

Abstract
Ring-constrained adenosine analogues have been designed to act as dualagonists at tissue-protective A1 and A3 adenosine receptors (ARs). 9-Ribosides transformed into the ring-constrained (N)-methanocarba-2-chloro-5′-uronamides consistently lost affinity at A1/A2AARs and gained at A3AR. Among 9-riboside derivatives, only N6-cyclopentyl and 7-norbornyl moieties were extrapolated for mixed A1/A3 selectivity and rat/human A3AR equipotency. Consequently, 2 was balanced in affinity and potency at A1/A3ARs as envisioned and dramatically protected in an intact heart model of global ischemia and reperfusion.
There are four subtypes of adenosine receptors (ARs): A1, A2A, A2B, and A3, and their selective agonists are under development as therapeutic agents.1–3 Activation of one or more of the ARs and receptor overexpression have been shown to have a cytoprotective role in ischemic models.3–8 Specifically, activation of either A1 or A3ARs in cardiac myocytes in several species has been shown to mimic the cardioprotective effect of ischemic preconditioning.9–11 The co-activation of A1 or A3ARs in the cardiac myocyte has been shown to be protective to a greater degree than activation of either subtype alone. In vivo experiments have also demonstrated the cardioprotective effects of A1 and A3ARs in certain species.12 Activation of A1 and A3ARs leads to activation of PLC and PLD (phospholipase C and D), respectively, in cultured cardiac myocytes.9 PLC and PLD converge on activation of protein kinase C (PKC), which mediates cardioprotection.9,13 In the brain, activation of A1 or chronic activation of A3ARs has been shown to protect neurons against ischemia in a variety of models.14,15 In a model of global ischemia in gerbils, the A1 agonists CPA (N6-cyclopentyladenosine) and ADAC (N6-[4-[[[4-[[[(2-aminoethyl)amino]carbonyl]methyl]anilin]carbonyl]-methyl]phenyl]-adenosine) and the chronically administered A3 agonist IB-MECA (1-[6-[[(3- iodophenyl)methyl]amino]-9H-purin-9-yl]-1-deoxy-N-methyl-β-D-ribofuran uronamide) were cytoprotective at very low doses.
The concurrent activation of A1 and A3ARs has been carried out either by co- administering selective agonists for each subtype, or using novel conjugates of functionalized congeners of A1 and A3 agonists.16 Some agonists of balanced potency have been reported,17–19 however they are often partial agonists and of selectivities limited to a particular species. The careful design and covalent joining of these functionalized congeners provide binary conjugates that are balanced in their ability to activate A1 and A3ARs. However, due to the high molecular weights (in excess of 1000) and the presence of multiple hydrogen bond donors, these molecules do not satisfy the criteria proposed by Lipinski for prediction of oral bioactivity20 and are of limited application in vivo.
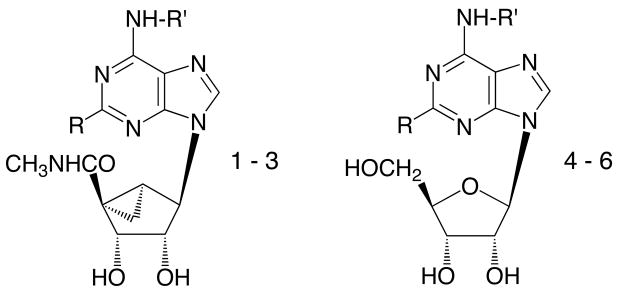
We have taken a new approach, i.e. based on differential effects on ARs of the ring-constrained (N)-methanocarba ring system,21 to design dual acting A1/A3AR agonists of relatively low molecular weight. The new agonists (1 – 3, Scheme 1) were synthesized subsequent to careful SAR analysis of a large number of adenosine derivatives at ARs with respect to both binding affinity and relative efficacy.22-24 We have incorporated specific molecular features that provide a balance in potency at A1 and A3ARs and that also maintain full efficacy at the A3AR.21,22 In the present study, several derivatives have been shown in binding and functional assays to be dual acting at the two receptor subtypes. Furthermore, a potent anti–ischemic cardioprotective effect in an intact mouse heart model of global ischemia and reperfusion injury was demonstrated.6,13 This mammalian heart model has been shown to express both AR subtypes.
Scheme 1.

