

Abstract
One approach to control colorectal cancer (CRC) is its preventive intervention by dietary agents or those consumed as supplements. However, since most of these products are often consumed by patients as an alternative and complementary medicine (CAM) practice, a scientific base such as efficacy, mechanism and standardized preparation, needs to be developed. Grape seed extract (GSE) is one such supplement widely consumed by humans for its several health benefits. We reported recently that GSE inhibits CRC cell HT29 growth in culture and nude mice xenograft. Since GSE is available commercially through different vendors, here we assessed whether GSE from two different manufacturers produces comparable biological effects in a panel of human CRC cell lines. Our results show that irrespective of source, GSE strongly inhibits LoVo, HT29 and SW480 cell growth, with a G1 arrest in LoVo and HT29 cells, but an S and/or G2/M arrest in SW480 cell cycle progression. GSE also induced Cip/p21 levels in all three cell lines. Furthermore, an induction of apoptosis was observed in all three cell lines by GSE. Taken together, our findings suggest that GSE could be an effective CAM agent against CRC possibly due to its strong growth inhibitory and apoptosis inducing effects.
Keywords: grape seed extract, colorectal cancer, cell cycle, apoptosis, chemoprevention, Cip1/p21
Introduction
According to the American Cancer Society 2007 report, colorectal cancer (CRC) is the third most common cancer in both men and women; the overall mortality due to this malignancy accounts for 10% of all the cancer-associated deaths (1). CRC is one of the disease conditions where dietary habits and life style are major etiological factors. Diets rich in fat, animal proteins, and low in fiber are often considered risk factors for developing CRC, whereas those rich in fruits, vegetables, and whole grains are often recommended for reducing the risk of this malignancy (2). Once a full blown clinically evident case of colorectal cancer emerges, treatment options include surgery, chemotherapy, and radiotherapy; however, most of the time, the cancer resurfaces later and the patients have to undergo additional rounds of radiotherapy or chemotherapy. Because of the severe toxicity and side-effects associated with current treatment regimens for CRC, the patients often also drift towards using complementary and alternative medicine (CAM) practices, such as consumption of health/dietary supplements, chiropractic treatments, massage therapy, herbs, etc, in order to relieve the symptoms of pain and to have better overall health. In the past few years, the increased popularity of CAM, especially among cancer patients, has been well-identified and recognized (3). With regard to health/dietary supplements, a wide number of herbs and/or herbal extracts, such as milk thistle extract, triptolide, Chinese herb extract, brucea fruit, European mistle toe, green tea extract, soy isoflavones, and grape seed extract (GSE), are being frequently consumed by both healthy people and patients as a CAM practice (4), mostly because of the strong belief that these agents provide several health benefits.
One of the focuses of our research program is to determine the biological efficacy and associated mechanisms of GSE in preventing CRC in pre-clinical models. GSE is rich in proanthocyanidins, which are largely responsible for the spectrum of biological activities exhibited by this extract. For example, GSE exerts anti-inflammatory, anti-bacterial, anti-viral, anti-nociceptive, and other health beneficial effects in human disease conditions such as atherosclerosis, hyperglycemia, etc (5-13). Also due to its high proanthocyanidins content, GSE exhibits strong antioxidant activity, which is even more potent than known antioxidants such as vitamin E and ascorbic acid (14). Regarding its efficacy against malignancy, several studies during the last ten years from our laboratory as well as by others have convincingly demonstrated that GSE exerts strong anti-cancer and cancer chemopreventive potential against different cancers in both cell culture and animal models (15-29). However, the major concern with GSE studies had been a lack of availability of uniformly standardized preparation to be used in biological efficacy studies. Even in our own studies, we have initially used the GSE prepared in the laboratory (30) followed by that obtained from different vendors (19, 27), which raises an important question whether different GSE preparations have comparable biological activity and associated mechanism of action in a given biological system. Employing a panel of well-defined human CRC cell lines, namely HT29, SW480 and LoVo, the present study was designed to address this important issue. The selection of CRC as a cancer model was based on our recently published studies showing that GSE (from Kikkoman Corporation, Japan) causes strong growth inhibitory and apoptotic effects in HT29 human CRC cells in culture and nude mice xenografts (19). Regarding the selection of a panel of human CRC cell lines for the present study, the HT29 cell line was chosen based on already completed studies with GSE in this cell line (19) and the fact that it is a widely used human CRC cell line possibly because it retains biochemical and physiological characteristics of normal colorectal epithelial cells (31). The selection of other cell lines was based on the degree of their differentiation and invasion, where the SW480 cell line is moderately differentiated and invasive, and the LoVo cell line represents a poorly differentiated, but highly invasive human CRC (32). We anticipated that by selecting these three different cell lines, we would also be able to assess and establish both selectivity and specificity of GSE effects on selected biological and molecular endpoints in different human CRC cells. Our results show that irrespective of its source, GSE causes strong growth inhibitory and cell death effects in all three human CRC cell lines employed in the study. Furthermore, the growth inhibitory effects of GSE were possibly due to a strong cell cycle arrest together with an induction in the levels of cyclin-dependent kinase inhibitors (CDKIs), Cip1/p21 (in all three cell lines) and Kip1/p27 (in LoVo and HT29 cell lines), whereas its cell death effect was mostly apoptotic in nature.
Materials and Methods
Chemicals, Reagents and Cell Culture
GSE was procured from two different sources namely Traco Labs (Champaign, IL) and Kikkoman Corporation (Noda City, Japan) referred hereafter as GSE-I and GSE-II, respectively. Anti-Cip1/p21 antibody was from Calbiochem (Cambridge, MA), and anti-Kip1/p27 and anti-tubulin antibodies were from Neomarkers, Inc. (Fremont, CA). Annexin V-Vybrant apoptosis assay kit2 was from Molecular Probes (Eugene, OR). All the cell culture materials were from Invitrogen Corporation (Gaithersberg, MD) and fetal bovine serum was from Hyclone (Logan, UT). Human CRC cell lines LoVo, HT29 and SW480 were from American Type Culture Collection (Manassas, VA). LoVo cells were cultured in F-12 Nutrient Mixture (HAM) with 10% fetal bovine serum, HT29 cells in DMEM with 10% fetal bovine serum, and SW480 cells in Leibovitz's L-15 Medium with 10% fetal bovine serum under standard culture conditions (37°C, 95% humidified air and 5% CO2).
