

Abstract
Early microglial accumulation in Alzheimer's disease (AD) delays disease progression by promoting clearance of β-amyloid (Aβ) before formation of senile plaques. However, persistent Aβ accumulation despite increasing microglial numbers suggests that the ability of microglia to clear Aβ may decrease with age and progression of AD pathology. To determine the effects of aging and Aβ deposition on microglial ability to clear Aβ, we used quantitative PCR to analyze gene expression in freshly isolated adult microglia from 1.5-, 3-, 8-, and 14-month-old transgenic PS1-APP mice, an established mouse model of AD, and from their nontransgenic littermates. We found that microglia from old PS1-APP mice, but not from younger mice, have a twofold to fivefold decrease in expression of the Aβ-binding scavenger receptors scavenger receptor A (SRA), CD36, and RAGE (receptor for advanced-glycosylation endproducts), and the Aβ-degrading enzymes insulysin, neprilysin, and MMP9, compared with their littermate controls. In contrast, PS1-APP microglia had a 2.5-fold increase in the proinflammatory cytokines IL-1β (interleukin-1β) and tumor necrosis factor α (TNFα), suggesting that there is an inverse correlation between cytokine production and Aβ clearance. In support of this possibility, we found that incubation of cultured N9 mouse microglia with TNFα decreased the expression of SRA and CD36 and reduced Aβ uptake. Our data indicate that, although early microglial recruitment promotes Aβ clearance and is neuroprotective in AD, as disease progresses, proinflammatory cytokines produced in response to Aβ deposition downregulate genes involved in Aβ clearance and promote Aβ accumulation, therefore contributing to neurodegeneration. Antiinflammatory therapy for AD should take this dichotomous microglial role into consideration.
Keywords: microglia, Alzheimer's disease, transgenic mice, amyloid, phagocytosis, degradation
Introduction
The senile plaque is a pathological hallmark of Alzheimer's disease (AD) and is composed of β-amyloid (Aβ), activated microglia, astrocytes, and degenerating neurons (Dickson et al., 1988; Selkoe, 2000; Mott and Hulette, 2005). Microglia, the mononuclear phagocytes of the brain (Perry and Gordon, 1988), accumulate in senile plaques in AD patients and in animal models of AD (McGeer et al., 1987; Perlmutter et al., 1990; Frautschy et al., 1998; Dickson, 1999; Stalder et al., 1999). However, their exact role in the pathogenesis of AD remains to be elucidated. Aβ can activate microglia to produce cytokines and neurotoxins, hence promoting neurodegeneration (Meda et al., 1995; El Khoury et al., 1996, 2003; Coraci et al., 2002). In contrast, microglia also express receptors that promote the clearance and phagocytosis of Aβ, such as class A scavenger receptor, CD36, and receptor for advanced-glycosylation endproducts (RAGE) (Yan et al., 1996; El Khoury et al., 1998), and microglia can restrict senile plaque formation by phagocytosing Aβ (Simard et al., 2006). Indeed, our own data also show that decreased early microglial accumulation leads to increased Aβ deposition and early mortality in the transgenic swAPP (Tg2576) AD mouse model (El Khoury et al., 2007). In addition to phagocytosis of Aβ, microglia also secrete proteolytic enzymes that degrade Aβ, such as IDE (insulin-degrading enzyme), neprilysin, matrix metalloproteinase 9 (MMP9), and plasminogen (Leissring et al., 2003; Yan et al., 2006), further suggesting a neuroprotective role for these cells in AD.
The data supporting a role for microglia in Aβ clearance are compelling, but these data also raise an important question. Why does Aβ continue to accumulate, and why does AD pathology progress despite continued microglia recruitment? One possible explanation for the failure of microglia to stop AD progression would be that these cells become overwhelmed by the excess amount of Aβ produced and cannot keep up with the pace of Aβ generation. Another possibility would be that, as AD progresses, the phenotype of accumulating microglia changes and these cells become more proinflammatory and lose their Aβ-clearing capabilities, resulting in reduced Aβ uptake and degradation, and increased Aβ accumulation. To investigate this hypothesis, we developed a method to isolate fresh adult mouse microglia from the bigenic APPswe/PSEN1dE9 (PS1-APP) mice (Borchelt et al., 1997; Jankowsky et al., 2001), a robust mouse model of AD, and from their nontransgenic littermates at various ages and stages of AD-like pathology, and compared gene expression of Aβ-binding receptors and Aβ-degrading enzymes. Our data show that, as PS1-APP mice age, their microglia become dysfunctional and exhibit a significant reduction in expression of their Aβ-binding receptors and Aβ-degrading enzymes, but maintain their ability to produce proinflammatory cytokines. These cytokines may in turn act in an autocrine manner and further reduce expression of Aβ-binding receptors and Aβ-degrading enzymes leading to decreased Aβ clearance and increased accumulation.
Materials and Methods
Mice.
