

Abstract
Deleted in liver cancer-1 (DLC-1) is a RhoGAP domain containing tumor suppressor that is often down-regulated in various cancer types. Previously, we have demonstrated that DLC-1 is recruited to focal adhesions by binding to the SH2 (Src homology 2) domains of tensins and the focal adhesion localization is critical for DLC-1's tumor suppression activity. To investigate whether mutations in the focal adhesion targeting (FAT) region might occur and attenuate DLC-1's expression, localization, and function, we have first mapped the FAT region to the amino acid residues from 201 to 500, and then sequenced cDNAs and genomic DNAs encoding the FAT region from cancer patients. Several missesne and nonsense mutations were detected. All missense mutations were further examined for the potential effect on DLC-1's function. Although these mutations did not appear to affect DLC-1's focal adhesion localization, the activities of suppressing tumor cell growth were impaired in two mutants: T301K and S308I. In consistent with the fact that the RhoGAP activity of DLC-1 is essential for inhibiting tumor cell growth, the RhoGAP activities were significantly reduced in these mutants, suggesting that the FAT region also contains a regulatory element for its C-terminal RhoGAP domain. Our studies have demonstrated that mutations in DLC-1 may lead to loss of function and contribute to the tumorigenesis, and have revealed an allosteric regulation site for its RhoGAP activity.
Keywords: DLC, focal adhesion, mutation, RhoGAP
Introduction
Tumor suppressors are genes that reduce the chance of normal cells turning into tumor cells. The loss of function of one or more tumor suppressor genes is believed to be an important step of cancer development. Deleted in liver cancer 1 (DLC-1) was originally isolated as a potential tumor suppressor gene often deleted in hepatocellular carcinoma (1). Further studies have indicated that down-expression of DLC-1 either by genomic deletion or promoter methylation is associated with a variety of cancer types including prostate, lung, breast, kidney, colon, uterus, ovary, and stomach (2, 3). Re-expression of DLC-1 in DLC-1 null cancer cell lines is able to suppress cell growth, induce morphological change, decrease cell migration, and reduce tumorigenicity (2, 3). These data support a tumor suppressor role of DLC-1.
Human DLC-1 protein contains 1091 amino acid (aa) residues sharing a SAM (sterile alpha motif) at the N-terminus, a RhoGAP (RhoGTPase activating protein) domain and a START (steroidogenic acute regulatory (StAR)-related lipid transfer) domain at the C-terminus. The SAM domain is predicted to interact with proteins, RNA, or DNA (4), whereas the START domain may bind to lipid (5). The RhoGAP domain converts the active GTP-bound Rho protein to the inactive GDP-bound state and negatively regulates Rho GTPase. This RhoGAP activity is essential but not sufficient for many of DLC-1's anti-oncogenic activities (6-8). The C-terminal half of DLC-1 interacts with PLCδ1 and enhances the phosphatidylinositol 4, 5-bisphosphate (PIP2)-hydrolyzing activity of PLCδ1 (9). The C-terminal region also targets to caveolae presuming by interacting with caveolin-1 through several potential binding sites on DLC-1 (8, 10). However, the significance of caveolae localization of DLC-1 is not clear.
Recently, we have reported that the N-terminal half of DLC-1 localizes to focal adhesions by binding to the SH2 domains of tensins (11). This focal adhesion localization is required for DLC-1's tumor suppression activity, since the tensin-binding defective DLC-1 mutants do not localize to focal adhesions and can not suppress tumor cell growth (11, 12). Therefore, in addition to genomic deletion and promoter hypermethylation, mislocalization of DLC-1 protein may be another mechanism of acquiring tumorigenicity involving DLC-1 dysregulation.
In the present study, we report the analysis of the focal adhesion targeting (FAT) region, the discovery of mutations within the FAT region of DLC-1 gene, and the functional relevance of these mutations. Our finding further reveals a new allosteric regulatory site within the FAT region for DLC-1's RhoGAP activity.
