

Abstract
Although histamine plays an essential role in inflammation, its influence on cyclooxygenases (COX) and prostanoid homeostasis is not well understood. Here, we investigated the effects of histamine on the expression of COX-1 and COX-2 and determined their contribution to the production of prostaglandin-E2 (PGE2), prostacyclin (PGI2) and thromboxane A2 (TXA2) in human coronary artery endothelial cells (HCAEC). Incubation of HCAEC monolayers with histamine resulted in marked increases in the expression of COX-2 and production of PGI2 and PGE2 with no significant change in the expression of COX-1. Histamine-induced increases in PGI2 and PGE2 production were due to increased expression and function of COX-2 as gene silencing by siRNA or inhibition of the catalytic activity by a COX-2 inhibitor blocked prostanoid production. The effects of histamine on COX-2 expression and prostanoid production were mediated through H1 receptors. In addition to the direct effect, histamine was found to amplify LPS-stimulated COX-2 expression and PGE2 and PGI2 production. In contrast, histamine did not stimulate TXA2 production in resting or LPS-activated HCAEC. Histamine-induced increases in the production of PGE2 and PGI2 were associated with increased expression of mRNA encoding PGE2 and PGI2 synthases. The physiological role of histamine on the regulation of COX-2 expression in the vasculature is indicated by the findings that the expression of COX-2 mRNA, but not COX-1 mRNA, was markedly reduced in the aortic tissues of histidine decarboxylase null (HDC-/-) mice. Thus, histamine plays an important role in the regulation of COX-2 expression and prostanoid homeostasis in vascular endothelium.
Keywords: Mast cells, endothelial cells, inflammation, lipopolysaccharide, lipid mediator
INTRODUCTION
Histamine is one of the major constituents of the mast cell that is released into the extracellular milieu upon degranulation. Histamine and other mast cell mediators are known to regulate vasodilation and bronchoconstriction (1, 2), and to modulate the functions of other cell types including monocytes and macrophages (3, 4), eosinophils (5, 6), T lymphocytes (7), neutrophils (8), and endothelial cells (9). A direct association between mast cell-derived histamine and vascular inflammation is evident from the finding that the coronary arteries of patients with ischemic heart disease contain more mast cells and histamine than normal vessels (10) and patients with variant angina have elevated levels of histamine in the coronary circulation (11). The fact that histamine is able to stimulate endothelial cell production of such proinflammatory cytokines as IL-6 and IL-8 (9, 12, 13) indicates that this mast cell product can act as an important inflammatory agent in addition to its well-recognized vasoactive functions.
Cyclooxygenase-1 (COX-1) and COX-2 belong to a family of enzymes that catalyze the oxygenation of arachidonic acid to prostaglandin (PG) G2/H2, which are utilized by specific prostaglandin synthases to generate PGE2, PGD2, PGF2α, PGI2 and thromboxane A2 (TXA2). Among the prostaglandins, PGI2 and TXA2 have earned considerable interest because of their involvement in cardiovascular diseases (14, 15). Prostacyclin (PGI2) is a major prostaglandin produced by endothelial cells, and is a potent vasodilator and an inhibitor of leukocyte adhesion and platelet aggregation. Hence, PGI2 is thought to play a protective role in atherothrombosis. TXA2, on the other hand, is a potent inducer of vasoconstriction, platelet activation and platelet adhesion. Since PGI2 and TXA2 act on vascular endothelium in opposing manners, their relative concentrations in the microenvironment and systemic circulation are critical for vascular homeostasis and athrogenesis.
It is well-recognized that innate immune dysregulation plays an important role in the pathogenesis of many inflammatory diseases. Recently, we have shown that histamine stimulates Toll-like receptor-2 (TLR2) and TLR4 expression in endothelial cells and enhances inflammatory responses to Gram-positive and Gram-negative bacterial cell wall components (16). Thus, histamine is not only able to induce inflammatory responses directly but also has the potential to amplify infection-associated activation of endothelial functions (16). The objectives of this study were to determine the role of histamine in the regulation of COX-2 gene expression in vitro and in vivo and to evaluate its regulatory function on the production of PGE2, PGI2 and TXA2 in naïve and lipopolysaccharide (LPS)-stimulated HCAEC. This report demonstrates that histamine directly and synergistically with LPS stimulates the expression of COX-2 and amplifies the production of PGI2 and PGE2 without affecting basal or LPS-induced TXA2 production in HCAEC. In addition, the study presents first evidence that the aortic tissues of histidine decarboxylase null mice (HDC-/- mice) express markedly reduced levels COX-2 mRNA without change in the level of expression of COX-1 mRNA. These findings support the concept that histamine plays a key role in the regulation of COX-2 expression and prostanoid homeostasis in the vasculature.