PS1-APP transgenic mice [B6C3-Tg(APPswe, PSEN1dE9)85Dbo/J; stock number 004462] were purchased from The Jackson Laboratory and subsequently bred in the animal care facilities at Massachusetts General Hospital. These mice coexpress a “humanized” Swedish amyloid precursor protein mutation (APP695SWE) and a mutant exon 9-deleted variant of human presenilin 1 (PSEN1/dE9). The mutations in both genes are associated with familial AD. The APP and PSEN1 transgenes are integrated into a single locus and are independently under the control of separate mouse prion protein promoter elements, which direct expression of the transgenes predominantly to CNS neurons (Borchelt et al., 1997; Jankowsky et al., 2001). Mice were used in pairs of age-matched transgenic or wild-type (WT) littermates at 1.5, 3, 8, and 14 months of age and killed according to approved institutional procedures. All protocols were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and met National Institutes of Health guidelines for the humane care of animals. Brains were harvested and microglia were isolated (see below) and used fresh for flow cytometry or gene expression analysis.
Immunohistochemistry staining for total Aβ.
Brains from transgenic PS1-APP or WT littermates were harvested and fixed in 2% paraformaldehyde (PFA) in PBS (Mediatech), pH 7.5, overnight at 4°C. The fixed brains were then placed in 30% sucrose overnight at 4°C for cryoprotection. Brains were embedded in Tissue-Tek OCT compound (Sakura Finetek USA) and 10–14 μm frozen sections were cut. To stain for Aβ, sections were blocked in PBS/0.3% Triton X-100/2% goat serum for 30 min, and then incubated overnight at 25°C with rabbit anti-pan Aβ antibody (BioSource International) or control antibody, at 2 μg/ml in PBS containing 0.3% Triton X-100 and 2% goat serum. The slides were then processed using the Vectastain Elite ABC kit rabbit IgG (Vector Laboratories) according to the manufacturer's instructions. Briefly, slides were incubated with biotinylated anti-rabbit IgG for 30 min followed by incubation with the kit ABC reagents (avidin and biotinylated peroxidase) for 30 min. Vector NovaRed substrate kit for peroxidase (Vector Laboratories) was used to develop target-bound peroxidase for detection of Aβ in brain sections. Slides were counterstained with hematoxylin, mounted with VectaMount (Vector Laboratories), visualized by bright-field microscopy, and digitally photographed.
Immunohistochemistry/fluorescence costaining of brains for CD11b and Aβ plaques.
Frozen sections from PS1-APP or WT littermate mice were fixed in acetone for 10 min, washed in PBS, and then treated with 0.25% trypsin for antigen retrieval. Endogenous peroxidase activity was quenched with 0.3% H2O2 followed by blocking with 1.5% donkey serum in PBS. Sections were incubated overnight at 25°C with rat anti-CD11b (clone 5C6) (AbD Serotec) or rat IgG2b negative control (AbD Serotec) each at 1:100 dilution in PBS with 1.5% donkey serum. The slides were then processed using the Vectastain Elite ABC reagent (Vector Laboratories) according to the manufacturer's instructions followed by development with the NovaRed Peroxidase Substrate kit. Aβ-containing plaques were stained with 1% thioflavin-S (Sigma-Aldrich) for 5 min in the dark. Finally, the sections were counterstained with hematoxylin, mounted with VectaMount, and digitally photographed via bright-field microscopy to detect CD11b, and via fluorescence to visualize thioflavin-S.
Isolation of CD11b+ cells.
Transgenic PS1-APP mice or their WT littermates were killed and perfused with 30 cc of PBS without Ca2+ and Mg2+ (PBS=). Brains were then removed, rinsed in PBS= and placed separately into a 60 mm tissue culture dish with RPMI 1640 (no phenol red) containing 2 mm l-glutamine (Mediatech), dispase and collagenase type 3 (Worthington Biochemicals). Brains were minced with sterile razor blades and allowed to incubate in enzymes for 45 min at 37°C, followed by addition of DNase I grade II (Roche Applied Science) at a concentration of 40 U/ml and incubation for an additional 15 min. The enzymes were inactivated by addition of 20 ml of HBSS without Ca2+ and Mg2+ (HBSS=) (Mediatech) containing 2 mm EDTA and 2% fetal bovine serum (FBS) and the digested brain bits were triturated sequentially with a 25, 10, and 5 ml pipette (8–10 times each step). Between pipette sizes, the cells were allowed to settle for 1 min, and then the top 50% of the cell suspension was removed and passed over a 100 μm filter (Thermo Fisher Scientific). This process was continued until all tissue had been triturated and passed through the filter. Cells were centrifuged and resuspended in RPMI 1640/l-glutamine and mixed with physiologic Percoll (Sigma-Aldrich) and centrifuged at 850 × g for 45 min. The cell pellet was resuspended in RPMI 1640 and the cells were passed over a 70 μm filter (Thermo Fisher Scientific), washed, and then passed over a 40 μm filter (Thermo Fisher Scientific). The cells were then incubated with anti-mouse Cd11b-coated microbeads (Miltenyi Biotec) for 20 min at 12°C. The cell–bead mix was then washed to remove unbound beads. The bead–cell pellet was resuspended in PBS/0.5% BSA/2 mm EDTA and passed over a magnetic MACS Cell Separation column (Miltenyi Biotec). In this way, Cd11b-positive cells (i.e., cells that bound the beads, microglia/mononuclear phagocytes) were separated from Cd11b-negative cells (CD11bneg) (i.e., cells that did not bind the beads). Flowthrough was collected and the column was rinsed three times with PBS/BSA/EDTA. CD11b-positive cells (CD11b+) were eluted by removing the column from the magnetic holder and pushing PBS/BSA/EDTA through the column with a plunger. Cells were centrifuged and the pellets were lysed in RLT-Plus buffer from the RNeasy Plus mini kit (QIAGEN) to use for quantitative PCR (QPCR).