Cell Growth and Death Assay
Cells (LoVo, HT-29 or SW480) were plated at a cell density of 5000 cells/cm2 in 60mm plates under the standard culture conditions for 24h. Cells were treated subsequently either with DMSO alone or with varying concentrations of GSE-I or GSE-II (25, 50 and 100μg/ml). At the end of the respective treatment times (12, 24 or 48h), cells were harvested after brief trypsinization and counted using a hemocytometer. Trypan blue dye exclusion was used to differentiate live and dead cells.
Flow Cytometry Analysis for Cell Cycle Distribution
Cells (LoVo, SW480 or HT-29) were plated to 60% confluency overnight, and subsequently treated with either DMSO alone or various doses of GSE-I or GSE-II (25, 50 and 100μg/ml). After 12, 24 and 48h of treatment time, cells were collected and processed for cell cycle analysis. Briefly, 0.5 × 105 cells were suspended in 0.5 ml of saponin/PI solution (0.3% saponin (w/v), 25 μg/ml PI (w/v), 0.1 mM ethylenediaminetetraacetic acid and 10 μg/ml RNase A (w/v) in phosphate-buffered saline) and incubated overnight at 4°C in dark. Cell cycle distribution was then analyzed by flow cytometry using fluorescence-activated cell sorting analysis core facility of the University of Colorado Cancer Center (Aurora, CO).
Quantitative Apoptotic Cell Death Assay
To quantify GSE-induced apoptotic death of human CRC cells, annexin V and PI staining was performed followed by flow cytometry, as described recently (19). Briefly, cells (LoVo, SW480 or HT-29) were plated to 60% confluency overnight, and subsequently treated with either DMSO alone or various doses of GSE-I or GSE-II (25, 50 and 100μg/ml). After 24h of treatment time, adherent cells were harvested by brief trypsinization, and both adherent and non-adherent cells were collected together by centrifugation. Cells were then washed twice with ice-cold PBS and subjected to annexin V and PI staining using Vybrant Apoptosis Assay Kit2 (Molecular Probes, Oregon) following the step-by-step protocol provided by the manufacture. After the staining, FACS analysis, utilizing the core service of the University of Colorado Cancer Center (Aurora, CO), was performed for the quantification of the apoptotic cells.
Western Immunoblotting
Cells (LoVo, HT-29 and SW480) were cultured under their standard culture conditions in 100 mm culture dishes. At 60% confluency, cells were treated with either DMSO or GSE-I at the concentrations of 25, 50 and 100 μg/ml in DMSO for 12 or 24h. At the end of treatment time, cell lysates were prepared in non denaturing lysis buffer (10mM Tris-HCL, pH7.4, 150mM NaCL, 1% triton X-100, 1mM EDTA, 1mM EGTA, 0.3 mM phenylmethyl suflonyl fluoride, 0.2mM sodium orthovanadate, 0.5% NP-40, and 5 U/ml of aprotinin). Briefly, medium was aspirated from the culture dishes followed by two washings with ice cold PBS, the adherent cells were incubated in lysis buffer for 10 min over the ice, and then scraped and kept on ice for additional 30 min. Finally, cell lysates were cleared by centrifugation at 4°C for 30min at 14,000 rpm, and protein concentration in cell lysates was estimated using Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA).
Statistical Analysis
Statistical significance of differences between control and treated samples were calculated by Student's t-test (Sigma Stat 2.0, Jandel Scientific). P values of < 0.05 were considered statistically significant. Each assay/observation represents a mean of three independent values.
Results
GSE Inhibits Growth and Induces Cell Death in a Panel of Human CRC Cell Lines
Our first aim was to examine the effect of GSE procured from two different vendors on the growth of three different human CRC cell lines, namely LoVo, HT-29 and SW480. As shown in Table 1, both GSE-I and GSE-II inhibited the growth of LoVo cells in a concentration- and time-dependent manner. Whereas only the highest concentration of GSE-I showed strong inhibition (38%, P<0.05) following 12h treatment, GSE-I at 25, 50 and 100μg/ml doses resulted in 14-70% (P<0.01-0.001) and 48 to 87% (P< 0.05-0.001) growth inhibition of LoVo cells after 24 and 48h of treatment, respectively (Table 1). Concomitantly, there was a significant increase in the LoVo dead cell population [9-35% (P<0.01-0.001), 13-56% (P<0.05) and 14-44% (P<0.05)] at similar concentrations and treatment times of GSE-I (Table 1). With GSE-II also, there was a significant growth inhibition in LoVo cells ranging from 15-48% (P<0.01-0.001), 12-59% (P<0.01-0.001) and 20-85% (P<0.001) at 25, 50 and 100μg/ml concentrations after 12, 24 and 48h of treatment (Table 1). GSE-II treatments also caused a significant induction in dead cell population [8-47% (P<0.001), 12-55% (P<0.01-0.001) and 12-494% (P<0.001)] of LoVo cells under similar treatment conditions (Table 1).
Table 1.
