

Summary
The c-myc proto-oncogene, crucial for the progression of many human cancers, has been implicated in key cellular processes in diverse cell types, including endothelial cells that line the blood vessels and are critical for angiogenesis. The de novo differentiation of endothelial cells is known as vasculogenesis, while the growth of new blood vessels from pre-existing vessels is known as angiogenesis. To ascertain the function of c-myc in vascular development, we deleted c-myc in selected cell lineages. Embryos lacking c-myc in endothelial and hematopoietic lineages phenocopied those lacking c-myc in the entire embryo proper. At embryonic day (e)10.5, both mutant embryos were grossly normal, had initiated primitive hematopoiesis, and both survived until e11.5–12.5, longer than the complete null. However, they progressively developed defective hematopoiesis and angiogenesis. The majority of embryos lacking c-myc specifically in hematopoietic cells phenocopied those lacking c-myc in endothelial and hematopoietic lineages, with impaired definitive hematopoiesis as well as angiogenic remodeling. c-myc is required for embryonic hematopoietic stem cell differentiation, through a cell-autonomous mechanism. Surprisingly, c-myc is not required for vasculogenesis in the embryo. c-myc deletion in endothelial cells does not abrogate endothelial proliferation, survival, migration, or capillary formation. Embryos lacking c-myc in a majority of endothelial cells can survive beyond e12.5. Our findings reveal that hematopoiesis is a major function of c-myc in embryos and support the notion that c-myc functions in selected cell lineages rather than a ubiquitous manner in mammalian development.
Keywords: c-myc, angiogenesis, vasculogenesis, mouse development, hematopoiesis
Introduction
The c-myc proto-oncogene encodes a basic helix-loop-helix/leucine zipper transcription factor that is short-lived but rapidly induced upon serum stimulation. Its deregulation is associated with a wide range of human cancers (Adhikary and Eilers, 2005; Evan et al., 2005; Grandori et al., 2000; Pelengaris et al., 2002), and its overexpression is highly tumorigenic in many types of tissues in animals. A large body of work performed in cultured cells shows that c-Myc is expressed broadly and functions as a central regulator of normal cellular programs including cell proliferation, differentiation, growth, survival, and migration in many cell types (Grandori et al., 2000). While these studies have made tremendous contributions to our understanding of the cellular mechanisms underlying the oncogenic effects of c-Myc, the physiological function of this protein remains largely unknown.
c-myc null embryos exhibit severe developmental abnormalities in a wide range of organs and die early in gestation before 10.5 days post coitum (dpc) (Davis et al., 1993; Trumpp et al., 2001), supporting the notion that c-myc is essential for a broad range of organ development. Recent studies suggest that c-Myc is required for the proliferation of progenitor cells and the self-renewal of stem cells (Murphy et al., 2005). In the intestine, c-Myc is expressed in the proliferative zone of intestinal crypts, where putative intestinal stem cells reside, and is essential for the formation of these crypts (Bettess et al., 2005; Muncan et al., 2006). Similarly, in the skin epidermis, c-Myc is expressed in the proliferative basal layer and bulge region, where stem and progenitor cells are located (Bull et al., 2001). These new in vivo findings suggest that c-myc is uniquely required in the stem and progenitor cell compartments.
The role of c-myc in the development of the vascular system is of particular interest because it is critical not only for all aspects of normal tissue function but also for pathological tumor growth and survival. Endothelial cells (ECs) line blood vessels, and are the primary cell type responsible for blood vessel function and regeneration. Hematopoietic cells (HCs) give rise to the blood cells of the circulatory system. Differentiation of these two lineages first occurs in yolk sac blood islands, where ECs and HCs may arise from a common mesoderm-derived precursor, the hemangioblast (Cumano and Godin, 2007; Eichmann et al., 2002; Ema and Rossant, 2003). During vascular morphogenesis ECs coalesce to assemble a primitive vascular network composed of a capillary plexus with uniform caliber and honeycomb appearance. This formation of blood vessels by de novo EC differentiation is known as vasculogenesis (Adams and Alitalo, 2007; Carmeliet, 2005). The primitive capillary plexus subsequently undergoes growth and remodeling to shape the mature vascular tree. Angiogenesis is the process of new blood vessel growth from existing vessels (Folkman, 2006; Hanahan and Folkman, 1996; Thurston et al., 2007). c-Myc has been shown to regulate angiogenesis by promoting the expression of pro-angiogenic factors such as VEGF in stromal cells while inhibiting the expression of the anti-angiogenic factor thrombospondin-1 (Baudino et al., 2002; Dews et al., 2006; Knies-Bamforth et al., 2004; Mezquita et al., 2005; Shchors et al., 2006; Watnick et al., 2003). c-Myc is reportedly required for vasculogenesis during development, as c-myc null embryos have no detectable blood vessels (Baudino et al., 2002). However, whether c-Myc in ECs plays an essential role in vasculogenesis or angiogenesis is currently unknown.
To ascertain the cell type-specific role of c-Myc during vascular development, we generated conditional knockouts (CKs) of the c-myc gene in c-mycflox/flox mice using cell lineage-specific Cre lines. We were surprised to find that c-myc was not required for vasculogenesis, and that deleting c-myc in a majority of ECs was compatible with early embryo survival. In contrast, c-myc deletion was detrimental to hematopoietic lineages during development, and c-myc deletion in these lineages was sufficient to cause vascular developmental defects.
RESULTS
c-myc is essential for angiogenesis but not vasculogenesis
Gross abnormalities and developmental retardation by 9.5dpc and death by 10.5dpc have been previously observed in c-myc−/− embryos (Davis et al., 1993; Trumpp et al., 2001). Multi-organ failure, including circulatory defects, is believed to underlie this early developmental arrest, although the specific cell lineages affected are largely unknown. We therefore analyzed c-myc−/− embryos specifically for vascular defects, using whole mount immunostaining against CD31, an EC specific marker. As previously reported, these mutant embryos were developmentally retarded and displayed major developmental defects. However, we were surprised to find that major vessels such as the dorsal aorta had developed by 8.75dpc. Smaller inter-somitic and cranial vessels had also formed in the embryo proper, although they were underdeveloped compared to wild-type vessels (Fig. 1A,B). In the yolk sac, primitive vascular networks had formed, although the mutant yolk sac contained more vascular plexuses and less organized hierarchal branches than the control (Fig. 1C,D). At 10.25dpc, while the control vasculature had matured to an elaborate and well-organized system (Fig. 1E,G), the c-myc−/−embryos retained a primitive vasculature in the head (Fig. 1F) and the yolk sac (Fig. 1H). Nonetheless, embryo cross-sections revealed that like controls, c-myc−/−embryos had ECs lining their dorsal aortae, common cardinal veins, and other blood vessels (Fig. 1I,J). These results indicate that, in contrast to a previous report (Baudino et al., 2002), c-myc−/− embryos contained differentiated ECs that were capable of assembling a primitive vasculature. Subsequent vascular remodeling, however, was defective in the mutants.
Figure 1. c-myc null and Sox2-Cre-mediated c-myc deletion result in anemia, vascular defects, and lethality.

A–H) Anti-CD31-stained whole mount embryos (A,B,E,F) and yolk sacs (C,D,G,H) dissected at 8.75dpc (A–D) or 10.25dpc (E–H). (I and J) Cross-sections (5 μm) of anti-CD31-stained whole-mount embryos. K–N, Live embryos dissected at 11.5dpc. (O–T) Anti-CD31-stained whole-mount yolk sacs (O,P) and heads (Q–T) at 11.5dpc. Note the less developed vessels with reduced caliber (yolk sac) and number of branches (head) in the mutant animal. Da, dorsal aorta; ISV, intersomitic vessel; pp, primitive plexus; vv, vitelline vessels; CA, carotid artery; HV, primary head veins; CV, cardinal vein; nt, neural tube; Ph, pharynx. Scale bars, 200μm.