Reagents: a) RNH2, MeOH, triethylamine; b) CH3NH2, MeOH; c) TFA, H2O, MeOH, 70°C; d) 10% Pd/C/ H2, MeOH.
The selection of N6-, C2, and 5′-uronamide substituents in the target compounds has been based largely on our recent findings relating to AR affinity, selectivity, and relative efficacy for specific adenine-9-riboside derivatives as well as a series of adenine- (N)-methanocarba-5′-alkyl uronamide derivatives.21,22 At the A3AR, in contrast to the A1AR, relative efficacy is easily diminished by substitution of the N6 and C2 positions while preserving affinity. This reduction in efficacy is readily overcome by a flexible 5′-methyluronamide moiety.22 The desired analogues would be balanced in high affinity at both human and rat A1 and A3ARs and would display full agonism. According to published SAR findings (correlated in Figure 1) the substitution of adenine-9-ribosides to obtain the corresponding 2-chloro-(N)-methanocarba-5′-alkyl uronamides produced consistent effects on AR affinity. Upon undergoing these modifications for various hydrophobic N6-substituents there was roughly one order of magnitude loss of binding affinity at the A1AR and slightly less loss at the A2AAR. The effect of this transformation on binding affinity at the A3AR resulted in either equal or greater affinity (up to 14-fold), due to the conformational preference of the A3AR binding site.21 Thus, we examined a large published series of seventy-four adenosine derivatives,23,24 mono-substituted at N6, for the ideal candidates predicted to have balanced binding affinity when adapted to the 2-Cl-(N)-methanocarba series. The following criteria were sought: 1) equipotency at rat and human A3ARs; 2) roughly two orders of magnitude greater affinity at A1ARs in comparison to A3ARs; and 3) selectivity for A1 and A3ARs in comparison to both A2A and A2BARs. Few N6-substituents satisfied all criteria; for example, although the affinities at the rat and human A1ARs were generally similar,24 at the A3ARs the species difference was as high as 1100-fold.23 The most likely candidates identified were N6- cycloalkyl groups of the A1-selective agonists 4 – 6.
Figure 1.

Correlation of Ki values at human A1 (■), A2A (◆), and A3 (▲) receptors of adenosine derivatives in two structural series were compared. The two series compared are: mono-substituted adenosine (9-riboside) derivatives, and tri-subsituted 2-chloro-(N)-methanocarba derivatives. In each case, pairs of compounds in which both members have the same N6-sustitution (as indicated) are correlated. The five (N)-methanocarba analogues depicted in this graph contained N6-benzyl and phenylethyl-type groups, however the general effects on affinity at each of the three adenosine receptor subtypes were generalized to design new N6-cycloalkyl analogues having desired pharmacological properties. For the N6 substitutions shown, there was consistent loss of affinity at A1 and A2AARs and the effect at the A3AR ranged from no change to a 14-fold gain of affinity. The (N)-methanocarba compounds shown here were all selective for the A3 receptor, while the desired property in the new analogues was balanced affinity at A1 and A3 receptors.
The synthetic route to three adenosine agonists 1 – 3 that were designed for highaffinity at the A1 and A3 adenosine receptors and low affinity at the A2 receptors is shown in Scheme 1. The synthesis of the target cyclopentyl derivative 2 and the 7-norbornyl derivative 3, was according to the general route presented for 5′-uronamido-(N)-methanocarba derivatives.21 The synthetic approach of Joshi et al.25 has been followed to incorporate the 5′-uronamido-(N)-methanocarba ring system. The requisite 7-norbornylamine was prepared in three steps from 7-norbornyl bromide using a Curtius rearrangement. Accordingly, the 2,6-dichloro 5′-ester 721 was treated first with a cycloalkylamine, which substituted selectively at the 6-position. Subsequent treatment with a large excess of methylamine converted the ester group to the corresponding amide. The final step was acidic deprotection of the isopropylidene protecting group at the 2′,3′-hydroxyl groups. Both 2 and 3 contain the 2-chloro and 5′-uronamido-(N)-methanocarba substituents. The 2-Cl group in 2 was hydrogenolyzed to give 1 in good yield.