Flow cytometry of CD11b+ cells.
CD11b+ cells isolated by magnetic beads (above) were centrifuged and cell pellets were resuspended in PBS/2% FBS containing 1:100 dilution of mouse seroblock (AbD Serotec) to block Fc receptors on microglia. Cells were maintained on ice for 10 min then stained with allophycocyanin (APC)-labeled anti-CD11b (2 μg/ml) or APC-labeled isotype control (2 μg/ml) (both from BD Biosciences Pharmingen) for 1 h on ice followed by fixation with 2% PFA. Fluorescence intensity was measured using a FACSCalibur (BD Biosciences) flow cytometer as described previously (Coraci et al., 2002; El Khoury et al., 2007).
Quantitative real-time PCR.
Total RNA from each sample of CD11b+ or CD11bneg cells (7.5–15 × 105 cells) or N9 cells was isolated using the RNeasy Plus mini kit for RNA isolation (QIAGEN) according to the manufacturer's instructions and all of each sample was reverse transcribed using Multiscribe reverse transcriptase (Applied Biosystems). Dilutions of each cDNA prep were used to assess β2-microglobulin RNA levels, and samples were then adjusted to give equivalent levels of β2-microglobulin per well in subsequent QPCRs for other genes. The QPCR was performed with the MX4000 unit (Agilent Technologies) using SYBR Green to detect the amplification products as described previously (El Khoury et al., 2003, 2007). The following cycles were performed: initial denaturation cycle of 95°C for 10 min, followed by 40 amplification cycles of 95°C for 15 s and 60°C for 1 min and ending with one cycle at 25°C for 15 s. Analysis was performed on the data output from the MX4000 software (Agilent Technologies) using Microsoft Excel XP. Relative quantification of mRNA expression was calculated by the comparative cycle method described by the manufacturer (Agilent Technologies). Primer sequences for the following murine genes can be found in supplemental Table 1 (available at www.jneurosci.org as supplemental material): β2-microglobulin, GAPDH, CD11b, CD36, scavenger receptor A (SRA), SRB1, macrophage receptor with collagenous domain (MARCO), RAGE, interleukin 1β (IL-1β), tumor necrosis factor α (TNFα), insulysin, neprilysin, and MMP9.
Staining of N9 microglia.
N9 mouse microglia (a gift from Dr. P. Riciarrdi-Castagnoli, University of Milan, Bicocca, Italy) (El Khoury et al., 1996) were plated on 24-well plates coated with 1 μg of fibronectin/cm2 (Sigma-Aldrich), and grown overnight in RPMI 1640 supplemented with10% FBS, l-glutamine (2 mm), penicillin (10 IU/ml), and streptomycin (10 μg/ml). The following day, TNFα (PeproTech) was added to a concentration of 50 ng/ml and cells were incubated overnight. For staining, cells were lifted from the plate with CellStripper (Mediatech) and resuspended in PBS/1% BSA/2% FBS containing 10 μg/ml Fc block (AbD Serotec) and allowed to sit 10 min before addition of primary antibodies. APC-labeled anti-mouse CD11b (1 μg/ml) (BD Biosciences Pharmingen) or APC-labeled hamster anti-mouse CD36 clone HM36 (1 μg/ml) (BioLegend) or Alexa647-labeled anti-CD204 (SRA) clone 2F8 (2.5 μg/ml) (AbD Serotec) or isotype-matched control antibodies (same concentrations as primary antibodies) were added and incubated on ice for 1 h. Cells were then fixed by the addition of 2% PFA. Fluorescence intensity was measured using a FACSCalibur (BD Biosciences) flow cytometer.
Aβ uptake.
For uptake of Aβ, N9 microglia were grown on fibronectin-coated 24-well plates and treated overnight with TNFα or medium alone. Adherent N9 microglia were rinsed once in PBS/1% BSA, followed by the addition of Aβ(1–42) labeled with the fluorescent dye HiLyte Fluor488 (AnaSpec) or with FITC as described previously (El Khoury et al., 2007) at a concentration of 5 μg/ml in PBS/1% BSA or diluent alone and incubated at 37°C for 2 h. Cells were then rinsed gently three times in PBS with divalent cations and then lifted from wells using CellStripper. Cells were fixed in 2% PFA and cell-associated fluorescence was analyzed by flow cytometry.