Comparative biological activity of GSE-I and GSE-II in human CRC cell linesa
| LoVo Cells (GSE-I) | LoVo Cells (GSE-II) | |||||||
| Total cell number X 105 | Total cell number X 105 | |||||||
| Time | Control | 25 μg/ml | 50 μg/ml | 100μg/ml | Control | 25 μg/ml | 50 μg/ml | 100μg/ml |
| 12h | 3.4±0.6 | 3.7±0.4 | 3.6±0.3 | 2.1±0.2* | 2.7±0.2 | 2.3±0.3 | 1.9±0.1# | 1.4±0.2$ |
| 24h | 9.6±1.0 | 8.3±1.3 | 6.0±0.7# | 2.9±1.0$ | 4.9±0.2 | 4.3±0.7 | 3.0±0.3# | 2.0±0.2$ |
| 48h | 15.0±2.9 | 7.8±0.8# | 4.7±0.8$ | 1.9±0.2$ | 12.5±1.1 | 10.0±1.5 | 2.5±0.1$ | 1.9±0.4$ |
| Percent Dead Cell Population | Percent Dead Cell Population | |||||||
| Time | Control | 25 μg/ml | 50 μg/ml | 100μg/ml | Control | 25 μg/ml | 50 μg/ml | 100μg/ml |
| 12h | 4.2±1.8 | 9.1±4.2 | 18.0±1.6$ | 35.0±7.4# | 3.4±0.7 | 8.2±0.2 | 21.9±0.8$ | 47.3±1.1$ |
| 24h | 9.8±2.4 | 12.7±1.1 | 23.6±6.3 | 56.0±15* | 5.6±1.4 | 12.0±4.1 | 30.0±2.3$ | 55.0±12# |
| 48h | 13.6±4.1 | 14.1±6.1 | 26.2±4.8* | 44.0±5.9* | 8.9±0.9 | 12.0±2.8 | 35.8±4.7$ | 49.4±4.7$ |
| HT29 Cells (GSE-I) | HT29 Cells (GSE-II) | |||||||
| Total cell number X 105 | Total cell number X 105 | |||||||
| Time | Control | 25 μg/ml | 50 μg/ml | 100μg/ml | Control | 25 μg/ml | 50 μg/ml | 100μg/ml |
| 12h | 2.4±0.5 | 1.9±0.4 | 1.6±0.3 | 1.5±0.1# | 2.0±0.2 | 1.6±0.4 | 1.7±0.3 | 1.7±0.2 |
| 24h | 4.0±0.34 | 4.0±0.4 | 4.0±0.6 | 2.4±0.3# | 3.3±0.2 | 2.8±0.5 | 2.2±0.3 | 1.9±0.3# |
| 48h | 8.0±1.4 | 8.3±1.8 | 6.5±0.2 | 3.2±0.7# | 6.4±0.2 | 5.6±0.5 | 5.0±0.4# | 1.4±0.2$ |
| Percent Dead Cell Population | Percent Dead Cell Population | |||||||
| Time | Control | 25 μg/ml | 50 μg/ml | 100μg/ml | Control | 25 μg/ml | 50 μg/ml | 100μg/ml |
| 12h | 1.4±0.6 | 1.7±0.7 | 4.1±0.9 | 5.6±1.9* | 3.3±1.4 | 4.0±0.7 | 8.3±1.3* | 12.0±1.4# |
| 24h | 3.7±2.2 | 4.8±1.6 | 6.1±2.5 | 14.0±2.4* | 5.1±1.6 | 5.5±1.0 | 10.4±1.8 | 26.8±4.1# |
| 48h | 4.8±1.4 | 6.6±3.3 | 11.0±2.7 | 18.0±8.5* | 4.7±0.1 | 10.0±1.4 | 17.0±3.8# | 33.0±4.5$ |
| SW480 Cells (GSE-I) | SW480 Cells (GSE-II) | |||||||
| Total cell number X 105 | Total cell number X 105 | |||||||
| Time | Control | 25 μg/ml | 50 μg/ml | 100μg/ml | Control | 25 μg/ml | 50 μg/ml | 100μg/ml |
| 12h | 1.2±0.1 | 1.3±0.2 | 1.3±0.1 | 1.2±0.2 | 1.2±0.1 | 0.9±0.1 | 0.9±0.1 | 0.7±0.2# |
| 24h | 2.0±0.3 | 2.2±0.4 | 1.9±0.5 | 1.4±0.3$ | 1.3±0.2 | 0.9±0.1 | 0.7±0.1# | 0.6±0.1$ |
| 48h | 3.0±0.3 | 2.0±0.1# | 1.8±0.2$ | 1.3±0.2$ | 2.2±0.4 | 0.8±0.1$ | 0.8±0.1$ | 0.7±0.1$ |
| Percent Dead Cell Population | Percent Dead Cell Population | |||||||
| Time | Control | 25 μg/ml | 50 μg/ml | 100μg/ml | Control | 25 μg/ml | 50 μg/ml | 100μg/ml |
| 12h | 2.7±2.4 | 3.4±2.4 | 6.6±2.3 | 14.0±5.3* | 4.3±0.3 | 5.4±0.6* | 16.0±4.1# | 20.5±4.2# |
| 24h | 4.6±1.4 | 13.2±9.2 | 11.3±6.0 | 30.0±7.0# | 3.8±0.4 | 15.0±2.6# | 29.0±4.2# | 40.0±9.6# |
| 48h | 4.4±0.8 | 10.0±3.6 | 15.0±0.8# | 33.6±10* | 4.2±1.1 | 14.6±3.6# | 25.4±2.2$ | 56.0±9.2$ |
The biological activity of GSE-I and GSE-II was measured in terms of their effects on total cell number and percentage of cell death. The data shown in each case are mean of three independent observations with standard deviation, and were reproducible in two additional independent experiments.
P< 0.05;
P<0.01;
P<0.001, control (DMSO) versus various GSE-I/GSE-II treatments as indicated in the Table.
In the case of HT29 cells, GSE-I had no significant effect on growth at 25 and 50μg/ml concentration levels at 12, 24 and 48h treatment time; however, a strong effect was observed at 100μg/ml concentration accounting for 38-60% (P<0.01) growth inhibition following 12-48h of treatment (Table 1). Under similar experimental conditions, the HT29 dead cell population also increased compared to vehicle controls accounting for 2-6% (P<0.001), 5-14% (P<0.001) and 7-18% (P<0.001) at 12, 24 and 48h of GSE-I treatment (Table 1). With GSE-II as well, a strong growth inhibition was observed at 100μg/ml dose level (42-78%, P<0.01-0.001) after 24 and 48h of treatment time only (Table 1). However, a better cell death effect of GSE-II, compared to GSE-I, was observed in HT29 cells accounting for 4-12% (P<0.05-0.01), 6-27% (P<0.01) and 10-33% (P<0.01-0.001) dead cell population following 25, 50 and 100μg/ml doses of GSE-II treatment for 12, 24 and 48h, respectively (Table 1).