Materials and Methods
Materials
HCAEC, endothelial cell growth medium (EGM-2 MV), trypsin-ethylenediaminetetraacetic acid (EDTA) and trypsin neutralizing solution were purchased from Cambrex (San Diego, CA). Enzyme immunoassay (EIA) kits for 6-keto PGF1α, PGE2 and TXB2; COX-2, COX-1, and goat anti-rabbit IgG HRP antibodies were obtained from Cayman Chemical Company (Ann Arbor, Michigan). Actin, rabbit anti-goat IgG antibodies and ECL chemiluminescence reagent were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Histamine dihydrochloride, fexofenadine hydrochloride, famotidine, and 2-[(3-trifluoromethyl) phenyl] histamine dimaleate, and Escherichia coli (0111:B4) LPS were purchased from Sigma-Aldrich, Inc (St Louis, MO). RNeasy minikit were purchased from Qiagen Inc, Valencia, CA. High-capacity cDNA Achieve kit, SYBR® Green PCR Master Mix kit, and all the gene specific primers were purchase from Applied Biosystems, Foster City, CA. RIPA lysis and extraction buffer was a product of Pierce Biotechnology Inc. (Rockford, IL). Transfection reagent siPort Amine, COX-2 siRNA, and negative control siRNA were obtained from Ambion, Austin, TX.
Culture of HCAEC
HCAEC were grown in EGM-2 MV containing 1 μg/ml hydrocortisone acetate, 50 ng/ml gentamycin, 50 μg/ml amphotericin B, and the recommended concentrations of human epidermal growth factor, vascular endothelial growth factor, human fibroblast growth factor-B, recombinant insulin-like growth factor-1, ascorbic acid and 5% fetal bovine serum as described previously (9, 16). At confluence, the cells were detached from the culture flasks using trypsin-EDTA, washed twice, and resuspended in fresh EGM-2 MV. The cells used in all experiments were between three and six passages.
EIA determinations of the production of PGI2, PGE2 and TXA2
HCAEC (1.5 × 104) were plated on to each of the wells of a 96-well cell culture plate and allowed to adhere for 16 to 18 h. Following adherence, selected concentrations of the activating stimuli or medium were added to the monolayers and incubated for indicated time periods in a final volume of 0.2 ml at 37°C in 5% humidified CO2. After the indicated incubation period, culture supernatants were harvested, appropriately diluted and assayed for 6-keto PGF1α (for PGI2), PGE2, and TXB2 (for TXA2) levels by EIA.
Transfection and siRNA
Gene-specific siRNA or negative control siRNA was introduced by using the siPORT Amine transfection reagent according to the manufacturer’s instructions. Briefly, 50 μl aliquot of the transfection reagent mixture containing 0.75 μl of siPORT Amine reagent, 100 nM of siRNA and OPTI-MEM I was added to each of the wells of a 24-well cell culture plate. An aliquot of the HCAEC cell suspension containing 5 × 104 cells were then added to the transfection mixture and incubated at 37°C in 5% CO2 for 24 h. After the incubation, medium containing the transfection reagent was discarded, and the cells were maintained in fresh medium for another 24 h. The cells were subsequently treated with histamine (10 μM) for 2 h. Total RNA was extracted from HCAEC monolayers for real-time PCR analysis. An aliquot of 12.5 μl above transfection reagent mixture and 1.3 × 104 cells were plated into each of the wells of a 96 well culture plate. The cells were treated the same as described above. Cell culture supernatants were collected after 12 h histamine treatment for PGI2 EIA analysis
Real-Time PCR
After incubating HCAEC monolayers with medium or the stimulating agents, supernatant was removed from the cell culture dish and total RNA was isolated using RNeasy mini kit, according to the manufacture’s protocol. Total RNA (1 μg) was reverse transcribed into first-strand cDNA using High-capacity cDNA Achive kit following the manufacturer’s procedure. The primers used for SYBR Green real-time RT-PCR were designed using the Primer Express Software v3.0 (Applied Biosystems, Foster City, CA). The sequences of primers used in the real-time PCR analyses of various genes are:
| COX-1: | forward, 5′-GCCAGTGAATCCCTGTTGTTACT-3′ reverse, 5′-GGCCGAAGCGGACACA-3′ |
| COX-2: | forward, 5′-AGGGTTGCTGGTGGTAGGAA-3′ reverse, 5′-GGTCAATGGAAGCCTGTGATACT-3′ |
| β-actin: | forward, 5′-CCAGCTCACCATGGATGATG-3′ reverse, 5′-ATGCCGGAGCCGTTGTC-3′ |
| PGI2 synthase: | forward, 5′-GCCACATAGCTCATAAGCTGTAGAAC-3′ reverse, 5′-AGTTGCTCATCCAGCATTTGC-3′. |
| mPGE2 synthase-1: | forward, 5′-CCTGGGCTTCGTCTACTCCTT-3′ reverse, 5′-AGTGCATCCAGGCGACAAA-3′ |
| TXA2 synthase: | forward, 5′- GTTGCCTGTTCCCTTTTCTACCT-3′ reverse, 5′- CTGATCTCGCCGCCCTTA- 3′. |
Real-time PCR was performed using the ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA) equipped with a 96-well optical reaction plate. The amplification reactions were performed in 25 μl total volume containing 20 μl of SYBR® Green PCR Master Mix and 5 μl of cDNA of each sample. All real-time experiments were run in triplicate and a mean value was used for the determination of mRNA levels. COX-2 or COX-1 mRNA levels from each treatment were normalized to the corresponding amount of β-actin mRNA levels. Negative controls, containing water instead of sample cDNA, were used in each real-time plate.
