

Abstract
Sulfinimine-derived N-sulfinyl δ-amino β-ketophosphonates are transformed via the enaminone to the phosphoryl dihydropyridones that selectively give trans-2,6-disubstituted 1,2,5,6-tetrahydropyridines on organocuprate addition and dephosphorylation.
Introduction
The piperidine ring has is a common motif found in natural products, drugs and drug candidates.1,2 Simple 2,6-disubstituted piperidines, isolated from fire ant venom, are reported to possess a broad range of activities (necrotic, insecticidal, antibacterial, antifungal, anti-HIV).3 Polyhydroxylated piperidines (azasugars) are potent inhibitors of carbohydrate-processing enzymes, which suggests they will find utility in treating viral infections, cancer, diabetes, and tuberculosis.4 Furthermore, piperidines serve as building blocks for the synthesis of more complex alkaloids including the indolizidine and quinolizidine ring systems, which in themselves exhibit a broad range of biological activities.5 Although cis-2,6-disubstituted piperidines are readily accessible, there are fewer methods for the synthesis of trans-2,6-disubstituted piperidines.6,7 Unfortunately, most of these procedures lack generality, are racemic7c or target specific syntheses.7d–n Few of these methods provide convenient access to enantiopure ring functionalized examples, with the majority of these being target specific.8 Among the more general methods for the synthesis of trans-2,6-disubstituted piperidines are the addition of silanes to chiral dihydropyridones,9 the hydroxyl directed reduction of 1,2-dehydropiperidines,10 the diastereoselective addition of organometallic reagents to chiral piperidine epoxides,11 and the synthesis of 2,4,6-trisubstituted piperidines via an azaelectrocyclization protocol.12
trans-2,6-Disubstituted 1,2,5,6-tetrahydropyridines 1 are potentially valuable chiral building blocks for asymmetric synthesis of polysubstituted piperidines because of the many methods available for functionalization of the ring carbon-carbon double bond (Scheme 1).13 Overman et al. developed an iminiuim ion-vinylsilane cyclization process to produce 1 with excellent stereocontrol.14 However, racemization via an aza-Cope process proved to be faster than cyclization. Panek and co-workers demonstrated that 1 could be prepared via cyclization of imines generated from α-allylsilane amino acids using Lewis acids.15 Reaction of Grignard reagents with oxazolidine derivatives derived from chiral pyridinium salts is also reported to give 1, in addition to the cis product.16 Employing ring-closing metathesis of chiral aminodienes the groups of Couty8i and Leberton17 prepared 1 having a 2-hydroxymethyl group. We describe here a new method for the asymmetric synthesis of trans-2,6-disubstituted 1,2,5,6-tetrahydropyridines 1 from N-sulfinyl δ-amino β-ketophosphonate enaminones, a new sulfinimine-derived chiral building block (Scheme 1). The application of this new protocol to the asymmetric synthesis of the quinolizidine alkaloid (−)-myrtine is presented.
Scheme 1.
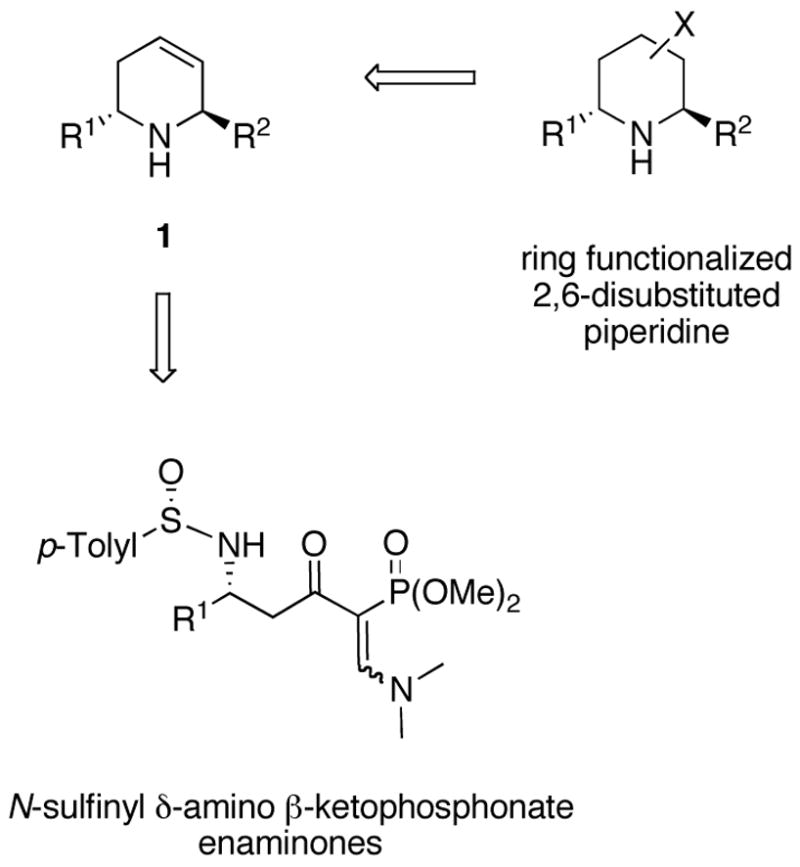
Results and Discussion
(SS,R)-(+)-N-Sulfinyl δ-amino β-ketophosphonate 2 was prepared as previous described,18 and treated with 20 equivalence of commercially available dimethylformamide dimethyl acetal at rt for 12 h.19 Removal of the solvent gave the crude N-sulfinyl δ-amino β-ketophosphonate enaminones 3 (Scheme 2). The absorptions appearing in the proton NMR of 3 at δ 8.02 and 2.63 ppm are attributed to the vinyl and N,N-dimethyl protons respectively, suggesting that a single isomer was formed of unknown stereochemistry. Because of the hydrolytic instability of 3, it was treated with 4 N HCl in dioxane without purification to give 4. Concentration and treatment of 4 with Cbz-Cl/DMAP gave 5-(dimethoxyphosphoryl) 2,3-dihydropyridone (2R)-(−)-5 in 55% yield for the five-step single flask reaction sequence (Scheme 2). As noted earlier, for the related δ-amino β-ketoester enaminone,19 the formation of 4 is consistent with an intramolecular Michael-type addition followed by retro-Michael elimination. The structure of (2R)-(−)-5 is supported by the chemical shift of the vinyl proton at δ 8.81 ppm and the phosphonate methoxy groups at δ 3.71 ppm in the 1H NMR. Comins and Ollinger prepared a related compound in racemic form via a nickel(II) cross-coupling reaction.9d
Scheme 2.

With dihydropyridone (−)-5 in hand, Michael addition of various cuprates was next explored. The expectation was that trans addition would predominate based on earlier observations by Comins et al., where it was suggested that the C-2 phenyl and C-5 phosphonate groups sterically inhibit axial attack via the lower energy chair conformation leading to the cis product.9d The cuprates were prepared by addition of methylmagnesium chloride, n-propylmagnesium chloride, and phenylmagnesium chloride to CuI at −78 °C in THF.20 The pale-white cuprate solutions were added via cannula to a −78 °C solution of (−)-5 to give the dihydropyridones 6 and 7 as separable trans/cis mixtures (Scheme 3). The chemical shift of the proton absorption at δ 10–11 ppm exchanges with MeOD and suggests that they exist 80 to 95% in the enol form. We attribute the major absorption at ca δ 25.5 ppm in the 31P NMR spectra of 6a-c to the enol forms. It was not possible to assign the stereochemistry of the major product to the trans isomer because of the complexity of the NMR spectra. However, the stereochemistry of the major product was assigned as trans by conversion into compounds of known absolute configuration as discussed below.