To determine whether cell-associated fluorescence was attributable to binding or endocytosis of labeled Aβ, N9 cells grown overnight on four-well glass slides were incubated with RPMI 1640/1% BSA alone or FITC-labeled Aβ(1–42) at 4 or 37°C for 4 h. Wells were then rinsed three times with ice-cold PBS to remove unbound Aβ and unfixed cells were analyzed by flow cytometry. To differentiate between bound versus endocytosed Aβ, trypan blue was added to some samples to quench fluorescence of FITC-Aβ on the outside of the cell as described previously (Thomas et al., 2000). Because cells are not fixed, the live cells are not permeable to trypan blue and any fluorescence detected would be intracellular.
Statistical analysis.
Statistical analysis was performed using Student's t test and one-way ANOVA provided in the OriginLab Origin 8 graphics and statistics software. Values of p < 0.05 were considered significant.
Results
Increased number of senile-like plaques and plaque-associated microglia with aging in transgenic PS1-APP mice
Frozen sections prepared from the brains of young (3 months) and old (14 months) PS1-APP mice were stained with an anti-pan-Aβ antibody. At 3 months of age, transgenic PS1-APP mice have sparse, but detectable Aβ deposits (Fig. 1A). By 14 months of age, Aβ deposits are distributed throughout the cortex and hippocampus (Fig. 1B). To visualize microglial distribution in young and old mice, sections were first stained with anti-CD11b antibody followed by costaining with thioflavin-S to detect fibrillar Aβ plaques. In 3-month-old mice, plaques are rarely seen and microglia are evenly distributed throughout the hippocampus region (Fig. 1C). In contrast, florid plaques are seen in transgenic mice at 14 months of age and intensely stained microglia are clustered around the plaques (Fig. 1D). These data indicate that significant microgliosis accompanies Aβ deposition in PS1-APP mice and confirm that these mice are a robust model to study the role of microglia in AD.
Figure 1.

Increased number of senile-like plaques and plaque-associated microglia with aging in transgenic PS1-APP mice. Top panels, Frozen sections from transgenic PS1-APP brains stained with pan-α-Aβ antibody (which recognizes all forms of β-amyloid) show (original magnification, 40×) sparse amounts of Aβ in a transgenic mouse at 3 months of age (A) and large numbers of Aβ deposits in a 14-month-old transgenic mouse distributed throughout cortex and hippocampus (B). The inset in B shows the plaque signified by an arrow magnified to 400× original magnification. Bottom panels, 400× original magnification. Microglia are stained using α-CD11b antibody (reddish brown Nova red development) and Aβ-containing plaques are stained with thioflavin-S (green). In a 3-month-old mouse, plaques are rarely seen and microglia are evenly distributed throughout the hippocampus region shown in C. D, In contrast, florid plaques are seen in a transgenic mouse at 14 months of age, and intensely stained microglia are clustered around the plaques. The staining was repeated in six mice in each age group with similar staining.
Freshly isolated microglia express CD11b and scavenger receptors, but not astrocyte or endothelial markers
We developed a method to isolate microglia (CD11b+ cells) from adult mouse brains and used freshly isolated cells to examine expression of Aβ receptors, Aβ-degrading enzymes, and proinflammatory cytokines. Figure 2A shows a diagram of our protocol for isolation of adult microglia, which is completed in <4 h. Immediately after brain removal, the brain is digested with dispase and collagenase, followed by trituration and filtration of large debris. Intact cells are enriched by centrifugation through Percoll. Microglia are then purified by binding to anti-CD11b-coated magnetic microbeads followed by separation from CD11bneg cells by passing through a magnetic column. Column-bound CD11b+ cells are eluted and centrifuged, and the cell pellet is immediately lysed and the total RNA isolated for use in QPCR. Because gene expression patterns of cultured microglia may differ from microglia in situ, the use of freshly isolated cells gives a “snapshot” of genes that are expressed in the environment in which the cells were residing just before isolation. To control for any changes in gene expression that may occur as a result of the isolation procedure itself, all mouse brains were isolated in pairs of transgenic and nontransgenic WT littermates harvested at the same time, under the same condition, using the same reagents.
Figure 2.

Gene expression profile of adult WT CD11b+ microglia and CD11bneg cells (endothelial cells and astrocytes) measured by QPCR. A, Protocol used for isolating CD11b+ cells from adult mouse brains. B, CD11b+ cells (black bars) have high levels of CD11b RNA and barely detectable levels of cell markers for astrocytes (GFAP) and endothelial cells (CD31) confirming high purity of the CD11b+ cells preparations. The CD11bneg population (gray bars) expresses CD31 and GFAP, but no CD11. C, CD11b+ cells isolated using our protocol expressed SRA, SRB1, and CD36 (3 known microglia receptors), some RAGE, and negligible levels of MARCO.