Because the detection of ECs in the c-myc−/− mutant was surprising, we examined the vasculature in another mutant embryo, in which c-myc is deleted in the entire embryo proper but retained in the placenta. We created a CK using the Sox2-Cre mouse line, in which Cre is active in all cells of epiblast origin, including the entire embryo proper, the yolk sac mesoderm, the amnion membrane, and the embryonic vessels in the placenta, but not the visceral endoderm or the extra-embryonic ectoderm (Vincent and Robertson, 2003). We verified Cre expression throughout the entire embryo by Cre reporter assay (data not shown). The Sox2-Cre;c-mycflox/−mutant progeny displayed growth retardation by 11.5dpc and died between 11.5–12.5dpc. These embryos, unlike the null mutants, appeared grossly normal without major organ defects before e10.5 (data not shown). However, at e11.5 they were anemic (Fig. 1L,N) and displayed abnormal vasculature (Fig. 1P,R,T). Anti-CD31 staining revealed abundant ECs in mutant yolk sacs and embryos proper (Fig. 1O–T), demonstrating that the mutant embryos were not defective in EC differentiation, although capillary remodeling was defective. Mutant yolk sac microvessels were more primitive, comprising disorganized capillary plexuses with larger intercapillary spaces compared with the controls. Major vitelline vessels were narrower and underdeveloped in the mutant (Fig. 1O,P). Similarly, head capillaries were more primitive. The carotid arteries were less elaborate and the primitive head veins were narrower compared to controls (Fig. 1Q–T). These findings confirmed that c-myc is not required for the onset of vasculogenesis in embryos. However, it is essential for subsequent vascular morphogenesis.
The Sox2-Cre;c-mycflox/− mutant embryos that maintained c-myc in their visceral endoderm and placenta died two days later than c-myc−/− embryos, suggesting that expression of c-myc in these extra-embryonic tissues is essential to the survival of the embryos at this stage. Consistent with this notion, c-myc is highly expressed in the ectoplacental cone starting at 6.5dpc (Downs et al., 1989) and c-Myc promotes trophoblast proliferation (Erlebacher et al., 2004). We found that by 8.25dpc, c-myc−/− placentas contained fewer trophoblasts, exhibiting defective placental morphology and cellular composition lacking trophoblast integrity. In addition, the mutant chorionic plate was thinner, and the ectoplacental cone was not integrated (Fig. S1). These findings suggest that c-myc is required for placental development, and the cellular function of c-myc in this organ requires future investigation. In this report, we have focused on the function of c-myc in the embryo proper.
Loss of c-myc in endothelial and hematopoietic cells induces similar vascular defects as loss of c-myc in the entire embryo proper
To determine if eliminating c-myc specifically from the circulatory system would result in developmental and vascular defects, we crossed c-mycflox/flox mice with Tie2-Cre;c-myc+/−mice, in which Cre is active in EC and HC lineages starting as early as 7.5dpc in the common progenitor of these two lineages (Braren et al., 2006). To assess the efficiency of Tie2-Cre-mediated c-myc deletion, we measured nuclear-specific c-Myc expression in isolated ECs by immunostaining (Fig. S3). Quantitative analysis showed that virtually no (0.62%) mutant ECs and 91.2% of control ECs had c-Myc staining by 10.5dpc (Fig. 2M). These experiments demonstrate the success of c-myc deletion.
Figure 2. Tie2-Cre-mediated c-myc deletion results in anemia, vascular defects, and lethality.

(A–F) Live embryos in yolk sac dissected at 9.5dpc (A–B), 10.5dpc (C–D), or 11.5dpc (E–F). (G–L) Anti-CD31-stained whole mount yolk sacs (G,H) and heads (I–L) at 11.5dpc. Note less developed vasculature in the mutant with reduced caliber and number of branches. Scale bar, 200μm. M–N, Efficient c-Myc deletion by Tie2-Cre did not increase N-Myc expression in 10.5dpc primary ECs. The percentage of c-Myc positive ECs is indicated in M. The percentage of N-Myc positive ECs is shown in N. “n” represents the number of cells counted in each assay. (O) Peripheral blood cell counts at 10.5dpc from control (n=19) and 7 Tie2-Cre;c-mycflox/− mutant (n=7) embryos. The numbers above the brackets show the fold decrease in the mutants over controls. *p<0.01, **p<0.05 by Student’s t test.
These embryos, like Sox2-Cre CKs, appeared grossly normal without major organ defects before e10.5 (data not shown). At 9.5dpc, the Tie2-Cre; c-mycflox/− embryos were similar to controls, with all embryos showing blood-filled vasculatures, although some were slightly paler (Fig. 2A,B). However, at 10.5dpc, the mutants appeared anemic (Fig. 2D). At 11.5dpc, the mutant yolk sacs and embryos were completely white whereas control yolk sacs displayed vessels filled with red blood cells (Fig. 2E,F). Mutant embryos were smaller than controls (Fig. 2F). No mutant embryos survived beyond 12.5dpc (Table S1). The gross abnormalities and the stage at which the phenotype occurred in Tie2-Cre CK closely resembled that of the Sox2-Cre CK embryos.
Anti-CD31 staining on 11.5dpc Tie2-Cre CK embryos also revealed a very similar vascular phenotype to the Sox2-Cre CK, with abundant ECs but more primitive capillaries and smaller major vessels in mutant yolk sacs and heads compared to the controls (Fig. 2G–L). Quantitative analysis of the yolk sac vascular defects is summarized in Fig. S2. These findings from Tie2-Cre CK embryos confirm that vasculogenesis occurred without c-myc, but further capillary remodeling was defective. Additionally, the allantois is vascularized by vasculogenesis, and we did not detect any apparent vascular defects in the Tie2-Cre CK allantoic explants (Fig. 3A,B), further suggesting that vasculogenesis occurs in the absence of c-myc.
Figure 3. Tie2-Cre-mediated c-myc deletion did not affect allantoic or placental vasculature but diminished hemogenic endothelial cells in the aorta.

(A–B) Allantoic explants stained for CD31 shows no obvious difference between the control and mutant. (C–D) Placental vasculature at 11.5dpc revealed by Tie2-LacZ reporter assay shows no obvious differences between the control and mutant. Arrowheads point to the blood vessels. (E–F) H&E stained cross-section of dorsal aorta shows hemogenic endothelial cells (arrows) seen in the control were never detected in the mutant. Scale bars, 50μm.
Expression of N-Myc, another Myc family member, driven by the c-myc promoter can functionally replace c-Myc activity in vivo (Malynn et al., 2000). To rule out the possibility that n-myc expression might compensate for the loss of c-myc in ECs, we stained purified ECs from 10.5dpc Tie2-Cre CKs with anti-N-Myc antibodies (Fig. S3). We found that N-Myc was expressed in a small fraction (7.1–8.4%) of ECs, but that this fraction was independent of the EC genotype (Fig. 2N). To rule out a possible compensatory effect from L-myc, we analyzed L-myc expression by real time RT-PCR in Sox2-Cre CK yolk sacs. While c-myc expression was significantly reduced, L-myc expression was unchanged in the mutant (data not shown). These results demonstrate that neither N-Myc nor L-Myc is likely to functionally compensate for c-Myc in ECs.