Scheme 3.

In order for the enols/ketophosphonates 6 to be useful building blocks methods need to be devised for the efficient removal of the phosphonate group. However, attempted conversion of the C-3 phosphonate into an enone using HWE reaction with acetaldehyde and DBU/LiCl or NaH failed, resulting in recovery of the starting material. Selective removal of the phosphonate groups in β-ketophosphonates has been described by reduction of the sodium enolate with lithium aluminum hydride.21 However, similar attempts to remove the C-3 phosphonate group in 6 resulted in decomposition.
Pagenkopf et. al. reported a procedure for conversion of acyclic β-hydroxy phosphonates into alkenes involving hydrolysis of the phosphonate and treatment with diisopropylcarbodiimide (DIC).22 Reduction of 6a–b with NaBH4 gave the alcohols 8a and 8b in 85 and 90 % yields, respectively (Scheme 4). All attempts to reduce 6c (R = Ph) failed. Hydrolysis of 8 to 9 was accomplished by reaction with 6 N NaOH/MeOH for 18 h. The crude acids 9 were not purified, but heated with DIC for 8 h to give the corresponding tetrahydropyridines 10a and 10b in 69 and 75% yields for the two-step sequence (Scheme 5).
Scheme 4.

Scheme 5.

Hydrogenation of (2S,6R)-(+)-10b was initially performed with Pd-C/H2 at 1 atm to reduce the double bond as well as to remove the Cbz group (Scheme 5). However, a 1:1 mixture of cis- and trans-isomers 11/12 resulted as determined by proton NMR. These results initially suggested that the double bond had migrated during the hydrogenation step. Selective hydrogenation of the carbon-carbon double bond in 10b was accomplished using Pt-C/H2, affording (+)-13 in 92% yield. Interestingly, when the hydrogenolysis of (+)-11 was carried out with Pd-C/H2 a 1:1 mixture of 11 and 12 was obtained suggesting that the source of the cis product 12 was isomerization at the C-2 phenyl group. The Cbz group was successfully removed without isomerization by treating (+)-13 with TMS-I, affording trans (+)-11 in 96% yield (Scheme 5).23 Similar results were obtained for 10a, resulting in trans-(+)-15 in 86 % yield. Since both (+)-1124 and (+)-1525 are known compounds, this confirms that the major product for the addition of organocuprates to dihydropyridone (−)-5 occurs trans to the C-2 phenyl group as predicted.
Asymmetric Synthesis of (−)-myrtine
(−)-Epimyrtine (16) and (+)-myrtine (17) are naturally occurring quinolizidine alkaloids isolated from Vaccinium myrtillus (Scheme 6).26 While a number of racemic syntheses27,28 of these compounds have been described, there are only three asymmetric synthesis of (−)-16.29 Two asymmetric syntheses of (+)-myrtine have been reported. Comins and LaMunyon described a three-step synthesis of this material starting from a chiral 4-methoxy-3-(triisopropylsilyl)pyridine.9a Gelas-Mialhe et al. employed (S)-2-(2-hydroxypropyl)allyltrimethylsilane and an intramolecular allylsilane N-acyliminium ion cyclization in their synthesis of (+)-17.8e However, this latter method also gave (−)-epimyrtine (16) resulting in poor selectivity.
Scheme 6.

We illustrate the utility of our new methodology for the asymmetric synthesis of trans-2,6-disubstituted 1,2,5,6-tetrahydropiperidines 1 with a total synthesis of (4S,10S)-(−)-myrtine (18), the unnatural myrtine isomer which has never been prepared. Our synthesis begins with β-amino ketoester (SS,S)-(+)-19 prepared earlier in our synthesis of (−)-16 (Scheme 7).29a The ester was treated with diethyl lithiummethylphosphonate to give an 85% isolated yield of (SS,S)-(+)-20. With DMF dimethyl acetal (+)-20 gave the intermediate enaminone, which was not purified, but treated with HCl-dioxane followed by trapping with (Boc)2O to give dihydropyridone (+)-21 in 55–60%. We found that hydrolysis of the enaminone intermediate with aqueous HCl improved the yield to 90%. Reaction of (+)-21 with the methyl cuprate at −78 °C afforded both the trans and cis piperidines in a ratio of 4:1 and an isolated yield of (2S,6S)-(−)-22 of 70%. The lower selectivity for this reaction is presumably due to the fact that the straight-chain C-2 alkyl substituent is not as sterically demanding as a phenyl group. trans-Tetrahydropyridine (2S,6S)-(+)-23 was prepared by reduction of (−)-22 with NaBH4, hydrolysis without purification with NaOH/MeOH, and treatment of the crude acid with DIC to give (+)-23 in 74% yield for the three steps (Scheme 7).
Scheme 7.

The 4-oxy group was introduced by oxymercuration of (+)-23 with Hg(NO3)2/NaBH4/NaOH to give a mixture of alcohols which were not isolated, but oxidized, using the Dess-Martin periodinane affording 4-oxo-trans-2,6-disubstituted piperidine (2S,6S)-(−)-24 in 78% yield for the three steps (Scheme 8).30 The N-Boc and benzyl-protecting groups were sequentially removed with TFA and subsequent hydrogenolysis with Pd(OH)2-C/H2 gave 4-oxo trans-2,6-disubstituted piperidine (2S,6S)-(−)-25 in 76% overall yield for the two steps. Cyclization to form the quinolizidine ring was readily accomplished using CCl4/PPh3/Et3N to give (4S,10S)-(−)-myrtine (18) in 68% yield (Scheme 7). Our synthesis of (−)-18 was accomplished in 14 steps (7 operations) with an overall yield of 17% from β-amino ester (+)-19. The alkaloid had spectral properties consistent with literature values.8e,9a,27f
In summary, new methodology for the asymmetric synthesis of 1,2,5,6-tetrahydropyridines, valuable chiral building blocks for the asymmetric synthesis of ring functionalized trans-2,6-disubstituted piperidines, has been devised. Highlights of the method include: 1) the one-pot conversion of sulfinimine-derived N-sulfinyl δ-amino β-ketophosphonates to 3-phosphoryl dihydropyridones; 2) the stereoselective addition of organocuprates to dehydropyridones to give the trans-2,6-disubstituted piperidines; and 3) the one pot dephosphorylation of the 3-phosphoryl piperidines to give the trans-1,2,5,6-tetrahydropyridine. The utility of this methodology was demonstrated in the synthesis of (−)-myrtine 18.
Experimental Section
(SS,R)-(+)-Dimethyl-4-(4-methylphenylsulfinamido)-2-oxo-4-phenylbutylphosphonate (2),18 and (SS,S)-(+)-methyl 7-(benzyloxy)-3-(4-methylphenylsulfinamido)heptanoate (19)29a were prepared as previously described.