Figure 2, B and C, shows a typical RNA expression profile of CD11b+ microglia and CD11bneg cells (mostly endothelial cells and astrocytes) prepared using our method. Analysis by flow cytometry showed that ≥96% of our CD11b+ populations isolated in this protocol stained with CD11b (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). RNA expression is shown as the ratio of copies of target RNA compared with β2-microglobulin RNA in the same sample. To determine the purity of our isolated cell populations, CD11b+ and CD11bneg cells were compared by QPCR for expression of cell markers for astrocytes (GFAP), endothelial cells (CD31), and microglia (CD11b). As expected, CD11b+ cells expressed high levels of CD11b, and the CD11bneg cells expressed CD31 and GFAP and no CD11b. Additional analysis showed that freshly isolated CD11b+ cells also express RNA for the three microglial scavenger receptors SRA, SRB1, and CD36 (El Khoury et al., 1998). Freshly isolated microglia express small but detectable levels of RAGE (Yan et al., 1996) and negligible levels of MARCO (Alarcón et al., 2005).
Microglia from old transgenic PS1-APP mice have reduced expression of Aβ-binding receptors
To determine whether aging and Aβ deposition affect expression of Aβ-binding receptors SRA, SRB1, CD36, RAGE, and MARCO, we measured expression of these receptors in CD11b+ cells isolated from PS1-APP mice and their age-matched wild-type littermates at 1.5, 3, 8, and 14 months of age (five to six mice per genotype per age group).
At 1.5 months of age, expression of SRA, SRB1, CD36, and RAGE RNAs were comparable between transgenic and WT mice. At 3 months of age, expression of SRA, CD36, and RAGE RNAs was 1.4- to 1.6-fold lower in PS1-APP mice than in their age-matched WT littermates, but the differences were not statistically significant. However, by 8 months of age, microglia from PS1-APP transgenic mice showed significantly reduced expression of SRA (1.9-fold decrease; p < 0.002), CD36 (2.6-fold decrease; p < 0.0002), and RAGE (twofold decrease; p < 0.042) compared with their age-matched WT littermates (Fig. 3A,C,D). At 14 months of age, microglial RNA levels for these genes had declined even further in PS1-APP transgenic mice compared with their WT littermates: SRA (2.5-fold decrease; p < 0.01), CD36 (fivefold decrease; p < 0.0003), and RAGE (sixfold decrease; p < 0.003) (Fig. 3A,C,D). There are no significant differences in expression of SRB1 observed between transgenic and WT mice at any age (Fig. 3B). These data indicate that microglia in mice with advanced AD-like pathology have decreased expression of Aβ receptors and suggest that they may have decreased capacity to bind and subsequently clear Aβ.
Figure 3.

Reduced expression of Aβ-binding receptors in microglia from old transgenic PS1-APP mice. Expression of Aβ-binding receptors in CD11b+ cells was compared between transgenic PS1-APP mice and their age-matched WT littermates at 1.5, 3, 8, and 14 months of age. Data represent the average values obtained by QPCR on six sets of mice for 3 and 14 months of age and on five sets of mice at 1.5 and 8 months of age. *Values of p are shown only if differences are significant (<0.05). A, B, D, At 8 and 14 months of age, PS1-APP transgenic mice show significantly reduced expression of SRA (A), CD36 (B), and RAGE (D) compared with their WT littermates. C, There are no significant differences in expression of SRB1 observed between transgenic and WT mice at any age. E, There was negligible expression of MARCO. Error bars indicate SEM.
Interestingly, WT mice also exhibited an age-related decrease in expression of SRA, CD36, and SRB1 RNAs in 8- and 14-month-old mice compared with younger WT mice. However, only reduction in CD36 was statistically significant (p < 0.03). Furthermore, the reduction in expression of SRA and CD36 was significantly more pronounced between microglia from transgenic mice and WT littermates than between WT mice of different age groups.
Microglia from old PS1-APP mice have reduced expression of Aβ-degrading enzymes
In addition to their ability to phagocytose Aβ, microglia can also clear Aβ by degradation via Aβ-degrading enzymes (Leissring et al., 2003; Yan et al., 2006). To determine whether aging and Aβ deposition affect microglial expression of major Aβ-degrading enzymes, we quantified by QPCR the mRNA levels of insulysin, neprilysin, and MMP-9 in CD11b+ cells from transgenic PS1-APP mice and their age-matched WT littermates at 1.5, 3, 8, and 14 months of age. Expression of RNA levels of the Aβ-degrading enzymes insulysin, neprilysin, and MMP9, were comparable in WT and transgenic mice at 1.5 and 3 months of age (Fig. 4A–C). However, in 8-month-old transgenic mice, insulysin RNA levels decreased to 50% (p < 0.001) of age-matched WT littermates. This level of reduction in transgenic mice is also observed at 14 months of age (50% of WT; p < 0.035) (Fig. 4A). At 8 months of age, transgenic mice express reduced levels of RNA for neprilysin and MMP9 compared with their WT littermates, but the differences were not statistically significant (Fig. 4B,C). However, by 14 months of age, microglial levels of both neprilysin (p < 0.015) and MMP9 (p < 0.05) in transgenic mice were reduced to only 20% of the levels seen in their age-matched WT littermates (Fig. 4B,C). These data show that microglia in mice with significant AD-like pathology have reduced expression of three major Aβ-degrading enzymes and suggest that these microglia may have become defective in their capacity to degrade Aβ.