Materials and methods
Mutation analysis
Prostate cDNA and colon genomic DNA samples were purchased from OriGene Technologies (Rockville, MD) and BioChain (Hayward, CA), respectively. DNA fragments were amplified by PCR using DLC-1 primers 5′-GGCAGCCTGCCCTCTCCCAAGGAA (forward) and 5′-CAGGGCTGAGTCCGAATCTCCCTC-3′ (reverse). The PCR products were purified by QIAquick PCR purification kit (Qiagen, Valencia, CA) and then subjected to DNA sequencing. Samples displayed mutations were subjected to another independent amplification and sequencing to rule out the PCR and DNA sequencing error. Mutations shown in this study were detected on both experiments.
Plasmid constructions and mutagenesis
The full-length and the region encoding residues 1-535 of human DLC-1 cDNA were constructed in the previous study (11). The truncated cDNA fragments encoding DLC-1 residues 279-535 and 201-500 were subcloned in frame into mammalian expression vector pEGFP-C2 (Clontech, Mountain View, CA). The site-specific mutation of P290L, T301K, S308I, S320I, Y338L, Y442F, or K714E was introduced into DLC-1 cDNA by site-directed mutagenesis. All constructs were verified by DNA sequencing.
Cell culture, gene transfection and colony formation assay
A549, HEK293T, and MDA-MB-468 cells purchased from American Type Culture Collection (Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with antibiotics and 10% fetal bovine serum. A549 and HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), whereas MDA-MB-468 cells were transfected using SuperFect transfection reagent (Qiagen) according to the manufacturer's introductions. MDA-MB-468 cells were used for colony formation assay as previously described (11).
Immunofluorescence microscopy
A549 cells grown on glass coverslips were transfected and incubated at 37°C in 5% CO2 for 16 hr before microscopic imaging. Cells were fixed with methanol at −20°C. After rinsing with PBS, cells were incubated with 1:100 anti-tensin3 rabbit polyclonal antibody for 1 h. Samples were then incubated with 1:800 Alexa Fluor 594-conjugated secondary antibody (Molecular Probes, Eugene, OR) for 1 h and visualized with a Zeiss LSM 510 confocal microscope.
RhoA activity assay
RhoA activities were measured with the Rho Activation Assay Biochem Kit (Cytoskeleton, Denver, CO). In brief, 24h after transfection, HEK293T cells were serum-deprived for another 24h and then treated with lysophosphatidic acid (5 μM, 30 min), which enhanced Rho-GTP levels. Cells were immediately washed with ice-cold PBS and lysed. Equal protein amounts of cell lysates were incubated with 30 μg GST-RBD (Rho binding domain of Rhotekin) beads for 1 h at 4°C. The beads were washed twice with washing buffer, and bound Rho proteins were analyzed by immunoblotting using an anti-Rho antibody (Cytoskeleton).
Results
Mutation analysis of the focal adhesion targeting region of DLC-1 from human patients
Recently, we have demonstrated that the focal adhesion localization of DLC-1 mediated by the SH2 domains of tensins is critical for DLC-1's function as a tumor suppressor (11). This finding has promoted us to investigate whether mutations occur in the focal adhesion targeting (FAT) region and whether these mutations lead to mislocalization of DLC-1. If so, even a high level of DLC-1 transcript is detected, the expressed protein may not function appropriately. Previous studies had found that the N-terminal half of DLC-1 localized to focal adhesion sites (11, 13). To further narrow down the region sufficient for DLC-1's focal adhesion targeting, we had examined the subcellular localization of several GFP-DLC-1 fusion constructs. As shown in figure 1, while GFP-DLC-1 (1-535) and GFP-DLC-1 (201-500) colocalized with tensin3 at focal adhesions, GFP-DLC-1 (279-535) was diffused in the cytoplasm, demonstrating that the fragment 201-500 contains sufficient FAT sequences. With this information, we then focused our DLC-1 mutation analysis on human cDNA and genomic DNA encoding this region.
Figure 1. Identification of the FAT region of DLC-1 and focal adhesion localization of DLC-1 mutants.