(R)-(−)-Benzyl 5-(dimethoxyphosphoryl)-4-oxo-2-phenyl-3,4-dihydropyridine-1-carboxylate (5)
In a 100-mL, oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed (SS,R)-(+)-2 (1.5 g, 3.66 mmol) in toluene (30 mL). To the solution was added DMF dimethyl acetal (5.2 mL, 36.6 mmol) by syringe at rt. The reaction mixture was stirred for 12 h, and concentrated to give a yellow solid. To the yellow solid was added 4 N HCl (20 mL) solution, the solution was stirred for 1 h; 6 N NaOH solution (12 mL) was added, and neutralized to pH 7 with sat. NaHCO3 solution. The solution was extracted with EtOAc (3 × 50 mL), the combined organic phases were washed with brine (20 mL), dried (Na2SO4), and concentrated. To the residue was added THF (20 mL), Et3N (1.53 mL, 11.0 mmol), DMAP (0.02 g), and benzyl chloroformate (1.03 mL, 7.3 mmol). The reaction mixture was stirred at rt for 3 h and quenched with sat. NH4Cl (10 mL). The solution was extracted with EtOAc (3 × 40 mL), the combined organic phases were washed with H2O (10 mL), brine (10 mL), dried (Na2SO4), and concentrated. Flash chromatography (EtOAc) afforded 0.84 g (55%) of a colorless oil; [α]20D = −93.0 (c 0.5, CHCl3); IR (neat) 3185, 1867, 1705 cm−1; 1H NMR (CDCl3) δ 8.81 (dd, J = 15.2, 1.2 Hz, 1 H), 7.38-7.28 (m, 8 H), 7.14 (m, 2 H), 5.77 (d, J = 7.2 Hz, 1 H), 5.28 (dd, J = 31.6, 12.0 Hz, 2 H), 3.71 (dd, J = 34.4, 11.6 Hz, 6 H), 3.16 (m, 1 H), 2.86 (m, 1 H); 13C NMR (CDCl3) δ 189.1, 152.7 (J = 18.9 Hz), 152.1, 137.5, 134.4, 129.2, 129.1, 128.9, 128.8, 128.6, 125.7, 105.7 (J = 194.4 Hz), 70.2, 56.7, 53.1 (J = 20.8, 6.2 Hz), 42.3 (J = 7.8 Hz); 31P NMR (CDCl3) δ 17.65. HRMS calcd for C21H23NO6P (M + H) 416.1263. Found 416.1273.
(2S,6R)-(+)-Benzyl 3-(dimethoxyphosphoryl)-4-hydroxy-6-phenyl-2-propyl-5,6-dihydropyridine-1-carboxylate (6b). Typical procedure
In a 100-mL, oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed (−)-5 (0.45 g, 1.08 mmol) in THF (20 mL), and the solution was cooled to −78 °C. In second 50-mL, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed CuI (0.825 g, 4.32 mmol) in THF (20 mL), the solution was cooled to −78 °C, and PrMgCl (2.2 mL, 2.0 M in THF, Aldrich). The solution was stirred for 45 min at this temperature and was transferred via cannula via to the −78 °C solution of (−)-9. The reaction mixture was quenched after 0.5 h with sat. NH4Cl (20 mL), extracted with EtOAc (3 × 40 mL), the combined organic phases were washed with brine (10 mL), dried (Na2SO4), and concentrated. Flash chromatography (EtOAc:hexane:acetic acid: 50:50:3) afforded 0.40 g (81%) of a light yellow oil; [α]20D +80.4 (c 0.8, CHCl3); IR (neat) 3442, 1769, 1700 cm−1; 1H NMR (CDCl3) δ 10.27 (broad, 1 H), 7.22-6.44 (m, 10 H), 5.05 (m, 1 H), 4.78 (m, 1 H), 4.67 (m, 2 H), 3.69 (m, 6 H), 2.87 (m, 1 H), 2.28 (d, J = 15.2 Hz, 1 H), 1.71 (m, 1 H), 1.42 (m, 1 H), 1.19 (m, 2 H), 0.69 (t, J = 7.2 Hz 3 H); 13C NMR (CDCl3) δ 201.6 (J = 42.4 Hz), 174.9, 169.6, 155.7, 143.2, 136.4, 129.1, 128.5, 128.3, 128.2, 127.8, 127.7, 127. 5, 127.4, 127.1, 126.2, 125.6, 125.3, 91.4 (J = 188.3 Hz), 67.3, 52.9, 52.4, 52.2, 38.8, 36.1 (J = 14.0 Hz), 18.9, 14.2; 31P NMR (CDCl3) δ 25.82, 22.01, 21.59. HRMS calcd for C24H31NO6P (M + H) 460.1889. Found 460.1893.
(2S,6R)-(+)-Benzyl 3-(dimethoxyphosphoryl)-4-hydroxy-2-methyl-6-phenyl-5,6-dihydropyridine-1-carboxylate (6a)
Yield: 50%; light yellow oil; [α]20D +78.8 (c 0.8, CHCl3); IR (neat) 3423, 3310, 1745, 1698, 1056 cm−1; 1H NMR (CDCl3) δ 10.6 (s, 1 H), 7.24-6.99 (m, 10 H), 5.30 (m, 1 H), 4.93 (m, 2 H), 4.72 (m, 1 H), 3.61 (m, 6 H), 3.03 (m, 1 H), 2.43 (d, 1 H), 1.35 (d, J = 6.4 Hz, 3 H); 13C NMR (CDCl3) δ 201.0 (J = 6.8 Hz), 168.8, 155.0, 142.8, 136.5, 129.0, 128.3, 127.7, 127.1, 125.6, 125.2, 92.2 (J = 189.5 Hz), 67.6, 67.1, 55.4, 53.7, 53.2, 52.8, 52.3, 48.9, 48.1, 44.7, 35.9 (J = 14.1 Hz), 22.4, 20.8; 31P NMR (CDCl3) δ 25.46, 21.80. HRMS calcd for C22H26NO6PNa (M + Na) 454.1395. Found 454.1400.
(2S,6R)-(+)-Benzyl 3-(dimethoxyphosphoryl)-4-hydroxy-2,6-diphenyl-5,6-dihydropyridine-1-carboxylate (6c)
Yield: 91%; light yellow oil; [α]20D +139.4 (c 1.36, CHCl3); IR (neat) 3430, 3298, 1730, 1683, 1087 cm−1; 1H NMR (CDCl3) δ 10.95 (s, 1 H), 7.50-6.97 (m, 13 H), 6.73 (m, 2 H), 5.55 (m, 2 H), 4.93 (m, 1 H), 4.78 (m, 1 H), 3.62 (d, J = 11.2 Hz, 3 H), 3.27 (m, 1 H), 3.01 (d, J = 11.6 Hz, 3 H), 2.58 (d, J = 16.0 Hz, 1 H); 13C NMR (CDCl3) δ 201.0 (J = 6.8 Hz), 169.2, 155.7, 143.0, 136.2, 128.7, 128.5, 127.8, 127.7, 127.4, 127.3, 125.7, 91.7 (J = 182.0 Hz), 67.5, 56.4 52.8, 51.9, 36.7; 31P NMR (CDCl3) δ 25.83. HRMS calcd for C27H28NO6PNa (M + Na) 516.1552. Found 516.1562.