The cell growth inhibitory effect of GSE-II was much stronger in SW480 cells as compared to GSE-I (Table 1). In this case, GSE-I treatment at all the concentrations studied did not produce any cell growth inhibitory effects in SW480 cells at 12h and 24h time-points, except in the 100μg/ml concentration at 24h treatment showing 30% (P<0.001) inhibition; however, the longer treatment time of 48h caused a dose-dependent effect accounting for 33% (P<0.01), 40% (P<0.001) and 57% (P<0.001) inhibition at 25, 50 and 100μg/ml concentrations, respectively (Table 1). Interestingly, GSE-I treatment at 100μg/ml concentration showed a strong cell death at all time-points studied accounting for 14% (P<0.05), 30% (P<0.01) and 37% (P<0.05) dead cell population in SW480 CRC cell line (Table 1). Regarding GSE-II, its treatment led to a stronger effect on SW480 cells accounting for 25-42% (P<0.01), 31-54% (P<0.01-0.001) and 64-68% (P<0.01-0.001) growth inhibition at 25, 50 and 100μg/ml doses following 12, 24 and 48h of treatment, respectively (Table 1). Importantly, GSE-II also caused a much stronger cell death effect in SW480 cells accounting for 5-21% (P<0.05-0.01), 15-40% (P<0.01) and 15-56% (P<0.01-0.001) dead cell population under same doses and treatment times (Table 1).
GSE Induces Cell Cycle Arrest in a Panel of Human CRC Cell Lines
Based on our results showing that both GSE-I and GSE-II concentration - and time-dependently inhibit the growth of a panel of human CRC cell lines, we next assessed whether an alteration in cell cycle progression by these extracts is possibly involved in causing cell growth inhibition. All the CRC cell lines were treated similarly as in cell growth assays with both GSE preparations, and the distribution of cells in different phases of the cell cycle was analyzed. In the case of LoVo cells, treatment with both GSE-I and GSE-II led to a marginal (though statistically significant) accumulation of cells in G2/M phase at higher concentration(s) following 12h of treatment time (Figure 1A and 1D). However, after 24 and 48h of similar treatments, a significant (P<0.001) arrest of the cells in G1 phase of the cell cycle was observed at all the dose levels with comparable response between two GSE preparations (Figure 1A-F). In the case of HT29 cells, both GSE-I and GSE-II had no effect on any phase of cell cycle at all the concentration levels tested after 12h of treatment time, except the highest concentration showing a marginal increase in S phase (in case of GSE-I) or G2/M phase (in case of GSE-II). However, similar to LoVo cell results, with an increase in treatment time to 24 and 48h, a strong G1 arrest was also observed in case of HT29 cells at 50 and 100μg/ml concentrations of both GSE-I and GSE-II, but to a lesser extent with GSE-I (Figure 2A-F). Unlike both LoVo and HT29 cell lines, treatment of SW480 cells with both GSE-I and GSE-II for 12h led to the arrest of the cells in the S phase of cell cycle at the highest concentration of 100μg/ml; however, a longer treatment time of 24 and 48h caused a significant arrest of the cells in G2/M phase of the cell cycle (Figure 3A-F). Together, these results clearly show that the observed cell growth inhibitory effects of both GSE preparations were associated with their cell cycle arrest inducing activity.
FIG. 1.

GSE-I and GSE-II preparations induce cell cycle arrest in LoVo cells. Cells were plated as described in the Materials and Methods and treated with either DMSO alone (control) or varying concentrations of GSE-I or GSE-II (25-100μg/ml) for 12, 24 and 48h. At the end of these treatments, cells were collected and incubated with saponin/PI solution at 4°C for 24 h in dark and subjected to FACS analysis as detailed in the Materials and Methods. Panels A, B and C represent data for GSE-I at 12, 24 and 48h, and panels D, E and F represent data for GSE-II at 12, 24 and 48h of treatment, respectively. The data shown are mean ± SD of three independent plates. *, P < 0.05; #, P < 0.01; and $, P < 0.001, control (DMSO) versus various GSE-I/GSE-II treatments as indicated in the figure.
FIG. 2.

GSE-I and GSE-II preparations induce cell cycle arrest in HT29 cells. Cells were plated as described in the Materials and Methods and treated with either DMSO alone (control) or varying concentrations of GSE-I or GSE-II (25-100μg/ml) for 12, 24 and 48h. At the end of these treatments, cells were collected and incubated with saponin/PI solution at 4°C for 24 h in dark and subjected to FACS analysis as detailed in the Materials and Methods. Panels A, B and C represent data for GSE-I at 12, 24 and 48h, and panels D, E and F represent data for GSE-II at 12, 24 and 48h of treatment, respectively. The data shown are mean ± SD of three independent plates. *, P < 0.05; #, P < 0.01; and $, P < 0.001, control (DMSO) versus various GSE-I/GSE-II treatments as indicated in the figure.
FIG. 3.
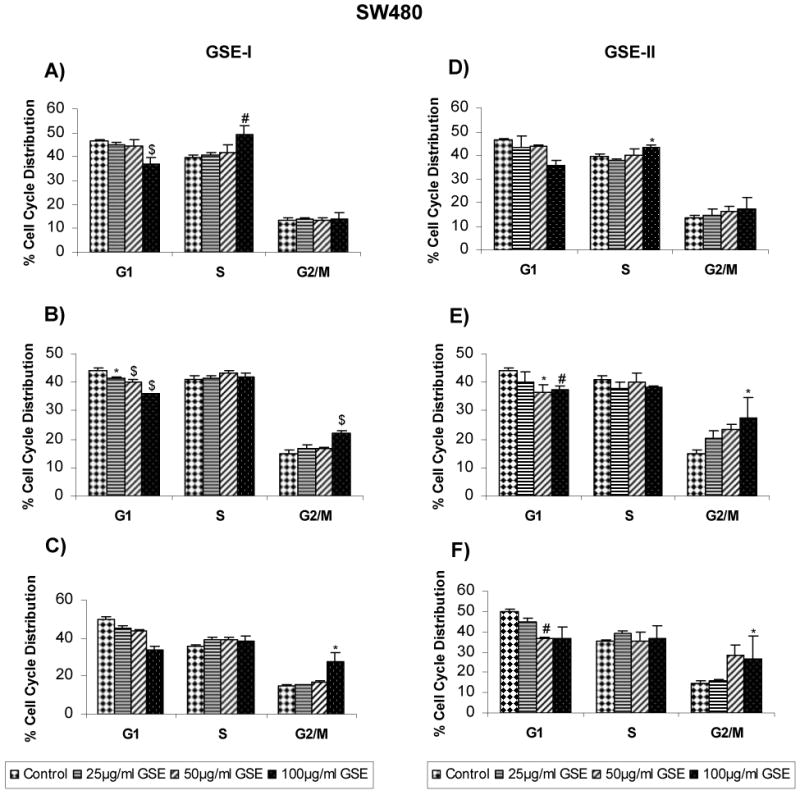
GSE-I and GSE-II preparations induce cell cycle arrest in SW480 cells. Cells were plated as described in the Materials and Methods and treated with either DMSO alone (control) or varying concentrations of GSE-I or GSE-II (25-100μg/ml) for 12, 24 and 48h. At the end of these treatments, cells were collected and incubated with saponin/PI solution at 4°C for 24 h in dark and subjected to FACS analysis as detailed in the Materials and Methods. Panels A, B and C represent data for GSE-I at 12, 24 and 48h, and panels D, E and F represent data for GSE-II at 12, 24 and 48h of treatment, respectively. The data shown are mean ± SD of three independent plates. *, P < 0.05; #, P < 0.01; and $, P < 0.001, control (DMSO) versus various GSE-I/GSE-II treatments as indicated in the figure.
