disease severity). Positive mice out of total are indicated within each bar.
Activation of iNKT cells ameliorates EAU but analogs of α-GalCer differ in their efficacy
We next examined whether functional triggering of iNKT cells using invariant TCR ligands can affect EAU. Five micrograms of α-GalCer incorporated into the IRBP/CFA emulsion ameliorated EAU severity and incidence in C57Bl/6 mice (Fig. 4a). To examine the effect of α–GalCer analogs, mice were treated with α-C-Gal-Cer and OCH. The data showed that OCH protected no better than α–GalCer, whereas α-C-GalCer was the most effective (Fig. 4b).
Fig. 4. Effect of iNKT cell activation on EAU.
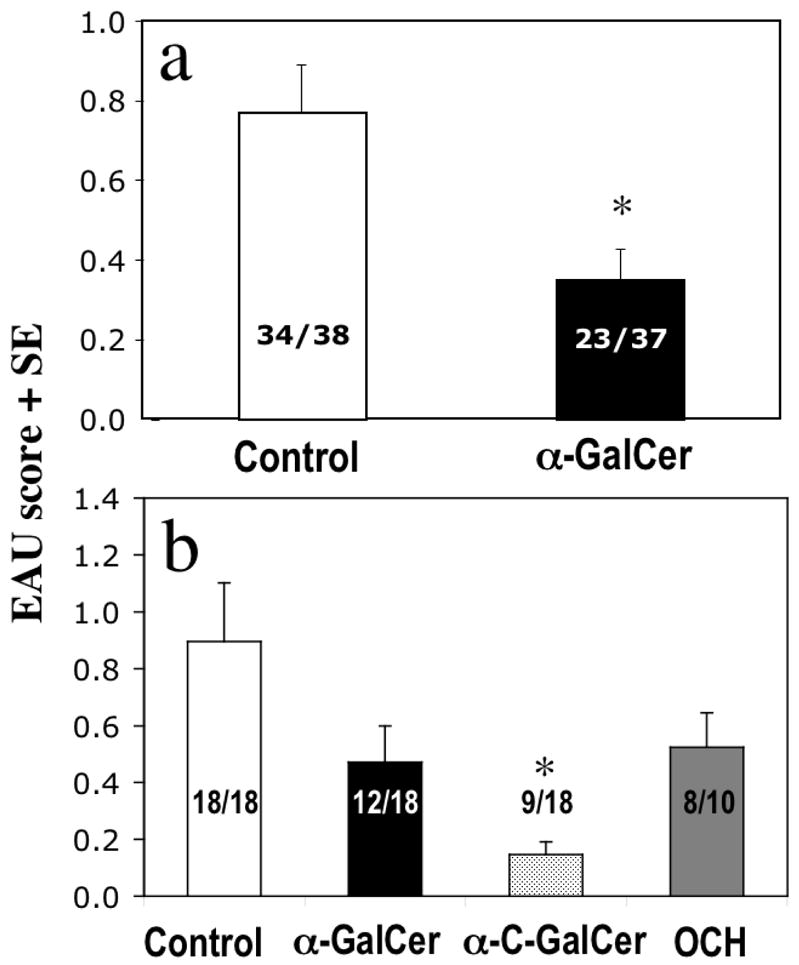
(a) Activation of iNKT cells ameliorates EAU. C57BL/6 WT mice were immunized with IRBP as described in Fig. 1a with (black column) or without (white column) 5 μg of the synthetic iNKT cell ligand α-GalCer. EAU scores on day 19 after immunization are shown. (p < 0.005); (b) Analogs of α–GalCer differ in their ability to ameliorate EAU. C57BL/6 WT mice were immunized as described in Fig. 1a without (white column) or with 5 μg of the synthetic iNKT cell ligand α-GalCer (black column, p vs. control < 0.09) or α-C-GalCer (grey column, p vs. control < 0.02) ) or OCH (dark grey column, p. vs. control < 0.22). Data combined from 3 experiments.
This pattern of protection was unexpected because OCH deviates the iNKT response to TCR ligation towards IL-4 production (whereas α–C-Gal-Cer skews towards IFN-γ) and has moreover been shown in experimental models of arthritis, diabetes in the NOD mouse and encephalomyelitis (EAE) to be more protective than α–GalCer in its original form through an IL-4 dependent mechanism (reviewed in 7, 22). We therefore examined whether our α–GalCer, α-C-Gal-Cer and OCH preparations had the expected effect on iNKT cytokine production. Data obtained by testing IL-4 and IFN-γ in serum of mice injected with the three α–GalCer analogs confirmed that indeed the three analogs elicited cytokine profiles that were in keeping with what has been reported by others (Fig 5). These data suggested that an IFN-γ-dominated cytokine profile elicited by α-C-Gal-Cer is more efficient in protecting from EAU than a deviation towards an IL-4 dominated profile in this model.