(2S,6R)-(+)-Benzyl 3-(dimethoxyphosphoryl)-4-hydroxy-6-phenyl-2-propylpiperidine-1-carboxylate (8b). Typical procedure
In a 25-mL, oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed (+)-6b (0.040 g, 0.087 mmol) in MeOH (10 mL). The solution was cooled to 0 °C and NaBH4 (0.02 g, 0.261 mmol) was slowly added. The reaction mixture was stirred for 0.5 h and quenched by addition of H2O (10 mL). At this time the solution was extracted with DCM (3 × 20 mL), the combined organic phases were washed with brine (10 mL), dried (Na2SO4), and concentrated. Flash chromatography (EtOAc) afforded 0.036 g (90%) of a colorless oil; [α]20D +47.5 (c 0.55, CHCl3); IR (neat) 3490, 1690, 1123 cm−1; 1H NMR (CDCl3) δ 7.24 (m, 8 H), 7.01 (m, 2 H), 4.89 (m, 2 H), 4.66 (m, 1 H), 4.39 (m, 2 H), 3.77 (dd, J = 10.8, 2.8 Hz, 6 H), 2.41 (m, 2 H), 2.18 (m, 2 H), 1.72 (m, 2 H), 1.50 (m, 2 H), 0.96 (t, J = 7.2 Hz, 3 H); 13C NMR (CDCl3) δ 157.0, 141.9, 136.3, 128.1, 127.9, 127.6, 126.5, 126.0, 66.9, 65.2 (J = 5.4 Hz), 53.5 (J = 9.2 Hz), 53.0 (J = 6.8 Hz), 52.7 (J = 5.9 Hz), 41.8 (J = 137.3 Hz), 36.1 (J = 8.8 Hz), 34.7 (J = 6.2 Hz), 19.7, 13.7; 31P NMR (CDCl3) δ 32.15. HRMS calcd for C24H32NO6PNa (M + Na) 484.1865. Found 484.1874.
(2S,6R)-(+)-Benzyl 3-(dimethoxyphosphoryl)-4-hydroxy-2-methyl-6-phenylpiperidine-1-carboxylate (8a)
Yield, 85%; colorless oil; [α]20D +54.4 (c 0.23, CHCl3); IR (neat) 3502, 3313, 1697, 1099 cm−1; 1H NMR (CDCl3) δ 7.29-6.98 (m, 10 H), 4.91 (m, 2 H), 4.63 (m, 2 H), 4.38 (m, 1 H), 3.78 (m, 6 H), 2.31 (m, 2 H), 2.12 (m, 1 H), 1.43 (d, J = 6.8 Hz, 3 H); 13C NMR (CDCl3) δ 156.8, 142.4, 136.3, 128.3, 128.0, 127.8, 126.6, 125.8, 67.1, 65.0, 54.5, 53.0, 52.7, 49.1, 43.6 (J = 136.2 Hz), 37.3, 19.2; 31P NMR (CDCl3) δ 31.5, 29.2. HRMS calcd for C22H28NO6PNa (M + Na) 456.1552. Found 456.1554.
(2S,6R)-(+)-Benzyl 2-propyl-6-phenyl-5,6-dihydropyridine-1-carboxylate (10b). Typical procedure for dephosphonylation
In a 25-mL, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum and an argon inlet was placed (+)-8b (0.160 g, 0.348 mmol) in MeOH (5 mL). To the solution was added 6N NaOH (5 mL), the solution was stirred for 16 h and brought to pH < 2 by addition of 1 N HCl (25 mL). The reaction mixture was extracted with DCM (3 × 30 mL), the combined organic phases were washed with brine (10 mL), dried (Na2SO4), and concentrated. The residue was dissolved in CHCl3 (20 mL) and DIC (0.43 mL), 2.77 mmol) was added. The reaction mixture was heated at 60 °C in an oil bath for 8 h and concentrated. Flash chromatography (EtOAc:hexane, 10:90) afforded 0.88 g (75%) of a colorless oil; [α]20D +207.2 (c 0.9, CHCl3); IR (neat) 3210, 2993, 1720 cm−1; 1H NMR (CDCl3) δ 7.24-7.11 (m, 8 H), 7.11-7.04 (m, 2 H), 6.01 (m, 1 H), 5.66 (m, 1 H), 5.24 (dd, J = 5.6, 1.6 Hz, 1 H), 4.97 (m, 2 H), 4.54 (m, 1 H), 2.72 (m, 1 H), 2.33 (m, 1 H), 1.86 (m, 1 H), 1.73 (m, 1 H), 1.37 (m, 2 H), 0.92 (m, 3 H); 13C NMR (CDCl3) δ 156.1, 145.2, 136.8, 130.6, 128.3, 128.1, 127.7, 126.5, 126.1, 123.0, 66.9, 55.2, 53.7, 37.9, 30.9, 19.0, 14.2. HRMS calcd for C22H25NO2Na (M + Na) 358.1783. Found 358.1773.
(2S,6R)-(+)-Benzyl 2-methyl-6-phenyl-5,6-dihydropyridine-1-carboxylate (10a)
Yield: 69%; colorless oil; [α]20D +184.7 (c 0.55, CHCl3); IR (neat) 3312, 2987, 1699 cm−1; 1H NMR (CDCl3) δ 7.23-7.06 (m, 8 H), 7.02 (m, 2 H), 5.90 (m, 1 H), 5.56 (m, 1 H), 5.25 (m, 1 H), 4.95 (m, 2 H), 4.52 (m, 1 H), 2.72 (m, 1 H), 2.28 (m, 1 H), 1.34 (d, J = 6.4 Hz, 3 H); 13C NMR (CDCl3) δ 156.0, 145.1, 136.9, 132.0, 128.4, 128.2, 127.7, 126.6, 126.1, 122.0, 67.0, 55.2, 49.4, 30.6, 21.4. HRMS calcd for C20H21NO2Na (M + Na) 330.1470. Found 330.1477.
(2S,6R)-(+)-Benzyl 2-propyl-6-phenylpiperidine-1-carboxylate (13). Typical hydrogenation procedure
In a 25-mL, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum was placed (+)-10b (0.034 g, 0.1 mmol) and 5% Pt/C (0.015 g) in MeOH (10 mL). The solution was stirred under 1 atm of H2 (balloon) for 0. 5 h, filtered, and concentrated. Flash chromatography (EtOAc:hexane, 10:90) afforded 0.031 g (92%) of a colorless oil; [α]20D +61.1 (c 0.36, CHCl3); IR (neat) 3269, 1743 cm−1; 1H NMR (CDCl3) δ 7.26-7.00 (m, 10 H), 5.07-4.95 (m, 3 H), 3.92 (m, 1 H), 2.06 (m, 2 H), 1.81-1.20 (m, 8 H), 0.84 (t, J = 7.2 Hz, 3 H); 13C NMR (CDCl3) δ 156.5, 143.3, 137.0, 128.4, 128.3, 127.9, 127.8, 126.3, 125.9, 66.9, 55.1, 52.7, 36.9, 27.4, 23.7, 20.4, 14.6, 14.2. HRMS calcd for C22H27NO2Na (M + Na) 360.1940. Found 360.1947.
(2S,6R)-(+)-Benzyl 2-methyl-6-phenylpiperidine-1-carboxylate (14)
Yield, 91%; colorless oil; [α]20D +66.0 (c 0.22, CHCl3); IR (neat) 3287, 1741 cm−1; 1H NMR (CDCl3) δ 7.25-7.06 (m, 10 H), 5.04 (m, 2 H), 5.01 (m, 1 H), 4.17 (m, 1 H), 2.09 (m, 2 H), 1.73 (m, 1 H), 1.57 (m, 1 H), 1.40 (m, 2 H), 1.28 (d, J = 6.8 Hz, 3 H); 13C NMR (CDCl3) δ 156.5, 143.6, 137.1, 128.4, 127.8, 126.3, 125.8, 66.9, 54.9, 48.1, 27.4, 26.7, 21.0, 14.1. HRMS calcd for C15H19NO2Na (M + Na) 332.1626. Found 332.1633.