Binding and functional assays were carried out at human A1, A2A and A3ARsexpressed in CHO (Chinese hamster ovary) cells. The results confirmed that 1 and 2were highly selective for A1 and A3ARs, and that the affinities were nearly balanced. However, 3 was considerably more potent in binding to the A3 than to the A1AR. For all three derivatives, the Ki values at the human A2AAR were at least several hundredfold greater than at the A1 or the A3AR. Compound 2 was equipotent in binding to human and rat A1ARs with Ki values of 18.3 and 17.4 nM, respectively. Also, the affinity of compound 2 was similar at human and rat A3ARs with Ki values of 3.7 and 5.8 nM, respectively.
In functional assays consisting of measuring inhibition of forskolin-stimulatedproduction of 3′,5′-cyclic-adenosine monophosphate (cAMP) in intact transfected CHO cells, single concentration determinations (Table 1) indicated that full A3AR agonism was maintained in compounds 1 – 3. Concentration response curves indicated that compound 2 was a dual acting full agonist with nearly equivalent functional potencies at human A1 (EC50 = 8.2 nM) and A3 (EC50 = 2.8 nM) ARs.
Table 1.
Potency of adenosine derivatives at human A1, A2A, and A3ARs and the rat A3AR a and maximal agonist effects at human A3ARs expressed in CHO cells.a
 | |||||||
|---|---|---|---|---|---|---|---|
| No. | N6-R′ | C2-R | Ki (hA1AR) nMa | Ki (hA2AAR) nMa | Ki (hA3AR) nMa | % Activation (hA3AR)b at 10 μM | Ki (rA3AR) |
| 1c | CP | H | 34.1 ± 6.1 | 6420 ± 630 | 13.1 ± 5.1 | 93 ± 7 | 10.2 ± 2.1 |
| 2c | CP | Cl | 18.3 ± 6.3g | 3250 ± 300 | 3.7 ± 0.9 | 101 ± 10 | 5.8 ± 1.6 |
| 3c | NB | Cl | 190 ± 37 | >10,000 | 14.6 ± 3.2 | 92 ± 6 | 9.6 ± 2.7 |
| 4d | CP | H | 0.45±0.04b | 462 | 240±36 | 72±12 | 97±4 |
| 5d | CP | Cl | 0.83e | 2270e | 38 ± 6 | 0 | 237±71f |
| 6d | NB | H | 0.48±0.01 | >10,000 | 229±76 | 112±25 | 103±1 |
All AR experiments were performed using adherent CHO cells stably transfected with cDNA encoding the human or rat ARs. Percent activation of the human A3AR was determined at 10 μM. Binding at human A1 and A2AARs in this study was carried out as described in Methods using [3H]R-PIA or [3H]CGS 21680 as a radioligand. Values from the present study are expressed as mean ± s.e.m., n = 3–5.
Percent activity at 10 μM, relative to 10 μM Cl-IB-MECA (A3).
1, MRS3706; 2, MRS3630; 3, MRS3638.
Data from Klotz et al.28
Data from van Galen et al.29
Ki at rat A1 AR is 17.4 ± 2.7 nM.
ND not determined. CP, cyclopentyl; NB, 7-norbornyl.
Compounds 1 – 3 were assayed for activation of the human A2BAR stably expressed in CHO cells.18 Each adenosine derivative was tested at a concentration of 10 μM. As for the ribosides 4 – 6, the EC50 values at the human A2BAR of the (N)- methanocarba-5′-uronamide N6-substituted nucleosides 1 – 3 were all >10 μM. Compound 2 also showed negligible effect in stimulation of adenylate cyclase at the murine A2A or A2BARs endogenously expressed in PC12 (rat) and NIH/3T3 cells (mouse), respectively.
Since 2 was the most potent and still nearly matched in binding affinity and in function at the two AR subtypes known to be cardioprotective, this compound was chosen for further pharmacological studies in an intact mouse heart model of ischemia and reperfusion.6,13 In this model, compound 2 at 30 nM exerted a potent anti-ischemic cardioprotective effect (Table 2). The mixed agonist was perfused until the induction of ischemia. The recovery of left ventricular developed pressure (LVPD), +dP/dt, -dP/dt and heart rate (HR) all improved significantly following treatment with the mixed agonist 2. The infarct size determined using computer morphometry26 after staining with triphenyltetrazolium chloride (TTC) was significantly reduced in the group treated with 2 (Figure 2). The percent necrosis in the group treated with 2 was 15 ± 7% compared to 23 ± 8% in the vehicle-treated controls, n = 6. In the same model, the classical A1AR agonist 5 at a higher concentration (100 nM) could also reduce myocardial infarct size. The percent necrosis following infusion of 5 was 15 ± 10%, n=15.