Fig. 5. Differing cytokine profiles are elicited by α-GalCer analogs in vivo.

C57BL/6 WT mice were injected i.p. with 5 μg of either (a) α–GalCer (b) α-C-GalCer or (c) OCH. Serum was collected from 3 mice at the indicated times (h after injection). The assay was performed on pooled serum samples, therefore, error bars could not be generated.
Protective effect of iNKT cells in EAU seems to require innate IFN-γ and is associated with reduced adaptive IFN-γ and IL-17 responses
As we have previously observed that systemically produced IFN-γ can have a protective role in EAU (19, 28, 29), we decided to examine whether the enhanced protective effect of α-C-Gal-Cer compared to the other analogs was due to its ability to induce enhanced production of IFN-γ by iNKT cells. We therefore injected mice immunized for EAU in the presence of α-C-Gal-Cer with neutralizing Abs to IFN-γ at the time of immunization, when innate production of IFN-γ induced by α-C-Gal-Cer would be occurring (according to the timeline established in Fig 5). EAU and adaptive IFN-γ and IL-17, representing pathogenic effector Th1 and Th17 cell responses were examined. Mice protected from disease with α-C-Gal-Cer had strongly reduced adaptive IFN-γ and IL-17 responses (Fig. 6). Neutralization of IFN-γ at the time of immunization restored EAU scores in αC-Gal-Cer treated mice, but had no effect on disease progression in the control group, demonstrating a specific requirement for IFNγ in the protection. In addition, subsequent Ag-specific production of IFN-γ and IL–17 was restored (Fig. 6b and 6c), supporting the notion that innate production of IFN-γ elicited by α-C-Gal-Cer had a role in the protection and inhibition of adaptive responses induced by this analog.
Fig. 6. Protective effect of iNKT cells in EAU seems to require innate IFN-γ and is associated with reduced adaptive IFN-γ and IL-17 responses.
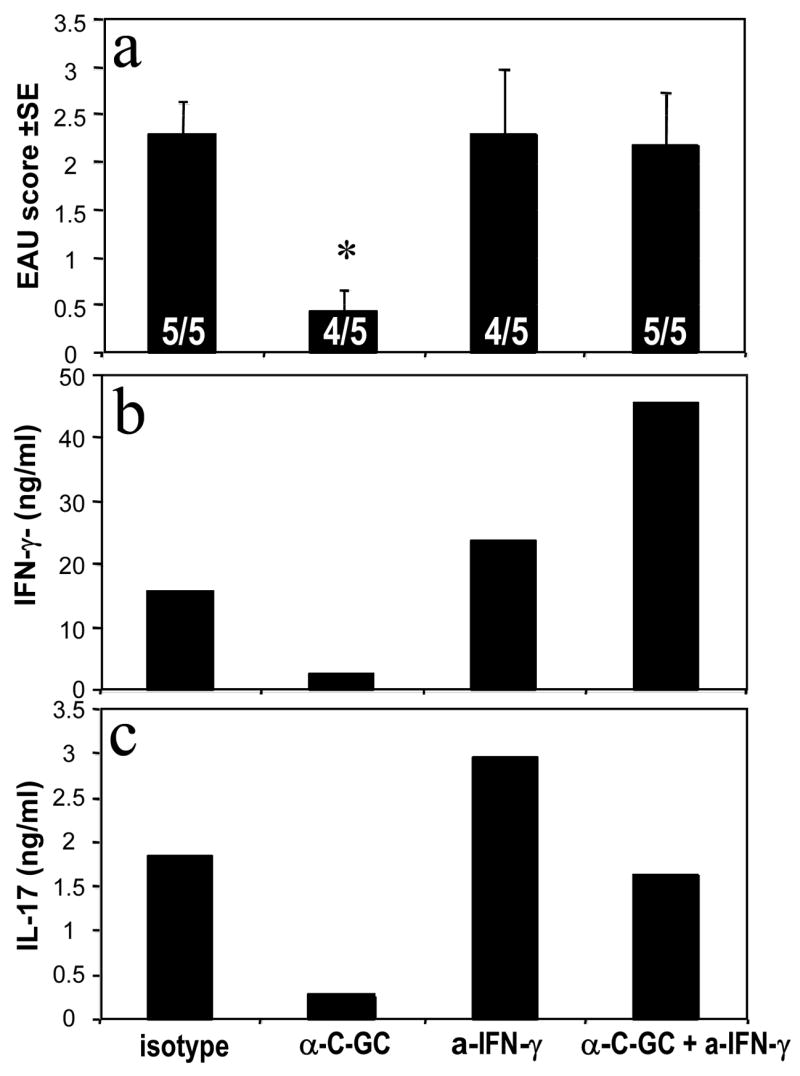
C57BL/6 WT mice were immunized as described in Fig. 1a with or without additional 5 μg of the iNKT cell ligand α-C-GalCer in the emulsion. Mice were injected with anti-IFN-γ or isotype on days –2, 0 and 2 relative to immunization. (a) EAU scores on day 19 after immunization. Positive out of total mice are shown within the bars. Only the α-C-GalCer treated group is significantly different from control (p < 0.028); (b) IFN-γ and (c) IL-17 titers in pooled supernatants of splenocytes from the mice in (a) obtained on day 21 after immunization and cultured for 48 h with 30 μg IRBP. Note that due to the fact that the assay was conducted on pooled supernatants the results represent an average of the group but no error bars could be generated. Cytokine levels were determined using Pierce Searchlight Technology. Control cultures were set up without IRBP (IFN-γ <200 pg/ml, IL-17 <15 pg/ml). Shown is a representative experiment of 2.
DISCUSSION