(2S,6R)-(+)-2-Propyl-6-phenylpiperidine (11)
In a 25-mL, single-necked round-bottom flask equipped with a magnetic stirring bar, and rubber septum was placed (+)-13 (0.034 g, 0.1 mmol) in MeCN (10 mL). To the solution was added TMS-I (57.0 mL, 0.4 mmol), the reaction mixture was stirred for 15 min, concentrated, and quenched with 1 N HCl (10 mL). The solution was extracted with ether (2 × 5 mL), the aqueous phase was neutralized 3 N NaOH until a white precipitate was formed. At this time the solution was extracted with DCM (3 × 10 mL), the combined DCM extracts were washed with brine (5 mL), dried (Na2SO4), and concentrated. Flash chromatography (EtOAc) afforded 0.2 g (96%) of a colorless oil; [α]20D +28.1 (c 0.2, acetone); IR (neat) 3280, 3060, 1231, 1033 cm−1; 1H NMR (CDCl3) δ 7.38 (m, 2 H), 7.32 (m, 2 H), 7.24 (m, 1 H), 3.96 (m, 1 H), 3.05 (m, 1 H), 2.01-1.21 (m, 11 H), 0.94 (d, J = 6.8 Hz, 3 H); 13C NMR (CDCl3) δ 145.3, 128.5, 126.9, 54.4, 52.1, 34.8, 33.6, 29.8, 20.4, 19.9, 14.3. HRMS calcd for C14H22N (M + H) 204.1752. Found 204.1753.
(2S,6R)-(+)-2-Methyl-6-phenylpiperidine (15)
Yield: 95%; colorless oil; [α]20D +34.2 (c 0.3, CHCl3) [lit.25 +33.4 (c 1.25, CHCl3)]; IR (neat) 3287, 3050, 1233, 1042 cm−1; 1H NMR (CDCl3) δ 7.38 (m, 2 H), 7.32 (m, 2 H), 7.23 (m, 1 H), 4.05 (m, 1 H), 3.30 (m, 1 H), 1.72 (m, 6 H), 1.42 (m, 1 H), 1.22 (d, J = 6.8 Hz, 3 H); 13C NMR (CDCl3) δ 145.3, 128.5, 126.9, 54.3, 47.5, 33.5, 31.5, 20.1, 20.0. HRMS calcd for C12H17N (M ) 175.1361. Found 175.1358.
(SS,S )-(+)-Diethyl 8-(benzyloxy)-4-(4-methylphenylsulfinamido)-2-oxooctylphosphonate (20)
In a 250 mL, oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon balloon was placed diethyl methyl phosphonate (5.12 mL, 35.4 mmol) in THF (100 mL). The solution was cooled to −78 °C and n-BuLi (2.5 M in hexane, 14.2 mL, 35.4 mmol) was slowly added via syringe. In a second 500 mL oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon balloon was placed (+)-19 (2.85 g, 7.07 mmol) in THF (120 mL) at −78 °C. The diethyl methyl phosphonate solution was transferred after 45 min to the −78 °C solution of (+)-19 and the reaction mixture was stirred for 2 h. At this time the reaction was quenched by addition of sat. NH4Cl (30 mL), warmed to rt, and diluted with H2O (10 mL). The solution was extracted with Et2O (50 mL), EtOAc (2 × 50 mL), and the combined organic phases were washed with brine (20 mL), dried (MgSO4) and concentrated. Flash chromatography (EtOAc) gave a colorless oil, which was subjected to Kugelrohr vacuum distillation (60 °C under 2.5 mm Hg), to remove the diethyl methylphosphonate, affording 3.15 g (85%) of a colorless oil; [α]20D +26.9 (c 3.3, CHCl3); IR (neat) 3420, 3180, 1721, 1226 cm−1; 1H NMR (CDCl3) δ 7.48 (m, 2 H), 7.31-7.13 (m, 7 H), 4.51 (d, J = 9.2 Hz, 1 H), 4.43 (s, 2 H), 4.05 (m, 4 H), 3.61 (m, 1 H), 3.42 (t, J = 6.4 Hz, 2 H), 2.98 (d, J = 22.8 Hz, 2 H), 2.88 (q, J = 5.2, 3.2 Hz, 2 H), 2.33 (s, 3 H), 1.66-1.34 (m, 6 H), 1.25 (m, 6 H); 13C NMR (CDCl3) δ 201.1 (J = 6.1 Hz), 142.8, 141.5, 139.0, 129.8, 128.6, 127.9, 127.8, 125.8, 73.2, 70.4, 62.9 (J = 6.3 Hz), 52.7, 50.0, 43.4 (J = 125.2 Hz), 35.8, 29.6, 23.1, 21.6, 16.6 (J = 6.0 Hz); 31P NMR (CDCl3) δ 19.53. HRMS calcd for C26H39NO6PS (M + H) 524.2236. Found 524.2239.
(S)-(+)-tert-Butyl 2-(4-(benzyloxy)butyl)-5-(diethoxyphosphoryl)-4-oxo-3,4-dihydropyridine-1-carboxylate (21)
In a 100-mL, oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed (+)-20 (1.92 g, 3.66 mmol) in toluene (30 mL). To the solution was added DMF dimethyl acetal (5.2 mL, 36.6 mmol), the reaction mixture was stirred for 12 h and concentrated to give a yellow solid. To the solid was added 4 N HCl (20 mL), the solution was stirred for 1 h, 6 N NaOH (12 mL) was added, and the solution neutralized to pH >10 with sat. NaHCO3 solution. The aqueous solution was extracted with EtOAc (3 × 50 mL), the combined organic phases were washed with brine (20 mL), dried (Na2SO4), and concentrated. The residue was dissolved in THF (20 mL), TEA (1.53 mL, 11.0 mmol), DMAP (0.20 g), and benzyl chloroformate (1.03 mL, 7.3 mmol) were added. After stirring the reaction mixture for 3 h, sat. NH4Cl (10 mL) was added. The solution was extracted with EtOAc (3 × 40 mL), the combined organic phases were washed with brine (20 mL), dried (Na2SO4), and concentrated. Flash chromatography (EtOAc) afforded 1.63 g (90%) of a colorless oil; [α]20D +31.2 (c 1.0, CHCl3); IR (neat) 3211, 1731, 1654, 1256 cm−1; 1H NMR (CDCl3) δ 8.49 (dd, J = 14.8, 1.2 Hz, 1 H), 7.36-7.24 (m, 5 H), 4.56 (m, 1 H), 4.47 (s, 2 H), 4.13 (m, 4 H), 3.42 (m, 2 H), 2.77 (m, 1 H), 2.48 (m, 1 H), 1.65-1.35 (m, 6 H), 1.53 (s, 9 H), 1.31 (q, J = 7.2 Hz, 6 H); 13C NMR (CDCl3) δ 190.5, 151.9 (J = 18.5 Hz), 150.7, 138.8, 126.7, 127.9, 127.8, 104.9 (J = 195.2 Hz), 85.3, 73.3, 70.2, 62.6, 40.2 (J = 8.9 Hz), 31.0, 29.7, 28.2, 22.8, 16.7; 31P NMR (CDCl3) δ 15.27. HRMS calcd for C25H39NO7P (M + H) 496.2464. Found 496.2461.