Table 2.
Recovery of left ventricular function in a mouse heart model of ischemia/reperfusion.a
| Parameter | Vehicleb | Compound 5c | Significance |
|---|---|---|---|
| LVDPd | 8.5 ± 5.3 | 26.0 ± 4.8 | t=7.1, P < 0.0001 |
| +dP/dtd | 6.3 ± 3.9 | 21.1 ± 4.9 | t=7.4, P < 0.0001 |
| -dP/dtd | 8.2 ± 3.7 | 24.6 ± 7 | t=7.22, P < 0.0001 |
| HRd | 37.1 ± 32.6 | 93.8 ± 19.3 | t=4.1, P < 0.0005 |
| % necrosis | 23.4 ± 7.8 | 15.0 ± 6.9 | t=2.32, Pe=0.029 |
Values were obtained after 35 global ischemia followed by reperfusion.
DMSO, n = 16.
A concentration of 30 nM 2 (initially dissolved in DMSO) was used, n = 6. During buffer perfusion of heart via the side port, the spontaneous heart rate dropped by 24 ± 8.2% (SEM) due to a decrease in the perfusion pressure. Perfusion of buffer containing 30 nM 2 was associated with a larger decrease in the heart rate of 57 ± 6.7%, likely because of a negative inotropic effect of 2.
% of baseline prior to ischemia/reperfusion.
Two-tailed.
Figure 2.

Anti-ischemic infarct-reducing effects of adenosine receptor agonists. Murine hearts were excised and subjected to normothermic global ischemia and reperfusion with or without (A) adenosine receptor agonists as described in Methods. The adenosine receptor agonists were: (B) A3 agonist Cl-IB-MECA, 30 nM; (C) A1 agonist 5, 100 nM; (D) Mixed A1/A3 agonist 2, 30 nM. Agonists were infused for five min till the induction of ischemia. The heart was stained with TTC after 35 min of ischemia and 120 min of reperfusion and the infarcted areas were visualized as TTC-negative (pale, white). The infarct was quantified by morphometry and normalized to the whole heart as % necrosis. Data were representative of 6 (adenosine receptor agonist-treated) and 16 (DMSO/vehicle-treated) mice. A vehicle control not subjected to ischemia showed no pale or TTC-negative area.
Thus, we have designed novel cardioprotective agents based on mechanistic andstructural considerations. The adenosine N6-substituents, cyclopentyl and 7-norbornyl,were selected based on predictions made from the binding affinities of the corresponding adenosine derivatives,22 and from the consistent effects on AR affinity of replacing the 9-riboside moiety with a 5′-uronamido-(N)-methanocarba-pseudoribose moiety in combination with the 2-Cl substituent. The sum of these effects on affinity at each of the three AR subtypes was generalized to design new N6-cycloalkyl analogues having desired pharmacological properties. The results of Tchilibon et al.,21 for substituted N6-benzyl and N6-phenylethyl derivatives suggested that in each case, in comparison to the corresponding adenine-9-riboside, the affinity at the human A1AR decreased by at least one order of magnitude while the affinity at the human A3AR tended to increase by typically one order of magnitude. In the case of N6-cyclopentyl-and N6-(7-norbornyl)-adenine 9-ribosides, 4 and 6, respectively,22 the affinity of each was similar at rat and human A3ARs, but the effects of such N6-cycloakyl substitution had not yet been probed in the 5′-uronamido-(N)-methanocarba series. The affinity of both 9-ribosides at the human A2A and A2BARs was weak, thus the corresponding 5′-uronamido-(N)-methanocarba derivatives were expected to be highly selective for A1 and A3ARs in comparison to A2A and A2BARs. We have confirmed the anticipated selectivity for the N6-cyclopentyl derivatives 1 and 2. Also, based on the 9-ribosides a large species difference at the A3AR common among N6-substituted adenosine derivatives24 was predicted to be absent in the new analogues. This prediction was confirmed in binding assays of all three newly-synthesized derivatives.