To examine whether placental vasculature is defective in this mutant, we analyzed the blood vessel structure in the placenta using a Tie2-LacZ reporter. By 11.5dpc while the mutant embryo was already pale and blood vessels in the yolk sac and embryo proper were already defective, the placental blood vessels appeared indistinguishable from the control (Fig. 3C,D). These data indicate that c-myc deletion in the ECs and HCs did not significantly affect the placental vasculature, suggesting that the death of this mutant embryo is unlikely to be due to a placental failure.
To characterize defects in the HC lineage, we counted the peripheral red blood cells (Ter119+), hematopoietic cells (CD45+), and myeloid cells (CD11b+) from yolk sacs and embryos and found that by 10.5dpc Tie2-Cre; c-mycflox/− mutants exhibited a 38-fold reduction in total blood cell number, a 13-fold reduction in CD45+ cells, and a 4-fold reduction in CD11b+ cells (Fig. 2O). H&E staining on paraffin sections also confirmed a nearly complete absence of blood cells by 11.5dpc (data not shown). Hemogenic ECs that reside in the ventral side of dorsal aorta at around 10.5dpc are thought to give rise to HSCs (de Bruijn et al., 2002; Taoudi and Medvinsky, 2007). We examined serial cross-sections of dorsal aortae from four pairs of embryos at 10.5dpc. Cells located at the ventral wall of the aorta and morphologically resembling hemogenic ECs were seen in all controls but none of the mutants (Fig. 3E,F). These findings suggest that removing c-myc from ECs and HCs is sufficient to induce the hematopoietic, angiogenic, and survival defects observed in embryos harboring a global c-myc deletion.
c-myc deletion in HCs is sufficient to induce vascular defects and embryonic lethality
To delineate the effect of c-myc deletion in hematopoietic lineages on vascular development, we examined embryos in which c-myc had been deleted specifically in HC lineages using Vav-iCre. Vav-iCre has been shown to mediate gene excision in adult HCs (de Boer et al., 2003). We thus analyzed Vav-iCre activity in embryos using a Rosa26R-LacZ reporter according to our established method (Braren et al., 2006). At 11.5dpc, we found that Vav-iCre was active almost exclusively in fetal liver HCs (Fig. S4A,C) and in some circulating blood cells in the yolk sac (Fig. S4B), but not in the endothelium or any other tissues (Fig. S4D). This result indicates that Vav-iCre mediated c-myc deletion in HCs without affecting ECs or any other cell types. We assessed Vav-iCre activity in individual embryos by FACS analysis in 11.5dpc fetal liver HCs using the Rosa26YFP reporter (Srinivas et al., 2001). The fraction of HCs that express Cre varied among individual embryos in all three HC lineages tested, with averages around 50% (Fig. S4K).
About 60% of Vav-iCre;c-mycflox/− mutants (27/43) appeared anemic at 11.5dpc (Fig. 4B), with paler and smaller fetal livers compared to controls (Fig. S4I, J). A similar % of mutants died around 12.5dpc, a stage similar to the Tie2-Cre and Sox2-Cre CKs. About 16% of mutants (7/43) showed various degrees of hemorrhaging in the embryo proper (Fig. S4E,G). About 21% of mutants (9/43) survived through 12.5dpc but developed anemia and died by 15.5dpc. None of the Vav-iCre mutants survived to birth. It is likely that non-uniform Cre activity among individuals resulted in these variable phenotypes (Fig. S4K). Nonetheless, deletion of c-myc solely in HCs resulted in complete embryonic lethality of all embryos examined.
Figure 4. Vav-iCre- mediated c-myc deletion results in cytopenia, vascular defects, and lethality.

(A–B) Live embryos at 11.5dpc. (C–H) Anti-CD31-stained whole-mount yolk sacs (C,D) and heads (E–H) at 11.5dpc. CA, internal carotid arteries; HA, primary head veins. Scale bars, 200 μm. (I) Peripheral blood cell counts of control (n=16) and Vav-iCre;c-mycflox/− mutant (n=8) embryos. The numbers above brackets show the fold decrease in the mutants over controls. *p<0.01 and **p<0.05 by Student’s t test. (J) Fetal liver hematopoietic cells.
To visualize the vasculature of the Vav-iCre;c-mycflox/− mutant embryos, we performed whole-mount anti-CD31 staining on 11.5dpc embryos. We found that the vascular defects in these anemic embryos resembled those in Tie2-Cre- and Sox2-Cre CKs (see Figs. 1P,R,T, 2H,J,L). While the overall vascular patterning in Vav-iCre;c-mycflox/− embryos proper and yolk sacs was in place, the vessels were narrower and the vascular network appeared underdeveloped and primitive compared to controls (Fig. 4D,F,H). Taken together, these results suggest that c-myc deletion in HC lineages alone likely accounts for the anemia, embryonic lethality, and vascular developmental defects induced by c-myc deletion in ECs and HCs combined.
To quantify the hematopoietic defects in 11.5dpc Vav-iCre CKs, we performed HC counts in peripheral blood (Fig. 4I) and fetal liver cells (Fig. 4J). We found a several fold decrease of cells (7.6 fold in Ter119+ cells, 4.3 fold in CD45+, and 4.6 fold in CD11b+ cells) in the mutant peripheral blood. The mutants’ fetal livers had cytopenia and contained 12.5 fold fewer cells than their littermate controls. In addition, we used Lin markers, containing CD3e, CD11b, CD45R/B220, Ter119, Ly-6G and Ly-6C to label the committed hematopoietic lineages, which include T lymphocytes, B lymphocytes, monocytes/macrophages, NK cells, erythrocytes, and granulocytes. We found the proportion of committed (Lin+) cells was reduced while that of uncommitted cells (Lin−) was increased in Vav-iCre;c-mycflox/− embryos. Among the Lin− cells, the proportion of KLS-HSCs (c-Kit+, Lin−/lo, Sca-1+) (Ivanova et al., 2002) in the Vav-iCre;c-mycflox/− fetal liver was increased, while the proportion of c-Kithi, Lin− cells was significantly decreased. In summary, these results demonstrate that elimination of c-myc in HCs by Vav-iCre compromised definitive hematopoiesis.
c-myc deletion in the majority of ECs and a subset of HCs results in partial survival
Since c-myc deletion in HCs alone using Vav-iCre resulted in defects similar to c-myc deletion from both HCs and ECs (with Sox2-Cre or Tie2-Cre), we investigated the effect of endothelial c-myc expression on vascular development. For this experiment, we needed a Cre line that would remove c-myc in the ECs but not the HCs, however, such a reagent is not currently available. We therefore chose the Tie1-Cre line, with Cre active in the majority of ECs and a subset of HCs by 10.5dpc (Gustafsson et al., 2001) (Fig. S5). We verified Tie1-Cre activity using the Rosa26R reporter and double immunostaining for CD31 and β-galactosidase. Approximately 80% of CD31 positive cells were Cre active at 10.5dpc (Fig. S5). We also characterized Tie1-Cre expression in HCs by FACS using the RosaYFP reporter. Cre was active in approximately 54% of myeloid cells, 26% of erythroid cells, and 15% of lymphocyte precursor cells at 11.5dpc (Fig. S6). These results suggest that Tie1-Cre mediated c-myc deletion in the majority of ECs and a subset of HCs.
By 12.5dpc, we did not detect any defects in Tie1-Cre;c-mycflox/− mutants. By 17.5 dpc we observed anemic, dying mutant embryos. Remarkably, half of the mutants survived to birth and a third to post-weaning (Fig. 5A). Surviving mutant adults appeared normal. These results demonstrate that c-myc deletion in a majority of ECs is compatible with embryo survival.