Figure 4.

Decreased expression of Aβ-degrading enzymes in microglia from old transgenic PS1-APP mice. Expression of Aβ-degrading enzymes in CD11b+ cells from transgenic PS1-APP mice was compared with their age-matched wild-type littermates at 1.5, 3, 8, and 14 months of age. Data are expressed for transgenic mice as percentage expression of WT. *Values of p are only shown if differences are significant. A–C, Expression of insulysin (A), neprilysin (B), and MMP9 (C), enzymes thought to degrade Aβ, is comparable across WT and transgenic microglia from mice at 1.5 and 3 months of age. A, In 8-month-old PS1-APP transgenic mice, there is a statistically significant reduction only in insulysin RNA levels. However, by 14 months of age, the reduction in expression of all three enzymes is statistically significant (A–C). The horizontal lines across the graphs indicate 100% WT levels. Error bars indicate SEM.
Microglia from old PS1-APP mice have increased expression of IL-1β and TNFα
Mouse microglia and macrophages stimulated with Aβ upregulate their expression of several proinflammatory chemokines and cytokines including IL-1β and TNFα (El Khoury et al., 2003). In addition, TNFα is upregulated in the cortex of triple transgenic AD mice (Janelsins et al., 2005). To determine the effects of aging and Aβ deposition on expression of TNFα and IL-1β in microglia, we measured RNA levels of these two cytokines in CD11b+ cells from transgenic PS1-APP mice and their WT littermates at 1.5, 3, 8, and 14 months of age using QPCR. IL-1β expression was comparable in microglia from transgenic and WT mice at 1.5 and 3 months (Fig. 5A). In contrast, at 8 months of age, microglial IL-1β RNA in transgenic mice had increased to 150% of their WT littermates (p < 0.015), and by 14 months, IL-1β RNA was increased to 250% of WT (p < 0.025) (Fig. 5A). Similarly, levels of TNFα RNA were also comparable between transgenic and WT mice at 1.5 and 3 months of age (Fig. 5B). Statistically significant increases in TNFα RNA were observed in transgenic microglia from 8-month-old mice (150% of WT; p < 0.015) and 14-month-old mice (200% of WT; p < 0.041), compared with their age-matched WT littermates. Together, these data show that microglia in aged AD mice retain their proinflammatory response in the presence of continued Aβ deposition. Because expression of these two cytokines are increased, these data also suggest that the decreased expression of Aβ receptors and Aβ-degrading enzymes observed in microglia from aging PS1-APP mice is not an indication of a generalized “shut down” in the functions of these microglia, but a rather selective defect in their Aβ-clearing pathways.
Figure 5.

Increased expression of IL-1β and TNFα in microglia from old transgenic PS1-APP mice. A, B, Expression of IL-1β (A) and TNFα (B) was measured by QPCR in CD11b+ cells from transgenic PS1-APP mice and their age-matched, nontransgenic littermates at 1.5, 3, 8, and 14 months of age. Data are displayed for transgenic mice as percentage expression of their WT littermates at the same ages. In young mice (1.5 and 3 months of age), expression of IL-1β (A) and TNFα (B) is comparable across all groups, but at 8 and 14 months of age there are significant increases (p < 0.05) in RNA expression of both IL-1β and TNFα in transgenic compared with WT mice. Error bars indicate SEM.
TNFα downregulates expression of SRA and CD36 and reduces uptake of Aβ in N9 microglia
Because microglia from old transgenic PS1-APP mice exhibited higher expression of proinflammatory cytokines that are potent regulators of gene expression in mononuclear phagocytes and microglia, we hypothesized that these cytokines regulate expression of Aβ receptors and Aβ-degrading enzymes. To test this hypothesis, we incubated murine N9 microglia with IL-1β, TNFα, or medium alone and measured expression of SRA, CD36, neprilysin, and insulysin by QPCR. We found that incubation with TNFα for 18 h significantly downregulated expression of SRA and CD36 to 65% (p < 0.0003) and 52.8% (p < 0.0001) of untreated N9 cells, respectively (Fig. 6A). In preliminary experiments, similar results were obtained with IL-1β, albeit to a lesser extent, and differences were not statistically significant (data not shown). TNFα-induced reduction in RNA levels corresponded to a reduction of both SRA and CD36 surface expression, as assessed by staining and flow cytometry. TNFα reduced SRA and CD36 surface expression to 54.7% (p < 0.008) and 64.5% (p < 0.00005) of untreated cells, respectively (Fig. 6B). Thus, treatment of N9 microglia with TNFα resulted in a significant decrease in expression of both SRA and CD36 RNAs and membrane protein. Interestingly, treatment with these proinflammatory cytokines did not affect expression of neprilysin and insulysin in N9 microglia, suggesting that the reduction in RNA levels of these enzymes observed in vivo may be mediated by a different mechanism(s).