(2S,6S)-(−)-tert-Butyl 6-(4-(benzyloxy)butyl)-3-(diethoxyphosphoryl)-4-hydroxy-2-methyl-5,6-dihydropyridine-1-carboxylate (22)
In a 100-mL, oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed (+)-21 (0.496 g, 1.08 mmol) in THF (20 mL) and the solution was cooled to −78 °C. In a second 50-mL, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed CuI (0.83 g, 4.32 mmol) in THF (20 mL). The reaction mixture was cooled to −78 °C, and MeMgCl (1.4 mL, 3.0 M in THF, Aldrich) was added. The reaction mixture was stirred for 45 min and cannulated to the solution of (+)-21, and after 0.5 h sat. NH4Cl (10 mL) was added. The solution was extracted with EtOAc (3 × 40 mL) and the combined organic phases were washed with brine (10 mL), dried (Na2SO4), and concentrated. Flash chromatography (EtOAc:hexane:acetic acid, 30:70:4) afforded 0.357 g (70%) of a light yellow oil; [α]20 D −7.5 (c 0.8, CHCl3); IR (neat) 3450, 1737, 1697, 1256, 1109 cm−1; 1H NMR (CDCl3) δ 10.90 (s, 1 H), 7.31-7.18 (m, 5 H), 4.42 (s, 2 H), 4.41 (m, 1 H), 4.04 (m, 4 H), 3.90 (m, 1 H), 3.39 (m, 2 H), 2.60 (m, 1 H), 2.24 (m, 1 H), 1.55 (m, 3 H), 1.40 (s, 9 H), 1.30-1.17 (m, 12 H); 13C NMR (CDCl3) δ 202.7 (J = 6.4 Hz), 170.3, 154.6, 139.1, 128.7, 127.9, 127.8, 127.7, 92.9 (J = 187.0 Hz), 80.0, 73.3, 70.7, 62.4, 52.5, 47.5 (J = 14.2 Hz), 34.8, 32.3 (J = 15.0 Hz), 30.0, 28.9, 23.8, 22.7, 16.5; 31P NMR (CDCl3) δ 24.84, 20.42, 20.32. HRMS calcd for C26H43NO7P (M + H) 512.2777. Found 512.2803.
(2S,6S)-(+)-tert-Butyl 6-(4-(benzyloxy)butyl)-2-methyl-5,6-dihydropyridine-1-carboxylate (23)
In a 25-mL, oven-dried, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum, and argon inlet was placed (+)-22 (0.178 g, 0.348 mmol) in MeOH (10 mL). The solution was cooled to 0 °C, NaBH4 (0.105 g, 1.39 mmol) was slowly added, and the reaction mixture was stirred for 0.5 h before addition of H2O (10 mL). The solution was extracted with DCM (3 × 20 mL), the combined organic phases were washed with brine (10 mL), dried (Na2SO4), and concentrated to give an oil. The oil, without further purification, was placed in a 25-mL, single-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum and an argon inlet and MeOH (5 mL) was added. To the solution was added 6 N NaOH (5 mL), and stirred 16 h. At this time the reaction mixture was brought to pH <2 with 1 N HCl (25 mL) and the mixture was extracted with DCM (3 × 30 mL). The combined organic phases were washed with brine (10 mL), dried (Na2SO4), and concentrated. To the residue was added CHCl3 (20mL), DIC (430 mL, 2.77 mmol) and the reaction mixture was refluxed at 60 °C for 8 h. Concentration and flash chromatography (EtOAc:hexane, 10:90) afforded 0.088 g (74%) of a colorless oil; [α]20D +55.0 (c 0.5, CHCl3); IR (neat) 3310, 2210, 1700, 1231 cm−1; 1H NMR (CDCl3) δ 7.34-7.26 (m, 5 H), 5.72 (m, 2 H), 4.49 (s, 2 H), 4.13 (m, 1 H), 4.00 (m, 1 H), 3.45 (t, J = 6.8 Hz, 2 H), 2.26 (m, 1 H), 2.08 (m, 1 H), 1.60 (m, 2 H), 1.50 (m, 2 H), 1.47 (s, 9 H), 1.31 (m, 2 H), 1.27 (d, J = 6.4 Hz, 3 H); 13C NMR (CDCl3) δ 156.1, 139.3, 132.2, 128.9, 128.1, 128.0, 122.7, 79.8, 73.4, 70.9, 52.1, 48.7, 33.9, 30.3, 29.1, 27.4, 24.3, 22.1. HRMS calcd for C22H33NO3Na (M + Na) 382.2358. Found 382.2365.
(2S,6S)-(−)-tert-Butyl 2-(4-(benzyloxy)butyl)-6-methyl-4-oxopiperidine-1-carboxylate (24)
In a 10 mL round-bottom flask fitted with a magnetic stirring bar was placed mercuric nitrate (0.210 g, 0.612 mmol) in THF (0.5 mL) and H2O (1.0 mL). In one portion was added (+)-23 (0.200 g, 0.556 mmol) in THF (0.5 mL). The reaction mixture was stirred for 10 min at rt and NaOH solution (6 M, 1 mL) was added, followed by NaBH4 solution (0.5 M in 3 M NaOH, 1 mL). At this time, EtOAc (5 mL) and solid NaCl were added, the solid mercury was allowed to settle, and the organic phase was separated, dried (MgSO4), and concentrated. The crude oil, without further purification, was placed in a 25-mL round-bottom flask equipped with a magnetic stirring bar and DCM (5 mL) was added. Dess-Martin periodinane (0.3 M in DCM, 0.69 mL, 0.334 mmol) was added, the reaction mixture was stirred for 2 h, and sat. Na2S2O3 (10 mL) solution was added. The solution was extracted with ether (3 × 10 mL), dried (MgSO4), and concentrated. Flash chromatography (EtOAc:hexane, 20:80) afforded 0.165 g (78%) of a colorless oil; [α]20D −85.0 (c 1.2, CHCl3); IR (neat) 3320, 1789, 1720, 1123 cm−1; 1H NMR (CDCl3) δ 7.34-7.26 (m, 5 H), 4.47 (s, 2 H), 4.34 (m, 1 H), 4.15 (m, 1 H), 3.44 (t, J = 6.0 Hz, 2 H), 2.75 (m, 2 H), 2.53 (m, 1 H), 2.35 (m, 1 H), 1.75 (m, 2 H), 1.60 (m, 2 H), 1.48 (s, 9 H), 1.35 (m, 2 H), 1.25 (d, J = 6.4 Hz, 3 H); 13C NMR (CDCl3) δ 208.2, 154.9, 138.9, 128.7, 127.9, 127.8, 80.3, 73.2, 70.4, 51.4, 46.9, 44.9, 41.6, 37.3, 29.9, 28.8, 23.8, 23.0. HRMS calcd for C22H33NO4Li (M + Li) 382.2570. Found 382.2555.