In the present study we demonstrate that iNKT cells can have a role in EAU regulation. Their role appears to be not in setting the threshold of susceptibility to EAU, as do the natural CD4+CD25+ regulatory cells whose function in deterring development of ocular autoimmunity we have characterized in recent studies (30, 31). Rather, they can inhibit developing disease following a pharmacological enhancement of their activity at or around the time of priming. In chronic autoimmunity, priming of new effector T cells is believed to be occurring on a continuous basis. Since endogenous ligands for iNKT cells exist in the body and can trigger iNKT activity (4, 5), it is conceivable that iNKT cells can participate in modulating the course of ocular autoimmune disease. Thus, there appears to be a “division of labor” between the natural CD4+CD25+ regulatory T cells and iNKT cells, with the former setting the threshold of susceptibility, and the latter possibly regulating the autoimmune response after that threshold has been passed.
The groups of Streilein and Stein-Streilein demonstrated that iNKT cells have a central role in ACAID, a prototypic regulatory phenomenon elicited by injection of Ag into the anterior chamber of the eye and its transport by eye-derived APC to the spleen (10, 11, 21). iNKT cells are recruited into the spleen via a mechanism involving MIP-2 and participate in priming the adaptive T regulatory cells typically associated with ACAID. Although prior elicitation of ACAID to IRBP can inhibit a subsequent episode of EAU (32), it is unlikely that the protection from EAU by iNKT that we observe here bears a relationship to their role in ACAID. In ACAID the eliciting Ag originates from the eye and the eye has to be perturbed (injected with Ag) in order for this phenomenon to be observed, and iNKT activation, if any, occurs without additional manipulation. In contrast, in our study, pharmacological activation of iNKT cells is needed and is applied when the eye is still intact. Thus, it is conceivable that iNKT cells may regulate ocular immune responses at more than one level.
Studies in the models of experimental arthritis, NOD diabetes and EAE (reviewed in 22) had indicated that activation of iNKT by OCH is more effective than by α–GalCer, which was attributed to its induction of IL-4 and Th2 skewing. We were therefore surprised to find that OCH was not more effective than α–GalCer in protecting from EAU, and that the most efficient protection followed administration of α-C-Gal-Cer, which induces an IFN-γ dominated iNKT cytokine response. Thus, effectiveness of protection paralleled the innate IFN-γ inducing ability of the invariant TCR ligand. The protection was accompanied by reduction in the IRBP-specific adaptive Th1 and Th17 pathogenic effector responses, as judged by production of their respective hallmark cytokines IFN-γ and IL-17 to in vitro recall with IRBP. The functional role of IFN-γ in the protective and regulatory effects of iNKT are strongly supported by direct evidence showing that neutralization of innate IFN-γ reversed the protective effect of α-C-Gal-Cer and restored the subsequent proinflammatory cytokine production of the adaptive response. This is not to say that the mechanism of protection is the same for all the three analogs. Our data do not negate the possibility that protection from EAU by OCH and by α–GalCer could involve IL-4, as was previously demonstrated in several other autoimmune disease models (22).