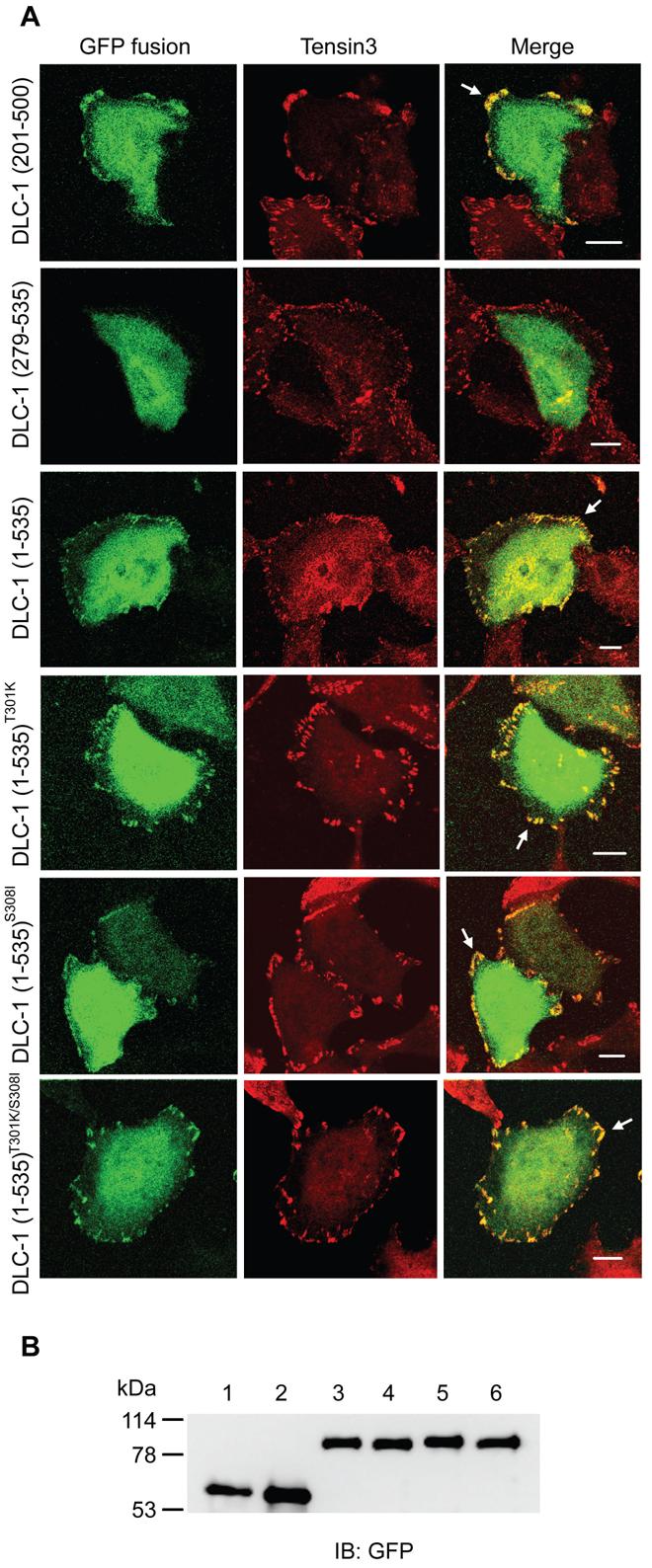
(A) A549 cells grown on coverslips were transfected with indicated GFP-fusion constructs. After labeling with anti-tensin3 antibodies followed by Alexa Fluor 594-conjugated secondary antibody, cells were visualized with a confocal microscope. Arrows indicate GFP-fusion protein and tensin3 colocalized at focal adhesions. Bar, 10 μm. (B) Cell lysate from DLC-1(201-500)(lane 1), DLC-1(279-535)(lane 2), DLC-1(1-535) (lane 3), DLC-1(1-535)T301K(lane 4), DLC-1(1-535)S308I (lane 5), or DLC-1(1-535)T301K/S308I (lane 6) GFP fusion transfectant was immunoprecipitated with anti-GFP and analyzed by immunoblotting with anti-GFP to show similar expression levels and correct sizes of recombinant proteins.