Figure 5. Tie1-Cre-mediated c-myc deletion results in partial survival.


(A) Animal survival rate. (B) Peripheral blood cell counts from control (n=20) and Tie1-Cre;c-mycflox/− mutant (n=6) embryos. The numbers above brackets show the fold decrease in the mutants over controls. *p<0.01 and **p<0.05 by Student’s t test. (C) Fetal liver hematopoietic cells.
To examine the HC development in these mutants, we performed HC counts from the peripheral blood (Fig. 5B) and fetal livers (Fig. 5C) of 11.5dpc Tie1-Cre CKs. We found a less than two-fold decrease in peripheral blood cell lineages (Ter119+, CD45+, CD11b+) in the mutants (Fig. 5B). The mutant fetal liver cell number, composed mainly of HCs, was half of that in the control. The proportion of committed (Lin+) and un-committed (Lin−) cells in fetal livers was similar between the mutant and control (Fig. 5C). Therefore, HC lineages were much less depleted in Tie1-Cre;c-mycflox/− than in Vav-iCre;c-mycflox/− embryos (Fig. 4K,L). The mild HC defects likely permitted the partial survival of the Tie1-Cre CKs.
c-Myc-depleted ECs show no significant cell biological defects
Most Tie1-Cre CKs examined at 11.5dpc showed no detectable abnormalities, suggesting that c-myc expression in the majority of ECs is not essential for vascular development. To verify whether c-myc is required in ECs for blood vessel formation, we examined the in vivo and in vitro behaviors of Tie2-Cre CK ECs.
We analyzed EC proliferation in vivo by BrdU incorporation and did not observe any significant difference in proliferation rates of aortic endothelium between control and mutant embryos (Fig. 6A). We also did not detect an obvious proliferative difference in isolated ECs from BrdU labeled embryos following maternal BrdU injection (data not shown).
Figure 6. c-myc deficient ECs exhibit no detectable defects in proliferation, survival, morphology, motility, or tube formation.
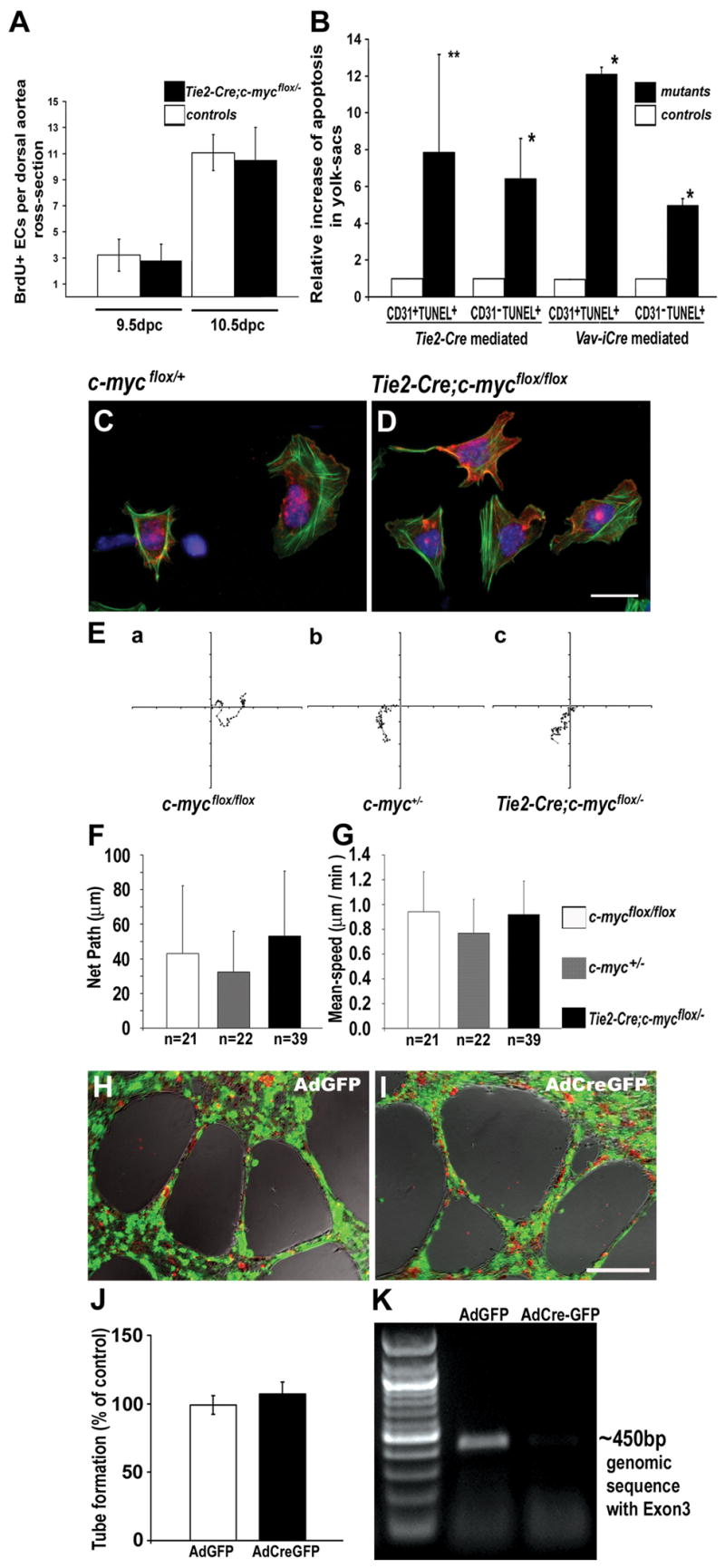
(A) BrdU+ ECs in the dorsal aortae. Cross sections were stained with anti-CD31 and anti-BrdU antibodies. (B) Fold increases in apoptosis in yolk sac ECs and non-ECs. Cross sections of Tie2-Cre;c-mycflox/− (at 10.5 dpc) and Vav-iCre;c-mycflox/− (at 11.5dpc) mutant (black bars) and control (white bars) yolk sacs were stained with anti-CD31 and for TUNEL. TUNEL+ CD31+ or TUNEL+ CD31− cells were quantified. **p<0.05, *p<0.01 by Student’s t test. Data were collected from 4 pairs of embryos from 2 different litters. (C and D) Phalloidin (green), anti-CD31 (red), and DAPI (blue) stained ECs isolated from 10.5dpc embryos. (E–G) Random movement of cultured ECs from 10.5dpc embryos monitored by timelapse microscopy. Representative EC migration paths are shown (E), and quantitative analysis demonstrated no significant difference either in the net-path of cell-migration (F, p=0.083) or in the average migration speed (G, p=0.090) in two independent experiments. The ANOVA two-tailed test was used in the statistical analysis. (H–K) No detectable defects in EC tube formation. mycflox/flox ECs were purified from adult vena cava. Cells 48 hours after infection with GFP fusion Cre-expressing adenovirus AdCreGFP or GFP expression control virus AdGFP were plated on Matrigel, and cultures were photographed 20 hours later (H,I). The results from four independent experiments were analyzed by two-tailed t-test (J, P=0.1916). Green, GFP; Red, DiI-AcLDL labeling ECs. Genomic DNA PCR analysis demonstrates the deletion of c-myc floxed sequence by AdCreGFP but not AdGFP (K). Scale bar, 10 μm (C and D); 200 μm (H and I).