We have examined the mixed A1/A3 agonist 2 in an intact mouse heart model of ischemia and reperfusion injury,6,13 in which either an A1- or A3-selective agonist acts as a potent cardioprotective agent. The initial findings validate the model for studying AR-dependent protection and illustrate the highly cardioprotective effect of 2. The role of cardiac A3ARs is complex, with protective effects demonstrated in models of preconditioning, delayed cardioprotection,13 and ischemia-reperfusion.6,8 The activation of the A3AR in the rat coronary circulation has been proposed to mediate vasodilation.27 Also, potential side effects of adenosine agonists, such as hypotension and sedation, must be considered.1 Therefore, additional pharmacological examination of 2 and similar mixed agonists will be needed.
In conclusion, we have used a semi-rational approach based on SAR analysis to focus on a small number (3) of candidate structures predicted to display the desired pharmacodynamic properties. Thus, a series of (N)-methanocarba nucleosides previously characterized as selective A3AR agonists has now been adapted to mixed AR selectivity desired for cytoprotection in a variety of tissue systems. Indeed, two of the three compounds synthesized reached this goal as judged by functional analyses and/or in vitro binding. Further chemical optimization of the structure to enhance the affinity and potency of mixed A1/A3 agonists is now feasible. One of the candidates was already shown to be highly cardioprotective in the mouse. These compounds may serve as prototypical examples for more detailed pharmacological studies leading to the development of novel dual acting cardioprotective AR agonists.
Supplementary Material
Experimental details for the synthesis and biological evaluation of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Dr. D. Eric Anderson (NIDDK) for mass spectral determinations. B.V. Joshi thanks Gilead Sciences (Foster City, CA) for financial support. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 2.Yao L, Burbiel JC, Maass A, Müller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerging Drugs. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 3.Liu GS, Richards SC, Olsson RA, Mullane K, Walsh RS, Downey JM. Evidence that the adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc Res. 1994;28:1057–1061. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- 4.Liang BT, Jacobson KA. A physiological role of the adenosine receptor:A3 sustained cardioprotection. Proc Natl Acad Sci USA. 1998;95:6995–6999. doi: 10.1073/pnas.95.12.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, Knight DR. Novel N6-substituted adenosine 5′-N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury. AmJ Physiol Heart Circ Physiol. 2003;285:H2780. doi: 10.1152/ajpheart.00411.2003. [DOI] [PubMed] [Google Scholar]
- 6.Peart J, Flood A, Linden J, Matherne GP, Headrick JP. Adenosine-mediated cardioprotection in ischemic-reperfused mouse heart. J Cardiovasc Pharmacol. 2002;39:117–129. doi: 10.1097/00005344-200201000-00013. [DOI] [PubMed] [Google Scholar]
- 7.Maddock HL, Mocanu MM, Yellon DM. Adenosine A3 receptor activation protects the myocardium from reperfusion/reoxygenation injury. Am J Physiol Heart Circ Physiol. 2002;283:H1307–H1313. doi: 10.1152/ajpheart.00851.2001. [DOI] [PubMed] [Google Scholar]
- 8.Cross HR, Murphy E, Black RG, Auchampach J, Steenbergen C. Overexpression of A3 adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am J Physiol Heart Circ Physiol . 2002;283:H1562–1568. doi: 10.1152/ajpheart.00335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parsons M, Young L, Lee JE, Jacobson KA, Liang BT. Distinct cardioprotective effects of adenosine mediated by differential coupling of receptor subtypes to phospholipases C and D. FASEB J. 2000;14:1423–1431. doi: 10.1096/fj.14.10.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shneyvays V, Leshem D, Zinman T, Mamedova LK, Jacobson KA, Shainberg A. Role of adenosine A1 and A3 receptors in regulation of cardiomyocyte homeostasis after mitochondrial respiratory chain injury. Am J Physiol Heart Circ Physiol. 2005;288:H2792–H2801. doi: 10.1152/ajpheart.01157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr CS, Hill RJ, Masamune H, Kennedy SP, Knight DR, Tracey WR, Yellon DM. Evidence for a role for both the adenosine A1 and A3 receptors in protection of isolated human atrial muscle against simulated ischaemia. Cardiovasc Res. 1997;36:52– 59. doi: 10.1016/s0008-6363(97)00160-0. [DOI] [PubMed] [Google Scholar]