Our data are in line with some previous reports, which revealed that protection from autoimmune disease by iNKT may not always involve IL-4 and Th2 skewing. Studies by Lehuen and her colleagues demonstrated that even in the absence of IL-4 iNKT cells can control EAE (33) and experimental type 1 diabetes (T1D) (34). This was associated with a decrease in Th1-associated pathogenic autoimmune responses without inducing Th2 responses, and was due at least in part to induction of anergy in the autoreactive T cells (35). Such a mechanism could also be involved in the prevention of EAU observed here. Although these studies did not directly implicate IFN-γ in these effects, participation of IFN-γ (rather than IL-4) in protection from EAE was suggested by Furlan et al. (27).
It should be noted that high systemic levels of IFN-γ early or late in the disease can be protective, but likely by different mechanisms. Initial production of IFN-γ would be mostly from NKT and NK cells, whereas later in disease Ag specific Th1 cells are a major source of IFN-γ. We previously showed that early upregulation of IFN-γ by injections of IL-12 inhibits development of EAU and associated immunological responses by aborting priming, through a process that involves induction of nitric oxide and apoptosis (29). The innate IFN-γ produced by α-C-Gal-Cer-triggered NKT cells may well work in a similar fashion. On the other hand, protective effects of IFN-γ later in the disease appear to be due to its role in elimination of spent effector cells by activation-induced cell death (36, 37). Thus, neutralization of systemic IFN-γ at that stage also enhances disease (19), although at that point it is not possible to distinguish between effects of IFN-γ produced by Ag specific T cells and iNKT cells.
In summary, we have demonstrated that iNKT cells can actively participate in regulating the autoimmune response to immunologically privileged retinal Ags. This apparently occurs at a different level than their role in induction of ACAID. The mechanism involves the induction of innate IFN-γ production through ligation of the invariant TCR and results in inhibited development of adaptive Th1 and Th17 responses that represent pathogenic effector mechanisms in uveitis.
Acknowledgments
The authors thank Dr. S. Yamano of the Kirin Brewery, Tokyo, Japan for providing α–GalCer (KRN7000) and Drs. Chi-Huey Wong and Douglas Wu of the Scripps Research Institute, La Jolla, CA, for synthesizing the OCH used in this study.
NONSTANDARD ABBREVIATIONS
- EAU
experimental autoimmune uveitis
- iNKT
invariant Natural Killer T cell
- IRBP
interphotoreceptor retinoid-binding protein
- α-GalCer
alpha galactosylceramide
- α-C-GalCer
alpha-C-galactosylceramide
- OCH
(2S,3S,4R)-1-O-(alpha-D-galactopyranosyl)-N-tetracosanoyl-2-amino-1,3,4-nonanetriol)
Footnotes
Funding sources: NIH Intramural funding;
References
- 1.Wilson SB, Delovitch TL. Janus-like role of regulatory iNKT cells in autoimmune disease and tumour immunity. Nat Rev Immunol. 2003;3:211–222. doi: 10.1038/nri1028. [DOI] [PubMed] [Google Scholar]
- 2.Rowe NJ, Tunstall J, Galbraith L, Wilkinson SG. Lipid composition and taxonomy of [Pseudomonas] echinoides: transfer to the genus Sphingomonas. Microbiology. 2000;146( Pt 11):3007–3012. doi: 10.1099/00221287-146-11-3007. [DOI] [PubMed] [Google Scholar]
- 3.Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 4.Tsuji M. Glycolipids and phospholipids as natural CD1d-binding iNKT cell ligands. Cell Mol Life Sci. 2006;63:1889–1898. doi: 10.1007/s00018-006-6073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tupin E, Kronenberg M. Activation of natural killer T cells by glycolipids. Methods Enzymol. 2006;417:185–201. doi: 10.1016/S0076-6879(06)17014-7. [DOI] [PubMed] [Google Scholar]