To determine whether loss of c-myc affected EC survival in vivo, we performed TUNEL assays on cryosections of yolk sacs. We found a significant increase in TUNEL-staining in Tie2-Cre CKs compared to the controls at 10.5dpc (Fig. 6B). However, increased TUNEL staining affected both ECs and non-ECs, even though non-ECs had a functional c-myc gene. These data suggest that the increase in EC death could be a secondary effect due to general poor embryo health rather than a direct result of c-myc deletion in these cells. In summary, we were unable to detect significant changes in cell proliferation and survival that could be ascribed to loss of c-myc specifically in ECs.
To examine the morphology and behavior of c-myc-deficient ECs, we isolated ECs from Tie2-Cre CKs at 10.5dpc and cultured them for 5 hours on fibronectin-coated culture plates. Using anti-CD31 and phalloidin staining, we observed neither morphological differences nor changes in the organization of the actin cytoskeleton between control and c-myc null ECs (Fig. 6C,D).
Normal angiogenesis relies on the ability of ECs to migrate (Beck and D’Amore, 1997). To assess the motility of c-myc-deficient ECs, we performed time-lapse video-microscopy of c-myc null and control ECs isolated from 10.5dpc embryos and cultured on fibronectin. The paths of both mutant and control ECs were random and indistinguishable from one another, as shown by our measurements of net path length and average speed (Fig. 6E,F,G). These results demonstrate that c-myc null ECs are not defective in cell motility in vitro.
To further address the angiogenic potential of c-myc null ECs, we tested their ability to form endothelial tubes in vitro (Fig. 6H,I). Because this assay requires a large number of ECs, we isolated ECs from the vena cava of adult c-mycflox/flox mice and removed their c-myc gene using an adenovirus-mediated Cre (AdCreGFP) deletion system. FACS analysis of GFP expression in the cultured ECs showed that the efficiency of adenovirus infection was 98% (Fig. 6J), and PCR analysis of genomic DNA confirmed that the c-myc gene was excised in the majority of the cells (Fig. 6K). However, we found no statistically significant difference in the number of branch points (data not shown) or the lengths of tubes formed by mutant and control ECs on the Matrigel surface (Fig. 6I,J). These findings suggest that c-myc null ECs are not defective in cell migration or capillary morphogenesis.
Deletion of c-myc in HCs leads to reduction of pro-angiogenic factors crucial for vascular morphogenesis
HCs have been increasingly recognized as significantly contributing to angiogenesis by modulating the production of proangiogenic factors (Tordjman et al., 2001; Kopp et al., 2006; Shojaei et al., 2007). We therefore examined the mRNA levels ofvarious proangiogenic factors, including pdgf-a, mmp2, and il-1β, in whole embryos, using Quantitative-PCR analysis. We found that mRNA levels of these genes were significantly decreased in the anemic Vav-iCre;c-mycflox/− embryos compared to their control littermates (Fig. 7A). In contrast, levels of VEGF transcripts were dramatically increased, indicating a hypoxic response in the embryos suffering anemia (see Fig. 4A,B). Similarly, elevated VEGF protein was found in Tie2-Cre;c-mycflox/−embryos (Fig. 7B). These results demonstrate that c-Myc deficiency-induced hematopoietic defects may have reduced expression of certain proangiogenic factors, thereby hindering normal vascular morphogenesis.
Figure 7. Expression of proangiogenic factors in Vav-iCre;c-mycflox/− and Tie2-Cre;c-mycflox/−mutant embryos.

(A) mRNA levels of proangiogenic factors in Vav-iCre;c-mycflox/− at 11.25dpc. Real-time PCR was performed using total RNA from 5 mutants and 15 control embryos (Vav-iCre;c-mycflox/+, c-myc+/−, or Vav-iCre;c-myc+/−). The statistical significance of the differences in means between controls and mutants are listed by their P values. (B) Increase in VEGF protein levels by ELISA in control and Tie2-Cre;c-mycflox/− embryos (n=5). VEGF levels in the Tie2-Cre mutant embryos were significantly elevated (*p<0.01).
Discussion
To ascertain the physiological functions of c-Myc in development, we examined embryos lacking c-myc completely, in the embryo proper but not in extra-embryonic tissues, in hematopoietic and endothelial lineages, and in hematopoietic cells specifically. Our findings demonstrate that c-myc is not required for vasculogenesis in the embryo but can indirectly control angiogenesis through its vital role in hematopoiesis. Deletion of c-myc in HCs alone is lethal, sufficient to elicit both hematopoietic and vascular defects.
c-Myc plays an essential role in embryonic hematopoiesis
Fetal hematopoiesis begins with primitive hematopoietic differentiation in blood islands of the yolk sac at 7.5dpc and lasts until 10.5dpc in mice. Definitive hematopoiesis, which generates enucleated erythrocytes among other hematopoietic lineages like HSCs, starts at 10.5dpc in the aorta-gonad-mesonephros region. Concurrently, HSCs colonize the developing fetal liver. The murine placenta also harbors HSCs during midgestation. Around birth, hematopoiesis translocates to the bone marrow (BM). Adult and fetal hematopoiesis differ in the types of niches in which they occur, as well as in the capacity of adult versus fetal HSCs to renew, proliferate, and differentiate (Cumano and Godin, 2007; Mikkola and Orkin, 2006; Wilson and Trumpp, 2006). The role of c-Myc in adult hematopoiesis in BM has been reported (Wilson et al., 2004). Our experiments demonstrate c-Myc’s essential function in fetal hematopoiesis.
Our data suggest that HCs undergo primitive differentiation in the absence of c-myc. At 9.5dpc, prior to definitive hematopoiesis, the majority of Tie2-Cre CKs were indistinguishable from the controls, with blood-filled circulatory systems. This phenotype is in sharp contrast to that of embryos lacking scl, a gene required for the differentiation of primitive hematopoietic cells. scl null embryos are devoid of blood cells with no sign of hematopoiesis at 9.5dpc (Robb et al., 1995). One concern is whether Tie2-Cre mediated c-myc deletion occurs early enough to assess its requirement in primitive hematopoiesis. We have previously reported that this Tie2-Cre is active early in the blood island precursors and mediates efficient gene excision by 9.5dpc (Braren et al., 2006). The presence of blood cells in both the c-myc null (Davis et al., 1993) and Sox2-Cre CKs (data not shown) further supports the idea that primitive hematopoiesis occurs in the absence of c-myc. From 9.5dpc, the Tie2-Cre CKs developed progressive cytopenia. The likely cause of the primitive hematopoietic failure is the reduced survival of HCs (Dubois et al. submitted). Thus, c-myc is not required for the initiation of primitive hematopoiesis but is essential to sustain primitive hematopoiesis.
Our data from Vav-iCre CKs provide evidence that c-Myc is also required for definitive hematopoiesis, through a cell-autonomous mechanism. The Vav1 promoter induces transgene expression in definitive but not primitive HCs (Ogilvy et al., 1999). Furthermore, we show that Vav-iCre mediates gene excision specifically in fetal liver HCs. The fact that the Vav-iCre CK developed severe cytopenia demonstrates that c-myc in HCs is essential for definitive hematopoiesis.