- 12.Kodani E, Bolli R, Tang XL, Auchampach JA. Protection of IB-MECA against myocardial stunning in conscious rabbits is not mediated by the A1 adenosine receptor. Basic Res Cardiol. 2001;96:487–96. doi: 10.1007/s003950170031. [DOI] [PubMed] [Google Scholar]
- 13.Zhao TC, Kukreja RC. Protein kinase C-delta mediates adenosine A3 receptor-induced delayed cardioprotection in mouse. Am J Physiol Heart Circ Physiol. 2003;285:H434–441. doi: 10.1152/ajpheart.00095.2003. [DOI] [PubMed] [Google Scholar]
- 14.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- 15.von Lubitz DKJE, Lin RC, Bischofberger N, Beenhakker M, Boyd M, Lipartowska R, Jacobson KA. Protection against ischemic damage by adenosine amine congener, a potent and selective adenosine A1 receptor agonist. Eur J Pharmacol. 1999;369:313–317. doi: 10.1016/s0014-2999(99)00073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobson KA, Xie R, Young L, Chang L, Liang BT. A novel pharmacological approach to treating cardiac ischemia: binary conjugates of A1 and A3 adenosine receptor agonists. J Biol Chem. 2000;275:30272–30279. doi: 10.1074/jbc.M001520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baraldi PG, Cacciari B, Pineda de las Infantas MJ, Romagnoli R, Spalluto G, Volpini R, Costanzi S, Vittori S, Cristalli G, Melman N, Park KS, Ji X-d, Jacobson KA. Synthesis and biological activity of a new series of N6-arylcarbamoyl-, 2-(ar)alkynyl-N6-arylcarbamoyl, and N6-carboxamido- derivatives of adenosine-5′-N-ethyluronamide (NECA) as A1 and A3 adenosine receptor agonists. J Med Chem. 1998;41:3174–3185. doi: 10.1021/jm980147p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Tilburg EW, van der Klein PAM, von Frijtag Drabbe Künzel J, de Groote M, Stannek C, Lorenzen A, IJzerman AP. 2,5′-Disubstituted adenosine derivatives: evaluation of selectivity and efficacy for the adenosine A1,A2A, and A3 receptor. J Med Chem. 2002;45:420–429. doi: 10.1021/jm010952v. [DOI] [PubMed] [Google Scholar]
- 19.Cappellacci L, Franchetti P, Pasqualini M, Petrelli R, Vita P, Lavecchia A, Novellino E, Costa B, Martini C, Klotz KN, Grifantini M. Synthesis, biological evaluation, and molecular modeling of ribose-modified adenosine analogues as adenosine receptor agonists. J Med Chem. 2005;48:1550–1562. doi: 10.1021/jm049408n. [DOI] [PubMed] [Google Scholar]
- 20.Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 21.Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. (N)- Methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao ZG, Blaustein J, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tchilibon S, Kim SK, Gao ZG, Harris BA, Blaustein J, Gross AS, Melman N, Jacobson KA. Exploring distal regions of the A3 adenosine receptor binding site: Sterically-constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg Med Chem. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi BV, Moon HR, Fettinger JC, Marquez VE, Jacobson KA. A new synthetic route to (N)-methanocarba nucleosides designed as A3 adenosine receptor agonists. J Org Chem. 2005;70:439–447. doi: 10.1021/jo0487606. [DOI] [PubMed] [Google Scholar]
- 26.Ichinose F, Bloch KD, Wu JC, Hataishi R, Aretz HT, Picard MH, Scherrer-Crosbie M. Pressure overload-induced LV hypertrophy and dysfunction in mice are exacerbated by congenital NOS3 deficiency. Am J Physiol Heart Circ Physiology. 2004;286:H1070–H1075. doi: 10.1152/ajpheart.00940.2003. [DOI] [PubMed] [Google Scholar]
- 27.Hinschen AK, Rose′Meyer RB, Headrick JP. Adenosine receptor subtypes mediating coronary vasodilation in rat hearts. J Cardiovasc Pharmacol. 2003;41:73–80. doi: 10.1097/00005344-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes –characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg′s Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 29.van Galen PJM, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details for the synthesis and biological evaluation of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.