Figure 6.
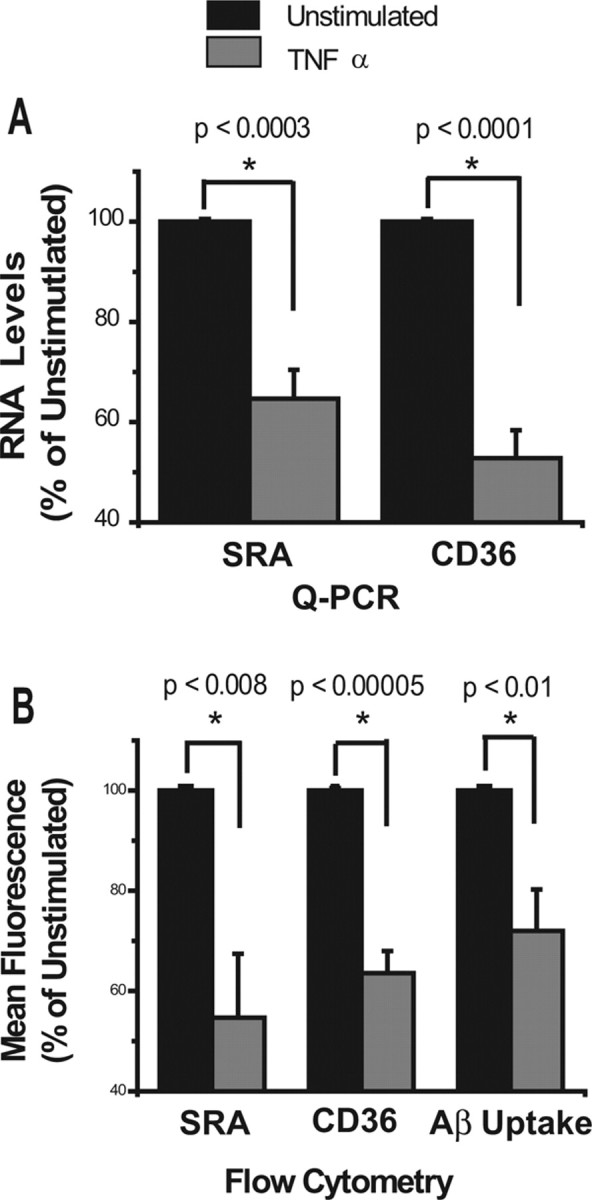
Treatment of N9 cells with TNFα decreases expression of SRA and CD36 and reduces uptake of Aβ. A, B, N9 microglia-like cells were treated with TNFα overnight, and expression of SRA and CD36 were measured by QPCR (A) and by cell surface staining and flow cytometry (B). Data represent the average of five separate experiments and are displayed for treated cells as percentage expression (%) of untreated controls (100%). B, Far right, Results of TNF-α treatment on binding/uptake of Aβ-labeled with Hylite fluor-488. TNF-α-treated or untreated N9 cells were incubated with fluorescent Aβ for 2 h, and then cell-associated fluorescence was measured by flow cytometry. Data represent an average of five experiments and are displayed for TNFα-treated cells as percentage (%) of mean fluorescence intensity of untreated cells. Error bars indicate SEM.
N9 microglia bind to and phagocytose fluorescently labeled Aβ (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To determine whether decreased expression of SRA and CD36 on N9 microglia in response to TNFα treatment affected the ability of these cells to bind and/or phagocytose Aβ, we incubated TNFα-treated and untreated N9 microglia with fluorescently labeled Aβ for 2 h, and measured cell-associated fluorescence intensity by flow cytometry. Cells treated with TNFα showed a 30% decrease in binding/uptake of labeled Aβ compared with untreated cells (p < 0.01) (Fig. 6B). These data show that TNFα reduces expression of genes involved in binding and phagocytosis of Aβ, resulting in decreased uptake of Aβ by N9 microglia. These data also suggest that the reduction in expression of Aβ-binding receptors observed in aged PS1-APP microglia may, in part, be mediated by TNFα
Discussion
Increasing evidence indicates that microglia may play a protective role in AD by mediating clearance of Aβ. Indeed, early microglial accumulation appears to delay progression of AD-like pathology, and bone marrow-derived microglia may restrict plaque formation in transgenic mice (Simard et al., 2006; El Khoury et al., 2007). As AD progresses, however, microglial accumulation appears to parallel disease progression, and the presence of increased numbers of microglia does not appear to prevent formation of plaques or AD development (Perlmutter et al., 1990; Dickson, 1999; Mott and Hulette, 2005). The data presented in this study show that, in the PS1-APP transgenic mouse model of AD, the phenotype of accumulating microglia changes as AD-like pathology progresses. Microglia continue to produce proinflammatory cytokines, but lose their Aβ-clearing capabilities. Expression of microglial Aβ receptors and Aβ-degrading enzymes is reduced, resulting in reduced Aβ uptake and degradation, and increased Aβ accumulation. This process is observed at the age of 8 months and appears to precede or parallel the increase in Aβ levels observed in PS1-APP mice at that age (Jankowsky et al., 2001, 2004). In contrast to their protective role early in the disease process, as AD-like pathology progresses, microglial dysfunction and their failure to phagocytose and/or degrade Aβ further contributes to disease progression. In support of this possibility, Fiala et al. (2005) found that monocytes and macrophages from AD patients exhibited ineffective phagocytosis of Aβ when compared with monocytes and macrophages from age-matched control non-AD patients.