The absence of c-Myc in definitive hematopoiesis seems to affect the differentiation of HSCs, resembling c-myc deficient adult hematopoiesis in BM (Wilson et al., 2004). The proportion of uncommitted HCs, KLS-HSCs and c-Kitlow progenitor cells increased in the Vav-iCre fetal liver. However, the proportion of c-Kithi progenitor cells decreased. This reduction could be a genuine decrease of c-Kithi progenitors in mutants or a loss of cell surface c-Kit expression on otherwise functional HSCs, as has been observed following myeloid ablation (Randall and Weissman, 1997). However, the proportion of the committed HCs, including Ter119+, CD45+, and CD11b+ lineages, decreased significantly in Vav-iCre CKs. In addition, the total number of isolated fetal liver cells, composed primarily of HCs, was decreased. These data suggest that the mutant HSCs and progenitor cells can survive and divide but subsequent differentiation into HCs are defective. Our findings provide crucial evidence that c-Myc is required in a cell-autonomous fashion for HSC differentiation. This finding is complementary to the finding in Sox2-Cre CKs (Dubois et. al. companion article).
The fact that mutant embryos in which c-myc is deleted in the entire embryo proper but not the placenta survived two additional days beyond the survival of the complete null embryo shows that c-myc is essential for placental development. It is currently unknown which specific cell lineage(s) c-Myc may function in and what precise role c-Myc may play in the placenta. However, this finding is intriguing in light of recent discoveries that the placenta is an active site for HSC development (Gekas et al., 2005; Mikkola et al., 2005). Given the crucial function of c-Myc in both fetal and adult hematopoiesis, it is plausible that c-Myc may function in placental HSCs. The c-Myc CKs described here may serve as an excellent model to elucidate the molecular control of placental hematopoiesis.
c-Myc is not required for embryonic vasculogenesis
Previous reports suggest that c-myc is required for vasculogenesis (Baudino et al., 2002), and down-regulation of c-myc in cultured ECs leads to cellular senescence (Guney and Sedivy, 2006). In contrast, we show here that vasculogenesis occurs in the absence of c-myc. Our method of gene excision is efficient, leading to the deletion of the entire coding region for c-Myc (Trumpp et al., 2001). We therefore respectfully disagree with this earlier conclusion. Gene disruption in the two studies was achieved by targeting a similar region of c-myc, so the reason for the discrepancy between our observations and those of Baudino et al. are currently unclear. However, we confirmed our results in three independent c-myc deficient mutants (c-myc null, Sox2-Cre and Tie2-Cre CKs), and the presence of ECs in c-myc deleted embryos was also verified in a different laboratory (Dubois et al. companion article). We conclude that c-myc is not required for vasculogenesis in embryos.
At the cellular level, c-myc-deficient ECs did not exhibit detectable defects in cell proliferation, survival, migration, or even capillary morphogenesis. These results are in contrast to the report that c-Myc is essential for EC proliferation in culture (Guney and Sedivy, 2006). We performed proliferation assays using primary ECs to closely mimic in vivo conditions. In addition, neither N- nor L-Myc compensated for the loss of c-Myc. We also show that about one third of Tie1-Cre CKs, in which c-Myc was deleted in the majority of ECs, survived into adulthood without apparent abnormalities. This result suggests that widespread deletion of c-myc in the endothelium is compatible with survival. Taken together, our observations suggest that abrogating c-Myc in ECs may not disrupt angiogenesis, and c-Myc likely regulates angiogenesis through a non-cell-autonomous fashion.
Hematopoietic abnormalities caused by c-myc deletion lead to defects in angiogenesis
Although a primitive vascular network formed in the absence of c-myc, its angiogenic remodeling into complex vascular tree was abnormal. Because c-Myc-deficient ECs appear to function normally, we propose that defective HCs cause the vascular defects observed in our c-myc CK embryos. Supporting this notion, vascular defects in the Vav-iCre CKs, where c-myc is deleted specifically in the hematopoietic lineage, resembled those in the Tie2-Cre CK embryos.
HCs are known to affect angiogenesis through hemodynamic influence (Lucitti et al., 2007) and oxidative stress, such as hypoxia (Jones et al., 2004; Ramirez-Bergeron et al., 2006), The reduced hematocrit in the mutants likely changes the viscosity of the blood, and hence alters the hemodynamic forces required for growth and maintenance of vessel size (Lucitti et al., 2007). Moreover, both Vav-iCre and Tie2-Cre CK embryos were anemic by 11.5dpc. Hypoxia was evident by elevated VEGF levels, a common consequence of embryos in hypoxia conditions. Hypoxia causes pan-tissue damage via apoptosis (Graven et al., 1993). Therefore, both low hematocrit and the hypoxia-mediated apoptosis could contribute to the vascular defects observed in the c-myc mutants.
However, lack of hemodynamic force and increased hypoxia stress are not the only explanation for the absence of vascular remodeling in c-myc mutants. HCs secrete factors capable of promoting angiogenesis in a paracrine manner (Okamoto et al., 2005; Okuda et al., 1996). Lack of proangiogenic factors from HCs are responsible for angiogenic defects in Arnt (Ramirez-Bergeron et al., 2006) and AML1 (Takakura et al., 2000) mutants. Since Tie-2Cre;c-mycflox/− embryos have hematopoietic defects, they might also lack HC-derived proangiogenic factors, which could explain their angiogenesis defects.
Consistent with this hypothesis, pale Vav-iCre;c-mycflox/− embryos exhibited a significant decrease of Il-1βand mmp2 mRNA at 11.5dpc. IL-1β is secreted primarily from monocytes and macrophages. In the anemic Vav-iCre;c-mycflox/− embryos, the significant decrease of CD11b+ myeloid cells in fetal livers and peripheral blood likely explains the decrease of IL-1β expression and secretion. IL-1β and MMPs were recently found to form an axis to regulate the bioavailability of VEGF in angiogenesis (Shchors et al., 2006). IL-1β mobilizes VEGF from the extracellular matrix (ECM) to ECs during active angiogenesis, via its ability to promote expression and proteolytic activation of stromal MMPs (Mountain et al., 2007; Shchors et al., 2006). MMPs not only modulate the ECM but also cleave the ECM binding domain of VEGF and release isoforms of VEGF to ECs (Bergers et al., 2000). Depletion of these and other paracrine factors originating from hematopoietic cells is likely to contribute to the impaired angiogenesis in the mutant, despite the elevated VEGF mRNA levels.
We therefore suggest that a combination of defects including reduction in hemodynamic stress and hypoxia-induced apoptosis with a shortage in proangiogenic factors contributes to the vascular defects in the mid-gestation c-myc mutant embryos. If these vascular defects resulted exclusively from the loss of HCs, then preserving c-myc in HCs while deleting it in ECs should allow the mutant embryos to survive beyond midgestation and develop normal vasculature. Indeed, when we deleted c-myc in the majority of ECs but only a subset of HCs using the Tie1-Cre line, all of these mutants survived to late gestation, well past the lethality of Vav-iCre;c-mycflox/− embryos. These results suggest that c-Myc regulates angiogenesis through its control over hematopoiesis and the production of paracrine factors.
The physiological function of c-myc may be restricted to hematopoietic lineages in the embryo and placenta
Retention of c-myc in the visceral endoderm and the extra-embryonic ectoderm of Sox2-cre mutants prevented the gross organ abnormalities seen in c-myc−/− embryos and extended the embryo survival, demonstrating that c-myc plays an essential function in these tissues. In the embryo proper, the severe Vav-iCre CKs phenocopied the Tie2-Cre and Sox2-Cre CKs, suggesting the possibility that c-myc in the HCs is most critical for the development and survival of the embryo at this stage.