GSE Modulates the Expression of p21 and p27 Proteins in Different Human CRC Cell Lines
Progression through different phases of the cell cycle is orchestrated by specific cyclins and CDKs associated with a particular phase of the cell cycle, and is negatively controlled by the inhibitors of CDKs; mostly the INK4 and CDKI family members (33, 34). Our recent studies have shown that GSE-II causes its in vitro and in vivo efficacy against human CRC HT29 cells mostly via a strong induction in Cip1/p21 levels (19). Based on these recent findings and the present results showing that both GSE preparations induce a strong cell cycle arrest in all the three CRC cell lines, we next also studied the effect of GSE-I on the protein levels of two CDKIs namely Cip/p21 and Kip1/p27. We observed that GSE-I treatment of LoVo and HT-29 cells increases the levels of both p21 and p27 with more prominent effect on p21 levels (Figure 4). In the case of SW480, an increase in the expression of p21 was also observed in time- and concentration-dependent manner; however, GSE-I caused a decrease in the levels of p27 at all the concentrations and treatment times (Figure 4).
FIG. 4.
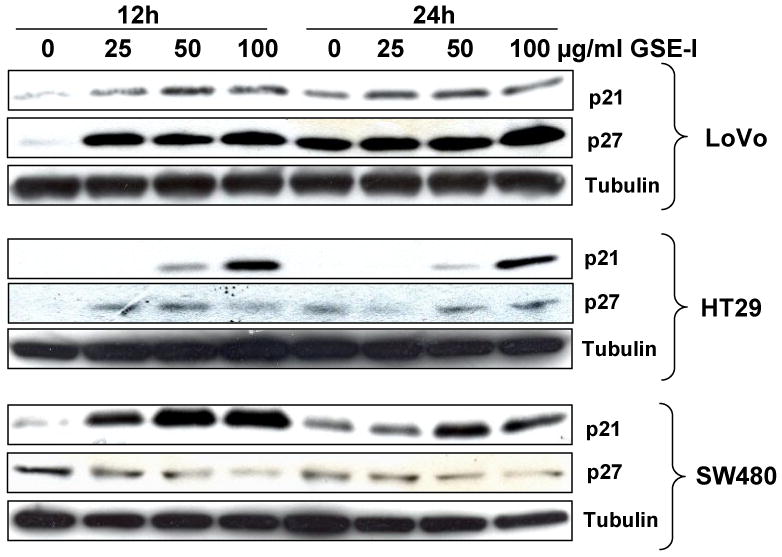
GSE-I modulates Cip1/p21 and Kip1/p27 levels in a panel of human CRC cell lines. LoVo, HT29 and SW480 cells were treated with either DMSO alone (control) or varying concentrations of GSE-I (25-100μg/ml) for 12 and 24 h. At the end of these treatments, total cell lysates were prepared and analyzed by western blotting for p21 and p27 levels as detailed in the Materials and Methods. Membranes were stripped and reprobed for tubulin as a loading control.
GSE Induces Apoptosis in Various Human CRC Cell Lines
Since we also observed significant cell death upon GSE treatment in all the three CRC cell lines, we next assessed whether this was apoptotic in nature. For this, all the three cell lines were treated with either GSE-I or GSE-II under similar conditions as in above mentioned studies, and the extent of apoptotic cell death was measured by flow cytometric analysis of annexin-V/ PI stained cells. As depicted in Figure 5A, GSE-I and GSE-II treatment of LoVo cells led to a significant increase in early and late apoptosis in a concentration-dependent manner. GSE-I treatment for 24h resulted in 7-35% cells in early apoptosis and 3-28% cells in late apoptosis (Figure 5A). Treatment of LoVo cells with GSE-II resulted in almost comparable early (7-31%; P<0.001) and late apoptotic cell population (3-47%; P<0.001, Figure 5B). In the case of HT29 cells, GSE-I treatment induced a significant early (4-22%; P<0.001) and late (3-6%; P<0.01) apoptotic cell death at highest concentration of 100μg/ml after 24h of treatment time (Figure 5C). However, with GSE-II, a significant increase in early apoptotic cells from 4% to 13% (P<0.001) was observed even at 50μg/ml concentration, which increased further to 34% (P<0.001) with the increase in concentration to 100μg/ml (Figure 5D). In the case of late apoptotic event, a significant increase (P<0.001) was observed at highest concentration of 100μg/ml GSE-II (Figure 5D). Treatment of SW480 cells with GSE-I induced a significant early (3-28%; P<0.001)) and late (3-21%; P<0.001)) apoptotic cell death only at 50 and 100μg/ml concentrations after 24h of treatment time (Figure 5E). Under similar conditions, GSE-II significantly induced both early (3-32%; P<0.001) and late (3-32%; P<0.001) apoptotic cell death at 25, 50 and 100μg/ml concentrations after 24h of treatment time (Figure 5F). Together, these results clearly show that the observed cell death effects of both GSE preparations were mostly apoptotic in nature.
FIG. 5.