Our data also suggest that failure of the microglia to perform their Aβ-clearing functions may be a direct result of the Aβ-induced inflammatory response. TNFα, a major cytokine produced by microglia in response to Aβ stimulation, reduced expression of the Aβ receptors SRA and CD36, similar to the reduction observed in microglia from aging PS1-APP mice. It is not clear what mechanism accounts for reduced expression of the degrading enzymes, but reactive oxygen species and a number of proinflammatory cytokines have been found to be upregulated in transgenic mice with AD-like pathology. It is possible that one or more of these cytokines is responsible for downregulation of the Aβ degrading enzymes.
Interestingly, in addition to downregulating Aβ clearance pathways, proinflammatory cytokines may also contribute to Aβ accumulation by another mechanism. Recently, it was shown that TNFα and interferon-γ upregulate β-secretase (BACE1), an enzyme involved in Aβ production (Yamamoto et al., 2007). TNFα, IL-1β, and interferon-γ were also found to stimulate γ-secretase-mediated cleavage of APP (Liao et al., 2004). Proinflammatory cytokines therefore may also contribute to AD-like pathology by promoting Aβ generation.
Our data provide evidence to support the paradigm that the inflammatory response in AD is a “double-edged sword.” Microglia are recruited to sites of Aβ deposition as part of the attempt of the brain to clear these neurotoxic peptides, and early microglial accumulation in AD delays disease progression. However, as AD mice age, their microglia become dysfunctional and show a significant reduction in expression of their Aβ-binding receptors and Aβ-degrading enzymes, but maintain their ability to produce proinflammatory cytokines. These cytokines may in turn act in an autocrine manner and promote Aβ production by stimulating β- and γ-secretases and/or reduce Aβ clearance by reducing expression of Aβ-binding receptors and Aβ-degrading enzymes. The convergence of both of these cytokine-mediated pathways leads to increased Aβ accumulation and contributes to disease progression. Antiinflammatory therapy that distinguishes between such dichotomous functions of microglia and promotes their ability to clear Aβ, while decreasing their ability to produce proinflammatory cytokines, may indeed be very helpful to delay or stop the progression of Alzheimer's disease.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS059005 and a grant from The Dana Foundation Neuroimmunology Program (J.E.K.). We thank David Borchelt for permission to use the PS1-APP mice.
References
- Alarcón R, Fuenzalida C, Santibáñez M, von Bernhardi R. Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J Biol Chem. 2005;280:30406–30415. doi: 10.1074/jbc.M414686200. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW. Microglia in Alzheimer's disease and transgenic models. How close the fit? Am J Pathol. 1999;154:1627–1631. doi: 10.1016/S0002-9440(10)65416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC. Microglia, scavenger receptors, and the pathogenesis of Alzheimer's disease. Neurobiol Aging. 1998;19:S81–S84. doi: 10.1016/s0197-4580(98)00036-0. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer's disease patients. J Alzheimers Dis. 2005;7:221–232. doi: 10.3233/jad-2005-7304. discussion 255–262. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer's disease mice. J Neuroinflammation. 2005;2:23. doi: 10.1186/1742-2094-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Mott RT, Hulette CM. Neuropathology of Alzheimer's disease. Neuroimaging Clin N Am. 2005;15:755–765. ix. doi: 10.1016/j.nic.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Barron E, Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett. 1990;119:32–36. doi: 10.1016/0304-3940(90)90748-x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988;11:273–277. doi: 10.1016/0166-2236(88)90110-5. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. The origins of Alzheimer disease: a is for amyloid. JAMA. 2000;283:1615–1617. doi: 10.1001/jama.283.12.1615. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CA, Li Y, Kodama T, Suzuki H, Silverstein SC, El Khoury J. Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J Exp Med. 2000;191:147–156. doi: 10.1084/jem.191.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR, Xiao Q, Hsu FF, Turk JW, Xu J, Hsu CY, Holtzman DM, Lee JM. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J Biol Chem. 2006;281:24566–24574. doi: 10.1074/jbc.M602440200. [DOI] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]