- 6.Schmieg J, Yang G, Franck RW, Tsuji M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-Galactosylceramide. J Exp Med. 2003;198:1631–1641. doi: 10.1084/jem.20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamura T, Miyamoto K, Illes Z, Pal E, Araki M, Miyake S. iNKT cell-stimulating synthetic glycolipids as potential therapeutics for autoimmune disease. Curr Top Med Chem. 2004;4:561–567. doi: 10.2174/1568026043451221. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal RK, Caspi RR. Rodent models of experimental autoimmune uveitis. Methods Mol Med. 2004;102:395–420. doi: 10.1385/1-59259-805-6:395. [DOI] [PubMed] [Google Scholar]
- 9.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. doi: 10.1084/jem.20071258. Epub 2008 Apr 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faunce DE, Sonoda KH, Stein-Streilein J. MIP-2 recruits iNKT cells to the spleen during tolerance induction. J Immunol. 2001;166:313–321. doi: 10.4049/jimmunol.166.1.313. [DOI] [PubMed] [Google Scholar]
- 11.Sonoda KH, Exley M, Snapper S, Balk SP, Stein-Streilein J. CD1-reactive natural killer T cells are required for development of systemic tolerance through an immune-privileged site. J Exp Med. 1999;190:1215–1226. doi: 10.1084/jem.190.9.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pepperberg DR, Okajima TL, Wiggert B, Ripps H, Crouch RK, Chader GJ. Interphotoreceptor retinoid-binding protein (IRBP). Molecular biology and physiological role in the visual cycle of rhodopsin. Mol Neurobiol. 1993;7:61–85. doi: 10.1007/BF02780609. [DOI] [PubMed] [Google Scholar]
- 13.Pepperberg DR, Okajima TL, Ripps H, Chader GJ, Wiggert B. Functional properties of interphotoreceptor retinoid-binding protein. Photochem Photobiol. 1991;54:1057–1060. doi: 10.1111/j.1751-1097.1991.tb02129.x. [DOI] [PubMed] [Google Scholar]
- 14.Sakai T, Naidenko OV, Iijima H, Kronenberg M, Koezuka Y. Syntheses of biotinylated alpha-galactosylceramides and their effects on the immune system and CD1 molecules. J Med Chem. 1999;42:1836–1841. doi: 10.1021/jm990054n. [DOI] [PubMed] [Google Scholar]
- 15.Silver PB, Rizzo LV, Chan CC, Donoso LA, Wiggert B, Caspi RR. Identification of a major pathogenic epitope in the human IRBP molecule recognized by mice of the H-2r haplotype. Invest Ophthalmol Vis Sci. 1995;36:946–954. [PubMed] [Google Scholar]
- 16.Moody MD, Van Arsdell SW, Murphy KP, Orencole SF, Burns C. Array-based ELISAs for high-throughput analysis of human cytokines. Biotechniques. 2001;31:186–190. 192, 184. doi: 10.2144/01311dd03. [DOI] [PubMed] [Google Scholar]
- 17.Snedecor GW, Cochran WG. Statistical methods. Iowa State University Press; 1967. [Google Scholar]
- 18.Caspi RR, Grubbs BG, Chan CC, Chader GJ, Wiggert B. Genetic control of susceptibility to experimental autoimmune uveoretinitis in the mouse model. Concomitant regulation by MHC and non-MHC genes. J Immunol. 1992;148:2384–2389. [PubMed] [Google Scholar]
- 19.Caspi RR, Chan CC, Grubbs BG, Silver PB, Wiggert B, Parsa CF, Bahmanyar S, Billiau A, Heremans H. Endogenous systemic IFN-gamma has a protective role against ocular autoimmunity in mice. J Immunol. 1994;152:890–899. [PubMed] [Google Scholar]
- 20.Sun B, Rizzo LV, Sun SH, Chan CC, Wiggert B, Wilder RL, Caspi RR. Genetic susceptibility to experimental autoimmune uveitis involves more than a predisposition to generate a T helper-1-like or a T helper-2-like response. J Immunol. 1997;159:1004–1011. [PubMed] [Google Scholar]
- 21.Nakamura T, Sonoda KH, Faunce DE, Gumperz J, Yamamura T, Miyake S, Stein-Streilein J. CD4+ iNKT cells, but not conventional CD4+ T cells, are required to generate efferent CD8+ T regulatory cells following antigen inoculation in an immune-privileged site. J Immunol. 2003;171:1266–1271. doi: 10.4049/jimmunol.171.3.1266. [DOI] [PubMed] [Google Scholar]
- 22.Miyake S, Yamamura T. Therapeutic potential of glycolipid ligands for natural killer (NK) T cells in the suppression of autoimmune diseases. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:315–322. doi: 10.2174/1568008054863772. [DOI] [PubMed] [Google Scholar]