(2S,6S)-(−)-2-(4-Hydroxybutyl)-6-methylpiperidin-4-one (25)
In a 25 mL oven-dried round-bottom flask equipped with a magnetic stirring bar, rubber septum and an argon inlet was placed (−)-24 (0.075 g, 0.2 mmol) in DCM (5 mL). The solution was cooled to 0 °C, TFA (1.5 mL) was added, and the reaction mixture was stirred at rt for 45 min. At this time the solution was concentrated and the salt was placed in a second 25 mL, one-necked round-bottom flask equipped with a magnetic stirring bar, rubber septum. To the solution was added Pd(OH)2 (20 mg, 20% on carbon), 2 drops of TFA in THF (5 mL) and the reaction mixture was stirred at rt for 3 h under an atmosphere of H2. At this time sat. NaHCO3 (5 mL) was added, the solution was extracted with EtOAc (3 × 10 mL), the organic phases were dried (MgSO4), and concentrated. Flash chromatography (MeOH:DCM, 10:90) afforded 0.029 g (85%) of a colorless oil; [α]20D −14.0 (c 0.3, CHCl3); IR (neat) 3485, 1760 cm−1; 1H NMR (CDCl3) δ 3.65 (t, J = 6.8 Hz, 2 H), 3.46 (m, 1 H), 3.35 (m, 1 H), 2.51 (m, 2 H), 2.18 (m, 2 H), 2.00 (br, 2 H), 1.61-1.38 (m, 6 H), 1.17 (d, J = 6.4 Hz, 3 H); 13C NMR (CDCl3) δ 209.7, 62.9, 53.0, 49.7, 48.2, 47.9, 34.6, 32.7, 22.6, 21.7; HRMS calcd for C10H20NO2 (M + H) 186.1494. Found 186.1493.
(4S,10S)-(−)-Myrtine (18)
In a 25-mL, one-necked, round-bottomed flask fitted with magnetic stirring bar, rubber septum, and argon balloon were placed (−)-25 (0.036 g, 0.19 mmol) in MeCN (3 mL). A solution of Et3N (0.028 mL, 0.20 mmol) in CCl4 (0.028 mL, 0.29 mmol) was added and the solution was cooled to 0 °C. At this time triphenylphosphine (0.047 g, 0.18 mmol) was added and the reaction mixture was stirred for 45 min, warmed to rt, and stirred for 20 h. To the reaction mixture was added sat. NaHCO3 (5 mL), and the solution was extracted with EtOAc (2 × 15 mL). The combined organic phases were washed with brine, dried (Na2SO4), and concentrated. Flash chromatography (EtOAc) gave 0.022 g (68%) of an oil; [α]20D −12.5 (c 0.4, CHCl3) [lit.26c +11.3 (c 2.7, CHCl3) and lit.9a +19.3 (c 1.85, CHCl3)] for its enantiomer); IR (neat) 2801, 1725 cm−1; 1H NMR (CDCl3) δ 3.40 (m, 1 H), 2.92-2.74 (m, 2 H), 2.65 (m, 1 H), 2.49 (t, J = 12.0, 3.6 Hz, 1 H), 2.32-2.15 (m, 3 H), 1.91-1.56 (m, 4 H), 1.48-1.17 (m, 2 H), 0.97 (d, J = 6.8 Hz, 3 H); 13C NMR (CDCl3) δ 209.5, 57.2, 53.6, 51.5, 48.7, 48.0, 34.3, 25.9, 23.4, 11.2. HRMS calcd for C10H18NO (M + H) 168.1389. Found 168.1389.
Supplementary Material
Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
This work was supported by grants from the National Institute of General Medical Sciences (GM57870 and GM51982)
References
- 1.(a) O’Hagan D. Nat Prod Rep. 2000;17:435. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]; (b) Amat M, Hidalgo J, Llor N, Bosch J. Tetrahedron: Asymmetry. 1998;9:2419. [Google Scholar]; (c) Mauger AB. J Nat Prod. 1996;59:1205. doi: 10.1021/np9603479. [DOI] [PubMed] [Google Scholar]
- 2.During a 10-year period Watson and co-workers state that there were thousands of piperidine compounds cited in clinical and preclinical trials. Watson PS, Jiang B, Scott B. Org Lett. 2000;2:3679. doi: 10.1021/ol006589o.
- 3.Leclercq S, Braekman JC, Daloze D, Pasteels JM. Prog Chem Org Nat Prod. 2000;79:115. doi: 10.1007/978-3-7091-6341-2_2. [DOI] [PubMed] [Google Scholar]
- 4.For leading references see: Davis BG, Maughan MAT, Chapman TM, Villard R, Courtney S. Org Lett. 2002;4:103. doi: 10.1021/ol016970o.Davis BG. Chem Rev. 2002;102:579. doi: 10.1021/cr0004310.
- 5.(a) Michael JP. Nat Prod Rep. 2001;18:520. doi: 10.1039/b005384h. [DOI] [PubMed] [Google Scholar]; (b) Mitchinson A, Nadin A. J Chem Soc, Perkin Trans 1. 2000:2862. [Google Scholar]; (c) Michael JP. Nat Prod Rep. 2000;17:579. doi: 10.1039/a904849i. [DOI] [PubMed] [Google Scholar]
- 6.For reviews see: Weintraub PM, Sabol JS, Kane JM, Borcherding DR. Tetrahedron. 2003;59:2953.Laschat S, Dickner T. Synthesis. 2002:1781.Pearson MSM, Mathe-Allainmat M, Fargeas V, Lebreton J. Eur J Org Chem. 2005:2159.Felpin FX, Lebreton J. Current Organic Synthesis. 2004;1:83.Buffat MGP. Tetrahedron. 2004;60:1701.
- 7.(a) Takahata H, Yotsui Y, Momose T. Tetrahedron. 1998;54:13505. [Google Scholar]; (b) Amat M, Llor N, Hidalgo J, Escolano C, Bosch J. J Org Chem. 2003;68:1919. doi: 10.1021/jo0266083. [DOI] [PubMed] [Google Scholar]; (c) Comins DL, Weglarz MA. J Org Chem. 1991;56:2506. [Google Scholar]; (d) Grierson DS, Royer J, Guerrier L, Husson HP. J Org Chem. 1986;51:4475. [Google Scholar]; (e) Najdi S, Kurth MJ. Tetrahedron Lett. 1990;31:3279. [Google Scholar]; (f) Kotsuki H, Kusumi T, Inoue M, Ushio Y, Ochi M. Tetrahedron Lett. 1991;32:4159. [Google Scholar]; (g) Comins DL, Hong H. J Am Chem Soc. 1993;115:8851. [Google Scholar]; (h) Jefford CW, Wang JB. Tetrahedron Lett. 1993;34:2911. [Google Scholar]; (i) Oppolzer W, Bochet CG, Merifield E. Tetrahedron Lett. 1994;35:7015. [Google Scholar]; (j) Takahata H, Inose K, Araya N, Momose T. Heterocycles. 1994;38:1961. [Google Scholar]; (k) Freville S, Bonin M, Celerier JP, Husson HP, Lhommet G, Quirion JC, Thuy VM. Tetrahedron. 1997;53:8447. [Google Scholar]; (l) David M, Dhimane H, Vanucci-Bacque C, Lhommet G. J Org Chem. 1999;64:8402. doi: 10.1021/jo9906476. [DOI] [PubMed] [Google Scholar]; (m) Takahata H, Ouchi H, Ichinose M, Nemoto H. Org Lett. 2002;4:3459. doi: 10.1021/ol0265620. [DOI] [PubMed] [Google Scholar]; (n) Girard N, Hurvois JP. Synthetic Communications. 2005;35:711. [Google Scholar]