GSE-I and GSE-II preparations induce apoptotic cell death in a panel of human CRC cell lines. LoVo, HT29 and SW480 cells were plated and treated with DMSO (control) or varying concentrations of GSE-I or GSE-II (25-100μg/ml) as detailed in the Materials and Methods. Following 24h of these treatments, adherent and non-adherent cells were collected by brief trypsinization and processed for FACS analysis following annexin V-PI staining. Panels A and B represent data for GSE-I and GSE-II treatment of LoVo cells; panels C and D represent data for GSE-I and GSE-II treatment of HT29 cells; and panels E and F represent data for GSE-I and GSE-II treatment of SW480 cells. The data shown are mean ± SD of three independent plates. *, P < 0.05; #, P < 0.01; and $, P < 0.001, control (DMSO) versus various GSE-I/GSE-II treatments as indicated in the figure.
Discussion
GSE is a commonly used dietary supplement marketed in the United States and Europe for its different health benefits including strong antioxidant activity (5-14). In the last ten years, several studies by our laboratory and by others have also convincingly documented the anti-cancer and cancer chemopreventive efficacy of GSE against various cancers (15-29); however, the major caveats had been the composition of various preparations of GSE being marketed under different names, and those being used under laboratory conditions. Furthermore, the lack of standardized preparations has also limited the validity and translational potential of the research findings obtained in the laboratory setting using different preparations/source of GSE. To address some of these issues, we compared the biological effects of GSE procured from two different vendors against three different human CRC cell lines. The central findings of our study are that irrespective of its source, GSE produces strong biological effects in different human CRC cell lines, which include growth inhibition, cell cycle arrest, induction of negative regulators of cell cycle progression namely Cip1/p21 and Kip1/p27, and apoptotic cell death.
The inhibition of cell growth in GSE-treated LoVo and HT-29 cells was possibly due to the induction of sustained G1 cell cycle arrest and a transient G2/M arrest during short treatment time. We observed that both GSE preparations produce similar effect in terms of an arrest at the particular phase of cell cycle in the context of the individual CRC cell line, though the extent of the effect and the GSE dose at which the effect was significant varied to some extent between the two preparations. In the case of SW480 cells, though we observed that GSE treatment results in S and/or G2-M arrest at the expense of the cells in G1 phase, the effect was comparable for both the preparations. The progression through various phases of cell cycle is a tightly regulated process involving the activities of various cyclins and CDKs, which are specific for different cell cycle phases (33). The activity of cyclin-CDK complexes, in a controlled cell cycle progression, is regulated by two different families of proteins known as INK4 and CDKI (34, 35). Cancer manifests itself as uncontrolled proliferation of cells wherein the tight regulation of cell cycle progression is compromised (36). In this regard, several studies have shown that both INK4 and CDKI family members are often non-functional in various malignancies including CRC, which results in an uncontrolled cell cycle progression and cancer growth (37, 38, 39). Therefore, the molecular players such as cyclins, CDKs and their inhibitors serve as potential targets to halt the uncontrolled proliferation (40-42). Specifically, it could be argued that the agents which induce the level and/or function of cell cycle inhibitory regulators (INK4 and Cip/Kip family members) might be useful in the control of various malignancies including CRC. In our recently published studies with GSE-II, we have reported that this extract inhibits cell cycle progression of human CRC HT29 cells mostly via an upregulation in the levels of p21 under both in vitro and in vivo conditions (19). In the present study, we employed only GSE-I to assess whether it produces similar effects on CDKI induction as observed with GSE-II in a recently published study (19). Our results clearly showed an induction in the levels of Cip1/p21 by GSE-I in all three cell lines, together with an increase in Kip1/p27 in the LoVo and HT29 cells, which is in line with the observed G1 arrest by this extract in these two CRC cell lines. Importantly, GSE-I caused a dose- and time-dependent decrease in the levels of Kip1/p27 in SW480 cells, which is consistent with its cell cycle arrest effect in S or G2/M phase in this cell line, as Kip1/p27 is mostly involved in G1 phase whereas Cip1/p21 is a universal CDKI (34). From these observations, it could be suggested that in case of SW480 cells, GSE causes the degradation of p27, by a mechanisms yet to be established, which drives the cells to S followed by G2/M phases of the cell cycle where an induction in Cip1/p21 (by GSE) arrests the cells in these cell cycle phases.
In addition to an aberrant cell cycle progression, loss of apoptotic function is a major contributor towards the resistance of cancer cells to cytotoxic chemotherapeutic agents (43-45), and accordingly, an induction of apoptosis under such circumstances is also highly desirable for preventive intervention of various malignancies including CRC. Consistent with this notion, our results, showing an induction of apoptotic death in all three human CRC cell lines by GSE, identify yet another anticancer mechanism (apart from cell cycle arrest) of this extract for its possible application in CRC control. Lastly, we would also like to emphasize here that the observed biological effects of both GSE preparations including an induction of Cip1/p21 seem independent of p53 involvement, as these responses were comparable in non-functional p53 harboring CRC cell lines (HT29 and SW480 cell lines) versus that carrying wild-type p53 (LoVo cell line). As mutation in p53 is a hallmark of CRC growth and progression in both clinical and pre-clinical settings (46), our results showing that GSE exerts its biological efficacy in human CRC cells independent of their p53 status are extremely important outcomes for future translational potential in preventive intervention of CRC in the clinic.
Acknowledgments
This work was supported by Grant RO1 AT003623 from the National Center for Complementary and Alternative Medicine, and the Office of Dietary Supplement, National Institutes of Health, Bethesda, MD. All authors have no personal or financial conflict of interest and have not entered into any agreement that could interfere with our access to the data on the research or on our ability to analyze the data independently, to prepare manuscripts, and to publish them.