For mutation analysis, we have amplified the DNA fragment encoding the FAT region by PCR from 48 prostate cDNA samples (7 normal, 11 benign prostatic hyperplasia, and 30 cancers) for DNA sequence analysis. No mutation was found in any of normal prostate samples. We did not detect the PCR products in 1 of 11 benign prostatic hyperplasia (BPH) samples and no mutation was detected in the remaining 10 BPH samples. Two of 30 prostate cancer samples did not produce PCR fragments and 4 point mutations (1 nonsense and 3 missense mutations) were found in the remaining 28 cancer samples (table 1). All mutations were re-confirmed by a second independent PCR amplification and DNA sequencing to eliminate potential PCR and sequencing error. Because the genomic DNAs of these samples were not available to us, we could not completely rule out the possibility that these mutations were derived during the reverse-transcription reaction. Therefore, we have analyzed genomic DNAs from 8 normal and 40 colon cancer samples. No mutation was found in normal samples; 3 cancer samples did not produce PCR fragments and 4 mutations (1 nonsense and 3 missense mutations) were detected in the remaining cancer samples (table 1). Meanwhile, the previously identified SNP (single nucleotide polymorphism) leading to V354M was found in our cDNA and genomic DNA mutation screening with a frequency of 35.6%.
Table 1.
Summary of the mutation analyses within the FAT region of human DLC-1 in various tissues
| In Prostate (cDNA) | Nucleotide | Normal | BPH | Tumor | Amino acid |
|---|---|---|---|---|---|
| Missense mutation | 869 C > T | 0 | 0 | 2 of 28 | P290L |
| 902 C > A | 0 | 0 | 1 of 28 | T301K | |
| 923 G > T | 0 | 0 | 3 of 28 | S308I | |
| Nonsense mutation | 929 C > A | 0 | 0 | 1 of 28 | S310Stop |
| Polymorphism | 1060 G > A | 7 of 7 | 5 of 10 | 12 of 28 | V354M |
| In Colon (genomic) | Nucleotide | Normal | Tumor | Amino acid | |
| Missense mutation | 923 G > T | 0 | 1 of 37 | S308I | |
| 959 G > T | 0 | 1 of 37 | S320I | ||
| 1013 AC > TA | 0 | 1 of 37 | Y338L | ||
| Nonsense mutation | 1016 T > A | 0 | 1 of 37 | L339Stop | |
| Polymorphism | 1060 G > A | 2 of 8 | 6 of 37 | V354M | |
The effect of mutations on the subcellular localization of DLC-1
The nonsense mutations (S310stop, L339stop) found in this study would result in truncated DLC-1 proteins containing only the first 309 or 338 of the 1091 residues. Based on the known domain function, both mutant proteins would not contain the complete FAT, RhoGAP, and START domains. Hence, these truncated proteins would not be functional. To determine the effect of missense mutations on the subcellular localization of DLC-1, we have generated mutations in GFP-DLC-1 (1-535) expression constructs. We used this truncated DLC-1 fragment because the full length DLC-1 often leads to cell morphology change due to its RhoGAP activity and it is difficult to observe the subcellular localization in rounded cells. We have examined P290L, T301K, S308I, S320I, and Y338L mutations and found all mutants localized to focal adhesions as effective as the wild type (WT) fragment (figure 1, and not shown). Even the T301K/S308I double mutations localized to focal adhesions, indicating that at least mutations identified in this study did not significantly affect focal adhesion localization of DLC-1.
T301K and S308I mutations render the tumor suppression activities by reducing the RhoGAP activities
We further investigated the potential effects of these mutations in the full length DLC-1 on their tumor suppression activities by colony formation assays. In agreement with previous studies, GFP-DLC-1WT was able to suppress tumor cell growth and both Y442F (defective in tensin binding) and K714E (RhoGAP dead) mutants lost the suppression activities by showing > 4.4-5.1 times more colonies than in GFP-DLC-1WT (figure 2). Surprisingly, the suppression activities were significantly reduced in T301K, S308I, and T301K/S308I mutants (figure 2). Since both the RhoGAP activity and focal adhesion localization of DLC-1 are required for suppressing tumor cell growths (11, 12) and focal adhesion targeting is normal in these mutants (figure 1), we tested whether the RhoGAP activities were altered in these mutant cells. As shown in figure 3, the RhoAGTP levels (the main target of DLC-1 RhoGAP domain) were significantly elevated in cells expressing GFP control, T301K, S308I, or T301K/S308I mutants comparing to the cells expressing DLC-1 WT, P290L, S320I or Y338L mutants, indicating that the RhoGAP activities were down-regulated in T301K, S308I, and the double mutants. These data demonstrate that some mutations identified in cancer patients are biological relevant and we unexpectedly discover a regulatory site around DLC-1 aa 301-308 for its C-terminal RhoGAP domain.