- 8.(a) Wuts PGM, Jung YW. J Org Chem. 1988;53:1957. [Google Scholar]; (b) Ciufolini MA, Hermann CW, Whitmire KH, Byrne NE. J Am Chem Soc. 1989;111:3473. [Google Scholar]; (c) Hamada T, Zenkoh T, Sato H, Yonemitsu O. Tetrahedron Lett. 1991;32:1649. [Google Scholar]; (d) Agami C, Couty F, Lam H, Mathieu H. Tetrahedron. 1998;54:8783. [Google Scholar]; (e) Gardette D, Gelas-Mialhe Y, Gramain JC, Perrin B, Remuson R. Tetrahedron: Asymmetry. 1998;9:1823. [Google Scholar]; (f) Chalard P, Remuson R, Gelas-Mialhe Y, Gramain JC. Tetrahedron: Asymmetry. 1998;9:4361. [Google Scholar]; (g) Korotayeva LM, Rubinskaya TY, Klimkina EV, Gultyai VP, Bubnov YN. Russian Chem Bull. 1999;48:754. [Google Scholar]; (h) Ratni H, Kundig EP. Org Lett. 1999;1:1997. doi: 10.1021/ol991158v. [DOI] [PubMed] [Google Scholar]; (i) Agami C, Couty F, Rabasso N. Tetrahedron. 2001;57:5393. [Google Scholar]; (j) Agami C, Comesse S, Kadouri-Puchot C. J Org Chem. 2002;67:2424. doi: 10.1021/jo010780+. [DOI] [PubMed] [Google Scholar]; (k) Goujon JY, Gueyrard D, Compain P, Martin OR, Asano N. Tetrahedron: Asymmetry. 2003;14:1969. [Google Scholar]; (l) Acherki H, Alvarez-Ibarra C, Guzman-Fernandez S, Quiroga-Feijoo ML. Tetrahedron: Asymmetry. 2004;15:693. [Google Scholar]; (m) Wang Q, Sasaki NA. J Org Chem. 2004;69:4767. doi: 10.1021/jo0496291. [DOI] [PubMed] [Google Scholar]
- 9.(a) Comins DL, LaMunyon DH. J Org Chem. 1992;57:5807. [Google Scholar]; (b) Comins DL, Kuethe JT. Org Lett. 2000;2:855. doi: 10.1021/ol0056271. [DOI] [PubMed] [Google Scholar]; (c) Comins DL, Sandelier MJ, Grillo TA. J Org Chem. 2001;66:6829. doi: 10.1021/jo015834u. [DOI] [PubMed] [Google Scholar]; (d) Comins DL, Ollinger CG. Tetrahedron Lett. 2001;42:4115. [Google Scholar]
- 10.Davis FA, Rao A, Carroll PJ. Org Lett. 2003;5:3855. doi: 10.1021/ol035390j. [DOI] [PubMed] [Google Scholar]
- 11.Lemire A, Charette AB. Org Lett. 2005;7:2747. doi: 10.1021/ol051022z. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi T, Hasegawa F, Tanaka K, Katsumura S. Org Lett. 2006;8:3813. doi: 10.1021/ol0614065. [DOI] [PubMed] [Google Scholar]
- 13.For a review on the synthesis of 2,6-dialkyl-1,2,5,6-tetrahydropyridines see ref. 6d.
- 14.(a) Daub GW, Heerding DA, Overman LE. Tetrahedron. 1988;44:3919. [Google Scholar]; (b) Castro P, Overman LE, Zhang X, Mariano PS. Tetrahedron Lett. 1993;34:5243. [Google Scholar]
- 15.(a) Sparks MA, Panek JS. J Org Chem. 1991;56:3431. [Google Scholar]; (b) Huang H, Spande TF, Panek JS. J Am Chem Soc. 2003;125:626. doi: 10.1021/ja028937i. [DOI] [PubMed] [Google Scholar]
- 16.(a) Compere D, Marazano C, Das BC. J Org Chem. 1999;64:4528. [Google Scholar]; (b) Guilloteau-Bertin B, Compere D, Gil L, Marazano C, Das BC. Eur J Org Chem. 2000:1391. [Google Scholar]; (c) Genisson Y, Marazano C, Mehmandoust M, Gnecco D, Das BC. Synlett. 1992:431. [Google Scholar]
- 17.(a) Felpin FX, Lebreton J. Tetrahedron Lett. 2003;44:527. [Google Scholar]; (b) Felpin FX, Boubekeur K, Lebreton J. Eur J Org Chem. 2003:4518. doi: 10.1021/jo035522m. [DOI] [PubMed] [Google Scholar]; (c) Felpin FX, Boubekeur K, Lebreton J. J Org Chem. 2004;69:1497. doi: 10.1021/jo035522m. [DOI] [PubMed] [Google Scholar]
- 18.Davis FA, Wu Y, Xu H, Zhang J. Org Lett. 2004;6:4523. doi: 10.1021/ol048157+. [DOI] [PubMed] [Google Scholar]
- 19.Davis FA, Zhang J, Li Y, Xu H, DeBrosse C. J Org Chem. 2005;70:5413. doi: 10.1021/jo050373o. [DOI] [PubMed] [Google Scholar]
- 20.(a) Gilman H, Jones RG, Woods LA. J Org Chem. 1952;17:1630. [Google Scholar]; (b) Yamamoto Y. Angew Chem Int Ed Engl. 1986;25:947. [Google Scholar]
- 21.Hong JE, Shin WS, Jang WB, Oh DY. J Org Chem. 1996;61:2199. [Google Scholar]
- 22.Reichwein JF, Pagenkopf BL. J Am Chem Soc. 2003;125:1821. doi: 10.1021/ja027658s. [DOI] [PubMed] [Google Scholar]
- 23.Jung ME, Lyster MA. J Chem Soc Chem Comm. 1978;315 [Google Scholar]
- 24.Korotayeva LM, Rubinskaya TY, Klimkina EV, Gultyai VP, Bobnov YN. Russ Chem Bull. 1999;48:754. [Google Scholar]
- 25.Poerwono H, Higashiyama K, Yamauchi T, Kubo H, Ohmiya S, Takahashi H. Tetrahedron. 1998;54:13955. [Google Scholar]
- 26.(a) Slosse P, Hootele C. Tetrahedron Lett. 1978:397. [Google Scholar]; (b) Slosse P, Hootele C. Tetrahedron. 1981;37:4287. [Google Scholar]; (c) Slosse P, Hotele C. Tetrahedron. 1979;20:4587. [Google Scholar]
- 27.For the syntheses of (±)-myrtine, see: King FDJ. Chem Soc, Perkin Trans 1. 1986:447.Comins DL, LaMunyon DH. Tetrahedron Lett. 1989;30:5053.Gelas-Mialhe Y, Gramain JC, Louvet A, Remuson R. Tetrahedron Lett. 1992;33:73.Pilli RA, Dias LC, Maldaner AO. Tetrahedron Lett. 1993;34:2729.Pilli RA, Dias LC, Maldaner AO. J Org Chem. 1995;60:717.Back TG, Hamilton MD, Lim VJJ, Parvez M. J Org Chem. 2005;70:967. doi: 10.1021/jo048284j.
- 28.For syntheses of (±)-epimyrtine see Comins DL, Brown JD. Tetrahedron Lett. 1986;27:4549.Comins DL, Weglarz MA, O’Connor S. Tetrahedron Lett. 1988;29:1751. (d) Ref. 29a. (e) Ref. 29c
- 29.(a) Davis FA, Zhang Y, Anilkumar G. J Org Chem. 2003;68:8061. doi: 10.1021/jo030208d. [DOI] [PubMed] [Google Scholar]; (b) Amorde SM, Judd AS, Martin SF. Org Lett. 2005;7:2031. doi: 10.1021/ol050544b. [DOI] [PubMed] [Google Scholar]
- 30.Krow GR, Fan DM. J Org Chem. 1974;39:2674. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org