Abbreviations
- CRC
colorectal cancer
- GSE
grape seed extract
- CAM
complimentary and alternative medicine
- CDKIs
cyclin-dependent kinase inhibitors
References
- 1.www.cancer.org
- 2.Campos FG, Logullo Waitzberg AG, Kiss DR, Waitzberg DL, Habr-Gama A, et al. Diet and colorectal cancer: current evidence for etiology and prevention. Nutr Hosp. 2005;20:18–25. [PubMed] [Google Scholar]
- 3.McEachrane-Gross FP, Liebschutz JM, Berlowitz D. Use of selected complementary and alternative medicine (CAM) treatments in veterans with cancer or chronic pain: a cross-sectional survey. BMC Complement Altern Med. 2006;6:34. doi: 10.1186/1472-6882-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.http://nccam.nih.gov/
- 5.Terra X, Valls J, Vitrac X, Mérrillon JM, Arola L, et al. Grape-seed procyanidins act as antiinflammatory agents in endotoxin-stimulated RAW 264.7 macrophages by inhibiting NFkB signaling pathway. J Agric Food Chem. 2007;55:4357–4365. doi: 10.1021/jf0633185. [DOI] [PubMed] [Google Scholar]
- 6.Smullen J, Koutsou GA, Foster HA, Zumbé A, Storey DM. The antibacterial activity of plant extracts containing polyphenols against Streptococcus mutans. Caries Res. 2007;41:342–349. doi: 10.1159/000104791. [DOI] [PubMed] [Google Scholar]
- 7.Vitseva O, Varghese S, Chakrabarti S, Folts JD, Freedman JE. Grape seed and skin extracts inhibit platelet function and release of reactive oxygen intermediates. J Cardiovasc Pharmacol. 2005;46:445–451. doi: 10.1097/01.fjc.0000176727.67066.1c. [DOI] [PubMed] [Google Scholar]
- 8.Uchida S, Hirai K, Hatanaka J, Hanato J, Umegaki K, et al. Antinociceptive effects of St. John's wort, Harpagophytum procumbens extract and Grape seed proanthocyanidins extract in mice. Biol Pharm Bull. 2008;31:240–245. doi: 10.1248/bpb.31.240. [DOI] [PubMed] [Google Scholar]
- 9.Nair MP, Kandaswami C, Mahajan S, Nair HN, Chawda R, et al. Grape seed extract proanthocyanidins downregulate HIV-1 entry coreceptors, CCR2b, CCR3 and CCR5 gene expression by normal peripheral blood mononuclear cells. Biol Res. 2002;35:421–431. doi: 10.4067/s0716-97602002000300016. [DOI] [PubMed] [Google Scholar]
- 10.Yamakoshi J, Kataoka S, Koga T, Ariga T. Proanthocyanidin-rich extract from grape seeds attenuates the development of aortic atherosclerosis in cholesterol-fed rabbits. Atherosclerosis. 1999;142:139–149. doi: 10.1016/s0021-9150(98)00230-5. [DOI] [PubMed] [Google Scholar]
- 11.Vinson JA, Mandarano MA, Shuta DL, Bagchi M, Bagchi D. Beneficial effects of a novel IH636 grape seed proanthocyanidin extract and a niacin-bound chromium in a hamster atherosclerosis model. Mol Cell Biochem. 2002;240:99–103. doi: 10.1023/a:1020611925819. [DOI] [PubMed] [Google Scholar]
- 12.Bagchi D, Sen CK, Ray SD, Das DK, Bagchi M, et al. Molecular mechanisms of cardioprotection by a novel grape seed proanthocyanidins extract. Mutat Res. 2003;523-524:87–97. doi: 10.1016/s0027-5107(02)00324-x. [DOI] [PubMed] [Google Scholar]
- 13.Pinent M, Blay M, Bladé MC, Salvadó MJ, Arola L, et al. Grape seed-derived procyanidins have an antihyperglycemic effect in streptozotocin-induced diabetic rats and insulinomimetic activity in insulin-sensitive cell lines. Endocrinology. 2004;145:4985–4990. doi: 10.1210/en.2004-0764. [DOI] [PubMed] [Google Scholar]
- 14.Bagchi D, Garg A, Krohn RL, Bagchi M, Tran MX, et al. Oxygen free radical scavenging abilities of vitamins C and E, and a grape seed proanthocyanidin extract in vitro. Res Commun Mol Pathol Pharmacol. 1997;95:179–189. [PubMed] [Google Scholar]
- 15.Bomser JA, Singletary KW, Wallig MA, Smith MA. Inhibition of TPA-induced tumor promotion in CD-1 mouse epidermis by a polyphenolic fraction from grape seeds. Cancer Lett. 1999;135:151–157. doi: 10.1016/s0304-3835(98)00289-4. [DOI] [PubMed] [Google Scholar]
- 16.Engelbrecht AM, Mattheyse M, Ellis B, Loos B, Thomas M, et al. Proanthocyanidin from grape seeds inactivates the PI3-kinase/PKB pathway and induces apoptosis in a colon cancer cell line. Cancer Lett. 2007;258:144–153. doi: 10.1016/j.canlet.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 17.Raina K, Singh RP, Agarwal R, Agarwal C. Oral grape seed extract inhibits prostate tumor growth and progression in TRAMP mice. Cancer Res. 2007;67:5976–5982. doi: 10.1158/0008-5472.CAN-07-0295. [DOI] [PubMed] [Google Scholar]
- 18.Agarwal C, Veluri R, Kaur M, Chou SC, Thompson JA, et al. Fractionation of high molecular weight tannins in grape seed extract and identification of procyanidin B2-3,3′-di-O-gallate as a major active constituent causing growth inhibition and apoptotic death of DU145 human prostate carcinoma cells. Carcinogenesis. 2007;28:1478–1484. doi: 10.1093/carcin/bgm045. [DOI] [PubMed] [Google Scholar]
- 19.Kaur M, Singh RP, Gu M, Agarwal R, Agarwal C. Grape seed extract inhibits in vitro and in vivo growth of human colorectal carcinoma cells. Clin Cancer Res. 2006;12:6194–6202. doi: 10.1158/1078-0432.CCR-06-1465. [DOI] [PubMed] [Google Scholar]
- 20.Kijima I, Phung S, Hur G, Kwok SL, Chen S. Grape seed extract is an aromatase inhibitor and a suppressor of aromatase expression. Cancer Res. 2006;66:5960–5967. doi: 10.1158/0008-5472.CAN-06-0053. [DOI] [PubMed] [Google Scholar]