Figure 2. Colony formation assays of DLC-1 mutants.

(A) A representative colony formation assay is shown. MDA-MB-468 cells were transfected with indicated constructs. After being cultured in media containing 0.8 mg/ml G418 for two weeks, G418-resistant colonies were stained with crystal violet. (B) The histogram shows the colony formation efficiency of each GFP-DLC-1 mutant comparing to the wild type (WT). (C) Cell lysate from each transfectant was immunoprecipitated with anti-GFP and analyzed by immunoblotting with anti-GFP to show similar expression levels and correct sizes of recombinant proteins.
Figure 3. RhoGAP activities of DLC-1 mutants.

HEK293T cells transfected with indicated GFP fusion constructs were treated with lysophosphatidic acid and then RhoA-GTP levels were analyzed by GST-RBD pull down assay followed by anti-RhoA blotting (top). The expression levels of endogenous RhoA (middle) and recombinant GFP-fusion proteins (bottom) were confirmed by immunoblotting with anti-RhoA and anti-GFP antibodies.
Discussion
Previous mutation screens of DLC-1 gene in tumor DNA samples have suggested that mutations in the coding region are rare events (3, 14-17). Only one exonic missense mutation (T522A) was identified from those studies (17). In addition, eight exonic single nucleotide polymorphisms (SNPs) were identified, and only one of them resulted in an amino acid change (V354M). In contrast, our mutation analyses have been focused on the coding region for the FAT domain (aa 201-500) and have identified 8 mutations and 1 SNP (V354M) in 96 genomic and cDNA samples, demonstrating that mutations in DLC-1 are not as rare as suggested. Although the missense mutations identified in this study did not appear to affect the focal adhesion localization, two mutations compromised DLC-1's tumor suppression activities. Our data indicate that further mutation screenings are highly warranted.
Numerous studies have described the down-regulation or absence of DLC-1 expression in a variety of cancers (2, 3). However, some of these studies also showed high frequencies of up-regulation of DLC-1 mRNA in cancers. For example, DLC-1 levels were up-regulated in 37%, 41%, and 39% of colon, stomach, and rectum cancers, respectively (18). Our identification of functional relevant mutations in DLC-1 offers a potential explanation for the up-regulation detected in cancer patients.
In this study, we have unexpectedly revealed a DLC-1 RhoGAP regulatory site away from its RhoGAP domain. Several GAPs are regulated by either protein-protein or protein-lipid interactions, and/or posttranslational modification (19). It is very likely that there is a binding site around residue 301-308 for a regulator that allosterically activate RhoGAP activity. When the recruitment of this regulator was abolished by mutations, RhoGAP activity was compromised. Alternatively, it is also possible that the RhoGAP activity is regulated by the phosphorylation status of this region that either leads to a conformational change or creates the binding site for the regulator. Interestingly, all missense mutations except one identified in this study show changes on amino acids containing a hydroxyl group, although it is currently unknown whether these residues are phosphorylated in vivo. Furthermore, the FAT region is serine rich with 55 (18%) serine residues within the fragment. Among them, two potential phosphorylation sites for AKT (RXRXXS/T), eight sites for PKC (S/TXR/K), and two sites for cAMP-dependent protein kinase (R/KR/KX S/T) were predicted using Motif Scan1. At least one phosphoserine site (S329) has been formally reported (20), suggesting that the posttranslational modification may contribute to the allosterical regulation. We are currently investigating potential regulatory mechanisms involving the RhoGAP activity.
Acknowledgments
This work is supported by grants from the National Institutes of Health (CA102537) and Shriners Hospital (8580) to SHL.