Therefore our genetic evidence suggests the possibility that c-myc functions restrictively in placenta and HCs but less so in other tissues. Supporting this notion, we found no significant cell autonomous requirement for c-myc in ECs. Other studies also indicate that c-Myc is dispensable for the homeostasis of the adult intestinal epithelium (Benitah et al., 2005; Oskarsson et al., 2006), postnatal hepatocyte proliferation (Baena et al., 2005), and liver regeneration (Li et al., 2006). While further investigation is required to delineate the precise physiological function of c-Myc, our data and the published findings raise the hypothesis that c-myc may be uniquely required in the hematopoietic lineage and placenta, playing a less critical role in other cell lineages in vivo.
Experimental Procedures
Mice
Tie2-Cre, Tie1-Cre, Sox2-Cre, Vav-iCre, Tie1-GFP, c-mycflox/flox, and c-myc+/− mice were previously described (Braren et al., 2006; de Boer et al., 2003; Gustafsson et al., 2001; Hayashi et al., 2002; Iljin et al., 2002; Trumpp et al., 2001). All animals were treated in accordance with the guidelines of the University of California San Francisco (UCSF) Institutional Animal Care and Use Committee.
Imaging of embryos, whole-mount immunofluorescence staining, and EC isolation and culture
We followed our previously established procedures (Braren et al., 2006).
Whole-mount LacZ staining
LacZ staining of embryos and yolk sacs was as previously described (Carpenter et al., 2005). Samples were fixed in 4% paraformaldehyde (PFA) overnight after LacZ staining. Specimens were then embedded in paraffin and sectioned to 5μms. Sections were stained with Eosin and visualized using a Zeiss Axioskop 2 Plus microscope (Zeiss, Thornwood, NY). Images were captured using a DC 300 camera and IM50 software (Leica, San Jose, CA).
Cell proliferation and TUNEL assays
Cell proliferation and apoptosis were evaluated as previously described (Braren et al., 2006). TUNEL+CD31+ ECs and TUNEL+CD31− cells were counted, and the ratio of TUNEL+CD31+ cells to total CD31+ ECs was obtained. Four pairs of embryos from two different litters were examined. Statistical analysis was performed using the t test.
EC motility assay
ECs isolated from 10.5dpc embryos were plated on a six-well plastic plate coated with 10 μg/ml fibronectin and cultured overnight, then labeled for three hours with Dil (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine perchlorate)-labeled, acetylated low-density lipoprotein (DiI-Ac-LDL, Biomedical Technologies, Stoughton, MA) at 2.5 μg/ml in medium. Time-lapse microscopy was performed using a Marianas time-lapse imaging system (Intelligent Imaging Innovations, Santa Monica, CA). EC migration was recorded for 4 hours at 6-min intervals. XY coordinates of individual ECs were tracked with Slidebook software (Intelligent Imaging Innovations, Santa Monica, CA).
Fetal liver cell-isolation and flow cytometry analysis
Fetal livers at 11.5dpc were dissociated mechanically and passed through a 40μm-nylon mesh. Cells were collected in 10% FBS/0.5% BSA/1X PBS-Calcium and Magnesium free (CMF). Cell viability was determined using a trypan blue dye exclusion assay. For flow cytometry analysis, cells (3–5×105) were diluted into 100 μl 5% FBS/0.5% BSA/1X PBS-CMF, and the antibody-cell suspension was incubated on ice for 30 min. A BD LSRII FACS Machine (BD Biosciences, San Diego, CA) was used to perform flow-cytometry, and FlowJo software was used for data analysis. Propidium iodide (1 μg/ml) staining was used to exclude dead cells.
Collection of embryonic peripheral blood cells
Embryonic peripheral blood (PB) was isolated by opening the embryonic vitelline vessels, dorsal aortae and the heart to completely release blood cells. The cells were passed through a 40μm-nylon mesh before use.
Immunofluorescence staining of sections and isolated cells
Rehydrated paraffin sections were blocked with 5% donkey serum in PBS for two hours at room temperature. They were then incubated with primary antibody at 4°C overnight, washed three times with PBS, followed by 1 hour secondary antibody incubation at 4°C in blocking solution (2% BSA, 3% normal donkey serum, 0.01% Triton X100, 1XPBS). Samples were washed with PBS and mounted with Vectashield containing DAPI (4′, 6 diamidino-2-phenylindole; Vector Laboratories, Inc. Burlingame, CA). Cells grown on 6-well plates were fixed in 4% PFA/PBS for 20 minutes and permeabilized in 0.1% TritonX-100/1 X PBS/2%BSA for 10 min at room temperature before blocking (2% BSA/0.01% of Triton X100/1X PBS). Images were captured using either air lenses or a 63X Anchroplan water immersion lens, and a Zeiss Axiovert2 Plus microscope equipped with a Sensicam CCD camera and Slidebook software (Intelligent Imaging Innovations, Santa Monica, CA).
Allantoic explants
Allantoic explant was performed as described (Braren et al., 2006). Allantoises were isolated at e8.0 at the 6- to 8-somite stage and cultured for 24 - 48 hrs on FN-coated dishes. Cultures were stained with anti-CD31 as described above.
Supplementary Material
Acknowledgments
We thank Dr. J.M. Bishop, in whose laboratory this project was first initiated; Drs. G. I. Evan and S. Kim for helpful comments of the manuscript; Drs. F. Chanut and C. Munkittrick for editorial advice; Dr. Y. H. Kim, P. A. Murphy, and other members of our laboratory for helpful discussions; the UCSF Liver Center Morphology Core supported by NIH P30-DK26743. This work was supported by funding from the Pacific Vascular Research Foundation, HHMI UCSF BRSP, HL075033, and Atorvastatin Award to R.W.; CA 44338 to J. M. Bishop and the George Williams Hooper Foundation.