- 21.Kaur M, Agarwal R, Agarwal C. Grape seed extract induces anoikis and caspase-mediated apoptosis in human prostate carcinoma LNCaP cells: possible role of ataxia telangiectasia mutated-p53 activation. Mol Cancer Ther. 2006;5:1265–1274. doi: 10.1158/1535-7163.MCT-06-0014. [DOI] [PubMed] [Google Scholar]
- 22.Hu H, Qin YM. Grape seed proanthocyanidin extract induced mitochondria-associated apoptosis in human acute myeloid leukaemia 14.3D10 cells. Chin Med J (Engl) 2006;119:417–421. [PubMed] [Google Scholar]
- 23.Veluri R, Singh RP, Liu Z, Thompson JA, Agarwal R, et al. Fractionation of grape seed extract and identification of gallic acid as one of the major active constituents causing growth inhibition and apoptotic death of DU145 human prostate carcinoma cells. Carcinogenesis. 2006;27:1445–1453. doi: 10.1093/carcin/bgi347. [DOI] [PubMed] [Google Scholar]
- 24.Singh RP, Tyagi AK, Dhanalakshmi S, Agarwal R, Agarwal C. Grape seed extract inhibits advanced human prostate tumor growth and angiogenesis and upregulates insulin-like growth factor binding protein-3. Int J Cancer. 2004;108:733–740. doi: 10.1002/ijc.11620. [DOI] [PubMed] [Google Scholar]
- 25.Kim H, Hall P, Smith M, Kirk M, Prasain JK, et al. Chemoprevention by grape seed extract and genistein in carcinogen-induced mammary cancer in rats is diet dependent. J Nutr. 2004;134:3445S–3452S. doi: 10.1093/jn/134.12.3445S. [DOI] [PubMed] [Google Scholar]
- 26.Ye X, Krohn RL, Liu W, Joshi SS, Kuszynski CA, et al. The cytotoxic effects of a novel IH636 grape seed proanthocyanidin extract on cultured human cancer cells. Mol Cell Biochem. 1999;196:99–108. [PubMed] [Google Scholar]
- 27.Agarwal C, Singh RP, Agarwal R. Grape seed extract induces apoptotic death of human prostate carcinoma DU145 cells via caspases activation accompanied by dissipation of mitochondrial membrane potential and cytochrome c release. Carcinogenesis. 2002;23:1869–1876. doi: 10.1093/carcin/23.11.1869. [DOI] [PubMed] [Google Scholar]
- 28.Sharma G, Tyagi AK, Singh RP, Chan DC, Agarwal R. Synergistic anti-cancer effects of grape seed extract and conventional cytotoxic agent doxorubicin against human breast carcinoma cells. Breast Cancer Res Treat. 2004;85:1–12. doi: 10.1023/B:BREA.0000020991.55659.59. [DOI] [PubMed] [Google Scholar]
- 29.Singletary KW, Meline B. Effect of grape seed proanthocyanidins on colon aberrant crypts and breast tumors in a rat dual-organ tumor model. Nutr Cancer. 2001;39:252–258. doi: 10.1207/S15327914nc392_15. [DOI] [PubMed] [Google Scholar]
- 30.Zhao J, Wang J, Chen Y, Agarwal R. Anti-tumor-promoting activity of a polyphenolic fraction isolated from grape seeds in the mouse skin two-stage initiation-promotion protocol and identification of procyanidin B5-3′-gallate as the most effective antioxidant constituent. Carcinogenesis. 1999;20:1737–1745. doi: 10.1093/carcin/20.9.1737. [DOI] [PubMed] [Google Scholar]
- 31.Von Kleist S, Chany E, Burtin P, King M, Fogh J. Immunohistology of the antigenic pattern of a continuous cell line from a human colon tumor. J Natl Cancer Inst. 1975;55:555–560. doi: 10.1093/jnci/55.3.555. [DOI] [PubMed] [Google Scholar]
- 32.Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. FASEB J. 2003;17:1640–1647. doi: 10.1096/fj.02-1011com. [DOI] [PubMed] [Google Scholar]
- 33.Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- 34.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 36.Golias CH, Charalabopoulos A, Charalabopoulos K. Cell proliferation and cell cycle control: a mini review. Int J Clin Pract. 2004;58:1134–1141. doi: 10.1111/j.1742-1241.2004.00284.x. [DOI] [PubMed] [Google Scholar]
- 37.Uchida T, Kinoshita T, Saito H, Hotta T. CDKN2 (MTS1/p16INK4A) gene alterations in hematological malignancies. Leuk Lymphoma. 1997;24:449–461. doi: 10.3109/10428199709055583. [DOI] [PubMed] [Google Scholar]
- 38.Shiohara M, Koike K, Komiyama A, Koeffler HP. p21WAF1 mutations and human malignancies. Leuk Lymphoma. 1997;26:35–41. doi: 10.3109/10428199709109155. [DOI] [PubMed] [Google Scholar]
- 39.Hemmati PG, Normand G, Verdoodt B, von Haefen C, Hasenjäger A, et al. Loss of p21 disrupts p14 ARF-induced G1 cell cycle arrest but augments p14 ARF-induced apoptosis in human carcinoma cells. Oncogene. 2005;24:4114–4128. doi: 10.1038/sj.onc.1208579. [DOI] [PubMed] [Google Scholar]
- 40.Li W, Sanki A, Karim RZ, Thompson JF, Soon Lee C, et al. The role of cell cycle regulatory proteins in the pathogenesis of melanoma. Pathology. 2006;38:287–301. doi: 10.1080/00313020600817951. [DOI] [PubMed] [Google Scholar]
- 41.Owa T, Yoshino H, Yoshimatsu K, Nagasu T. Cell cycle regulation in the G1 phase: a promising target for the development of new chemotherapeutic anticancer agents. Curr Med Chem. 2001;8:1487–1503. doi: 10.2174/0929867013371996. [DOI] [PubMed] [Google Scholar]
- 42.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 44.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 45.Melet A, Song K, Bucur O, Jagani Z, Grassian AR, et al. Apoptotic pathways in tumor progression and therapy. Adv Exp Med Biol. 2008;615:47–79. doi: 10.1007/978-1-4020-6554-5_4. [DOI] [PubMed] [Google Scholar]
- 46.Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectal cancer: genetics of development and metastasis. J Gastroenterol. 2006;41:185–192. doi: 10.1007/s00535-006-1801-6. [DOI] [PubMed] [Google Scholar]