- 23.Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, Nussenblatt RB, Caspi RR. Cutting edge: iNKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol. 2008;180:5167–5171. doi: 10.4049/jimmunol.180.8.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong S, Scherer DC, Singh N, Mendiratta SK, Serizawa I, Koezuka Y, Van Kaer L. Lipid antigen presentation in the immune system: lessons learned from CD1d knockout mice. Immunol Rev. 1999;169:31–44. doi: 10.1111/j.1600-065x.1999.tb01304.x. [DOI] [PubMed] [Google Scholar]
- 25.Singh AK, Wilson MT, Hong S, Olivares-Villagomez D, Du C, Stanic AK, Joyce S, Sriram S, Koezuka Y, Van Kaer L. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801–1811. doi: 10.1084/jem.194.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahng AW, Maricic I, Pedersen B, Burdin N, Naidenko O, Kronenberg M, Koezuka Y, Kumar V. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1789–1799. doi: 10.1084/jem.194.12.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furlan R, Bergami A, Cantarella D, Brambilla E, Taniguchi M, Dellabona P, Casorati G, Martino G. Activation of invariant iNKT cells by alphaGalCer administration protects mice from MOG35-55-induced EAE: critical roles for administration route and IFN-gamma. Eur J Immunol. 2003;33:1830–1838. doi: 10.1002/eji.200323885. [DOI] [PubMed] [Google Scholar]
- 28.Jones LS, Rizzo LV, Agarwal RK, Tarrant TK, Chan CC, Wiggert B, Caspi RR. IFN-gamma-deficient mice develop experimental autoimmune uveitis in the context of a deviant effector response. J Immunol. 1997;158:5997–6005. [PubMed] [Google Scholar]
- 29.Tarrant TK, Silver PB, Wahlsten JL, Rizzo LV, Chan CC, Wiggert B, Caspi RR. Interleukin 12 protects from a T helper type 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide, and apoptosis. J Exp Med. 1999;189:219–230. doi: 10.1084/jem.189.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avichezer D, Grajewski RS, Chan CC, Mattapallil MJ, Silver PB, Raber JA, Liou GI, Wiggert B, Lewis GM, Donoso LA, Caspi RR. An immunologically privileged retinal antigen elicits tolerance: major role for central selection mechanisms. J Exp Med. 2003;198:1665–1676. doi: 10.1084/jem.20030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grajewski RS, Silver PB, Agarwal RK, Su SB, Chan CC, Liou GI, Caspi RR. Endogenous IRBP can be dispensable for generation of natural CD4+CD25+ regulatory T cells that protect from IRBP-induced retinal autoimmunity. J Exp Med. 2006;203:851–856. doi: 10.1084/jem.20050429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hara Y, Caspi RR, Wiggert B, Chan CC, Wilbanks GA, Streilein JW. Suppression of experimental autoimmune uveitis in mice by induction of anterior chamber-associated immune deviation with interphotoreceptor retinoid-binding protein. J Immunol. 1992;148:1685–1692. [PubMed] [Google Scholar]
- 33.Mars LT, Laloux V, Goude K, Desbois S, Saoudi A, Van Kaer L, Lassmann H, Herbelin A, Lehuen A, Liblau RS. Cutting edge: V alpha 14-J alpha 281 iNKT cells naturally regulate experimental autoimmune encephalomyelitis in nonobese diabetic mice. J Immunol. 2002;168:6007–6011. doi: 10.4049/jimmunol.168.12.6007. [DOI] [PubMed] [Google Scholar]
- 34.Novak J, Beaudoin L, Griseri T, Lehuen A. Inhibition of T cell differentiation into effectors by iNKT cells requires cell contacts. J Immunol. 2005;174:1954–1961. doi: 10.4049/jimmunol.174.4.1954. [DOI] [PubMed] [Google Scholar]
- 35.Beaudoin L, Laloux V, Novak J, Lucas B, Lehuen A. iNKT cells inhibit the onset of diabetes by impairing the development of pathogenic T cells specific for pancreatic beta cells. Immunity. 2002;17:725–736. doi: 10.1016/s1074-7613(02)00473-9. [DOI] [PubMed] [Google Scholar]
- 36.Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:123–128. doi: 10.1084/jem.192.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalton DK, Haynes L, Chu CQ, Swain SL, Wittmer S. Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J Exp Med. 2000;192:117–122. doi: 10.1084/jem.192.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]