Footnotes
No potential conflicts of interest
References
- 1.Yuan BZ, Miller MJ, Keck CL, Zimonjic DB, Thorgeirsson SS, Popescu NC. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998;58:2196–99. [PubMed] [Google Scholar]
- 2.Liao YC, Lo SH. Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. Int J Biochem Cell Biol. 2008;40:843–7. doi: 10.1016/j.biocel.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durkin ME, Yuan BZ, Zhou X, et al. DLC-1:a Rho GTPase-activating protein and tumour suppressor. J Cell Mol Med. 2007;11:1185–207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiao F, Bowie JU. The many faces of SAM. Sci STKE. 2005;286:re7. doi: 10.1126/stke.2862005re7. [DOI] [PubMed] [Google Scholar]
- 5.Iyer LM, Aravind L, Koonin EV. Common origin of four diverse families of large eukaryotic DNA viruses. J Virol. 2001;75:11720–34. doi: 10.1128/JVI.75.23.11720-11734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sekimata M, Kabuyama Y, Emori Y, Homma Y. Morphological changes and detachment of adherent cells induced by p122, a GTPase-activating protein for Rho. J Biol Chem. 1999;274:17757–62. doi: 10.1074/jbc.274.25.17757. [DOI] [PubMed] [Google Scholar]
- 7.Wong CM, Yam JW, Ching YP, et al. Rho GTPase-activating protein deleted in liver cancer suppresses cell proliferation and invasion in hepatocellular carcinoma. Cancer Res. 2005;65:8861–8. doi: 10.1158/0008-5472.CAN-05-1318. [DOI] [PubMed] [Google Scholar]
- 8.Yam JW, Ko FC, Chan CY, Jin DY, Ng IO. Interaction of deleted in liver cancer 1 with tensin2 in caveolae and implications in tumor suppression. Cancer Res. 2006;66:8367–72. doi: 10.1158/0008-5472.CAN-05-2850. [DOI] [PubMed] [Google Scholar]
- 9.Homma Y, Emori Y. A dual functional signal mediator showing RhoGAP and phospholipase C-delta stimulating activities. EMBO J. 1995;14:286–91. doi: 10.1002/j.1460-2075.1995.tb07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaga M, Sekimata M, Fujii M, et al. A PLCdelta1-binding protein, p122/RhoGAP, is localized in caveolin-enriched membrane domains and regulates caveolin internalization. Genes Cells. 2004;9:25–37. doi: 10.1111/j.1356-9597.2004.00698.x. [DOI] [PubMed] [Google Scholar]
- 11.Liao YC, Si L, Devere White RW, Lo SH. The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. J Cell Biol. 2007;176:43–9. doi: 10.1083/jcb.200608015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian X, Li G, Asmussen HK, et al. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc Natl Acad Sci U S A. 2007;104:9012–7. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawai K, Yamaga M, Iwamae Y, et al. A PLCdelta1-binding protein, p122RhoGAP, is localized in focal adhesions. Biochem Soc Trans. 2004;32:1107–9. doi: 10.1042/BST0321107. [DOI] [PubMed] [Google Scholar]
- 14.Pang JC, Chang Q, Chung YF, et al. Epigenetic inactivation of DLC-1 in supratentorial primitive neuroectodermal tumor. Hum Pathol. 2005;36:36–43. doi: 10.1016/j.humpath.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 15.Hewitt C, Wilson P, McGlinn E, et al. DLC1 is unlikely to be a primary target for deletions on chromosome arm 8p22 in head and neck squamous cell carcinoma. Cancer Lett. 2004;209:207–13. doi: 10.1016/j.canlet.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 16.Zheng SL, Mychaleckyj JC, Hawkins GA, et al. Evaluation of DLC1 as a prostate cancer susceptibility gene: mutation screen and association study. Mutat Res. 2003;528:45–53. doi: 10.1016/s0027-5107(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 17.Wilson PJ, McGlinn E, Marsh A, et al. Sequence variants of DLC1 in colorectal and ovarian tumours. Hum Mutat. 2000;15:156–65. doi: 10.1002/(SICI)1098-1004(200002)15:2<156::AID-HUMU4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Ullmannova V, Popescu NC. Expression profile of the tumor suppressor genes DLC-1 and DLC-2 in solid tumors. Int J Oncol. 2006;29:1127–32. [PubMed] [Google Scholar]
- 19.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–77. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Hers I, Wherlock M, Homma Y, Yagisawa H, Tavare JM. Identification of p122RhoGAP (deleted in liver cancer-1) Serine 322 as a substrate for protein kinase B and ribosomal S6 kinase in insulin-stimulated cells. J Biol Chem. 2006;281:4762–70. doi: 10.1074/jbc.M511008200. [DOI] [PubMed] [Google Scholar]