References
- Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Baena E, Gandarillas A, Vallespinos M, Zanet J, Bachs O, Redondo C, Fabregat I, Martinez AC, de Alboran IM. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci U S A. 2005;102:7286–7291. doi: 10.1073/pnas.0409260102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16:2530–2543. doi: 10.1101/gad.1024602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck L, Jr, D’Amore PA. Vascular development: cellular and molecular regulation. Faseb J. 1997;11:365–373. [PubMed] [Google Scholar]
- Benitah SA, Frye M, Glogauer M, Watt FM. Stem cell depletion through epidermal deletion of Rac1. Science. 2005;309:933–935. doi: 10.1126/science.1113579. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettess MD, Dubois N, Murphy MJ, Dubey C, Roger C, Robine S, Trumpp A. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol Cell Biol. 2005;25:7868–7878. doi: 10.1128/MCB.25.17.7868-7878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006;172:151–162. doi: 10.1083/jcb.200506184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ, Muller-Rover S, Patel SV, Chronnell CM, McKay IA, Philpott MP. Contrasting localization of c-Myc with other Myc superfamily transcription factors in the human hair follicle and during the hair growth cycle. J Invest Dermatol. 2001;116:617–622. doi: 10.1046/j.1523-1747.2001.12771234.x. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Carpenter B, Lin Y, Stoll S, Raffai RL, McCuskey R, Wang R. VEGF is crucial for the hepatic vascular development required for lipoprotein uptake. Development. 2005;132:3293–3303. doi: 10.1242/dev.01902. [DOI] [PubMed] [Google Scholar]
- Cumano A, Godin I. Ontogeny of the hematopoietic system. Annu Rev Immunol. 2007;25:745–785. doi: 10.1146/annurev.immunol.25.022106.141538. [DOI] [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7:671–682. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs KM, Martin GR, Bishop JM. Contrasting patterns of myc and N-myc expression during gastrulation of the mouse embryo. Genes Dev. 1989;3:860–869. doi: 10.1101/gad.3.6.860. [DOI] [PubMed] [Google Scholar]
- Eichmann A, Pardanaud L, Yuan L, Moyon D. Vasculogenesis and the search for the hemangioblast. J Hematother Stem Cell Res. 2002;11:207–214. doi: 10.1089/152581602753658411. [DOI] [PubMed] [Google Scholar]
- Ema M, Rossant J. Cell fate decisions in early blood vessel formation. Trends Cardiovasc Med. 2003;13:254–259. doi: 10.1016/s1050-1738(03)00105-1. [DOI] [PubMed] [Google Scholar]
- Erlebacher A, Price KA, Glimcher LH. Maintenance of mouse trophoblast stem cell proliferation by TGF-beta/activin. Dev Biol. 2004;275:158–169. doi: 10.1016/j.ydbio.2004.07.032. [DOI] [PubMed] [Google Scholar]
- Evan GI, Christophorou M, Lawlor EA, Ringshausen I, Prescott J, Dansen T, Finch A, Martins C, Murphy D. Oncogene-dependent tumor suppression: using the dark side of the force for cancer therapy. Cold Spring Harb Symp Quant Biol. 2005;70:263–273. doi: 10.1101/sqb.2005.70.054. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- Gekas C, Dieterlen-Lievre F, Orkin SH, Mikkola HK. The placenta is a niche for hematopoietic stem cells. Dev Cell. 2005;8:365–375. doi: 10.1016/j.devcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- Graven KK, Zimmerman LH, Dickson EW, Weinhouse GL, Farber HW. Endothelial cell hypoxia associated proteins are cell and stress specific. J Cell Physiol. 1993;157:544–554. doi: 10.1002/jcp.1041570314. [DOI] [PubMed] [Google Scholar]
- Guney I, Sedivy JM. Cellular senescence, epigenetic switches and c-Myc. Cell Cycle. 2006;5:2319–2323. doi: 10.4161/cc.5.20.3348. [DOI] [PubMed] [Google Scholar]
- Gustafsson E, Brakebusch C, Hietanen K, Fassler R. Tie-1-directed expression of Cre recombinase in endothelial cells of embryoid bodies and transgenic mice. J Cell Sci. 2001;114:671–676. doi: 10.1242/jcs.114.4.671. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, McMahon AP. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev. 2002;119(Suppl 1):S97–S101. doi: 10.1016/s0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Iljin K, Petrova TV, Veikkola T, Kumar V, Poutanen M, Alitalo K. A fluorescent Tie1 reporter allows monitoring of vascular development and endothelial cell isolation from transgenic mouse embryos. Faseb J. 2002;16:1764–1774. doi: 10.1096/fj.01-1043com. [DOI] [PubMed] [Google Scholar]
- Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A stem cell molecular signature. Science. 2002;298:601–604. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- Jones EA, Baron MH, Fraser SE, Dickinson ME. Measuring hemodynamic changes during mammalian development. Am J Physiol Heart Circ Physiol. 2004;287:H1561–1569. doi: 10.1152/ajpheart.00081.2004. [DOI] [PubMed] [Google Scholar]
- Knies-Bamforth UE, Fox SB, Poulsom R, Evan GI, Harris AL. c-Myc interacts with hypoxia to induce angiogenesis in vivo by a vascular endothelial growth factor-dependent mechanism. Cancer Res. 2004;64:6563–6570. doi: 10.1158/0008-5472.CAN-03-3176. [DOI] [PubMed] [Google Scholar]
- Li F, Xiang Y, Potter J, Dinavahi R, Dang CV, Lee LA. Conditional deletion of c-myc does not impair liver regeneration. Cancer Res. 2006;66:5608–5612. doi: 10.1158/0008-5472.CAN-05-4242. [DOI] [PubMed] [Google Scholar]
- Malynn BA, de Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, Alt FW. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000;14:1390–1399. [PMC free article] [PubMed] [Google Scholar]
- Mezquita P, Parghi SS, Brandvold KA, Ruddell A. Myc regulates VEGF production in B cells by stimulating initiation of VEGF mRNA translation. Oncogene. 2005;24:889–901. doi: 10.1038/sj.onc.1208251. [DOI] [PubMed] [Google Scholar]
- Mikkola HK, Gekas C, Orkin SH, Dieterlen-Lievre F. Placenta as a site for hematopoietic stem cell development. Exp Hematol. 2005;33:1048–1054. doi: 10.1016/j.exphem.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Mikkola HK, Orkin SH. The journey of developing hematopoietic stem cells. Development. 2006;133:3733–3744. doi: 10.1242/dev.02568. [DOI] [PubMed] [Google Scholar]
- Mountain DJ, Singh M, Menon B, Singh K. Interleukin-1beta increases expression and activity of matrix metalloproteinase-2 in cardiac microvascular endothelial cells: role of PKCalpha/beta1 and MAPKs. Am J Physiol Cell Physiol. 2007;292:C867–875. doi: 10.1152/ajpcell.00161.2006. [DOI] [PubMed] [Google Scholar]
- Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, Sancho E, Cole AM, Gregorieff A, de Alboran IM, Clevers H, et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol. 2006;26:8418–8426. doi: 10.1128/MCB.00821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MJ, Wilson A, Trumpp A. More than just proliferation: Myc function in stem cells. Trends Cell Biol. 2005;15:128–137. doi: 10.1016/j.tcb.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, Adams JM. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood. 1999;94:1855–1863. [PubMed] [Google Scholar]
- Okamoto R, Ueno M, Yamada Y, Takahashi N, Sano H, Suda T, Takakura N. Hematopoietic cells regulate the angiogenic switch during tumorigenesis. Blood. 2005;105:2757–2763. doi: 10.1182/blood-2004-08-3317. [DOI] [PubMed] [Google Scholar]
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- Oskarsson T, Essers MA, Dubois N, Offner S, Dubey C, Roger C, Metzger D, Chambon P, Hummler E, Beard P, et al. Skin epidermis lacking the c-Myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21Cip1 gene. Genes Dev. 2006;20:2024–2029. doi: 10.1101/gad.381206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- Ramirez-Bergeron DL, Runge A, Adelman DM, Gohil M, Simon MC. HIF-dependent hematopoietic factors regulate the development of the embryonic vasculature. Dev Cell. 2006;11:81–92. doi: 10.1016/j.devcel.2006.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall TD, Weissman IL. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood. 1997;89:3596–3606. [PubMed] [Google Scholar]
- Robb L, Lyons I, Li R, Hartley L, Kontgen F, Harvey RP, Metcalf D, Begley CG. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci U S A. 1995;92:7075–7079. doi: 10.1073/pnas.92.15.7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchors K, Shchors E, Rostker F, Lawlor ER, Brown-Swigart L, Evan GI. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006;20:2527–2538. doi: 10.1101/gad.1455706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura N, Watanabe T, Suenobu S, Yamada Y, Noda T, Ito Y, Satake M, Suda T. A role for hematopoietic stem cells in promoting angiogenesis. Cell. 2000;102:199–209. doi: 10.1016/s0092-8674(00)00025-8. [DOI] [PubMed] [Google Scholar]
- Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7:327–331. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 2001;414:768–773. doi: 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- Vincent SD, Robertson EJ. Highly efficient transgene-independent recombination directed by a maternally derived SOX2CRE transgene. Genesis. 2003;37:54–56. doi: 10.1002/gene.10226. [DOI] [PubMed] [Google Scholar]
- Watnick RS, Cheng YN, Rangarajan A, Ince TA, Weinberg RA. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell. 2003;3:219–231. doi: 10.1016/s1535-6108(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, Trumpp A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747–2763. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.