Semi-quantitative reverse transcriptase-PCR analyses of the expression of COX-1 and COX-2 mRNA in aortic tissues of wild type and HDC-/- mice
Aortic tissues were harvested from two HDC-/- mice and two wild type controls. The protocols employed in this study were in accordance with the guidelines approved by the institutional review boards of the University of Kansas Medical Center and Vanderbilt University. After exsanguination, thoracic aortas were dissected and rinsed with cold phosphate buffered saline. Total RNA was extracted using TRIzol Reagent. Total RNA (1 μg) was used for RT reaction using random oligonucleotide primers according to the manufacturer’s instruction (Invitrogen, Carlsbad, CA). Aliquots of the reverse transcription product were PCR amplified using gene-specific sense and antisense primers. The PCR cycle parameters were as follows: 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds for the 1st cycle followed by 94°C for 5 min and 72°C for 10 min for 45 cycles. The PCR products were electrophoresed on agarose gels (1.5%) and stained with ethidium bromide for imaging. The sequences of primers used are:
| COX-1: | forward, AGGAGATGGCTGCTGAGTTGG reverse, AATCTGACTTTCTGAGTTGCC (product size 601 bp) |
| COX-2: | forward, ACCTCTGCGATGCTCTTCC reverse, CACCATAGAATCCAGTCCGG (product size 861 bp) |
| rpL7: | forward, TCAATGGAGTAAGCCCAAAG reverse, CAAGAGACCGAGCAATCAAG (product size 246 bp) |
Western blot analyses
Confluent HCAEC monolayers were incubated with selected stimuli for indicated times at 37°C. The cells were then washed with ice cold PBS twice and lysed in RIPA buffer containing 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, supplied with 1% (v/v) protease inhibitor cocktail at 4°C. Cell debris was removed by centrifugation of the lysate at 13, 000 g for 15 min at 4°C. Aliquots of supernatants normalized for protein concentrations were mixed with equal volumes of 2 × sodium dodecyl sulphate sample buffer and heated to 100°C for 5 min. Samples were resolved on 10% sodium dodecyl sulphate-polyacrylamide gel elcetrophoresis and transferred onto a nitrocellulose membrane. After blocking in TBST (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) containing 5% non-fat milk overnight, membranes were washed thrice in TBST and incubated with primary antibody for 1 h in blocking solution at room temperature. After washing thrice, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h, washed 3 times and the signals were detected using enhanced chemiluminescence reagents.
Immunoblot data quantitation
The chemiluminiscent signals were quantified by using Bio-Rad ChemiDoc XRS imager and COX-1 and COX-2 specific signals in each lane were normalized to the actin signal. For quantifying magnitude change in treated cells over untreated cell, the actin-normalized value for medium-treated cells was considered as 1 and fold increase over unstimulated cells was calculated.
Statistical analysis
Analyses were done using SPSS 14.0 (SPSS, Chicago, IL). For overall comparisons among treatment groups a significant factorial analysis of variance was followed up using Student-Newman-Keuls post-hoc tests. For multiple comparisons with a control (e.g., medium alone) we used Dunnett’s test. The test for synergy was declared significant if the mean value obtained for the combined-treatment group was significantly greater than the sum of the upper limits of the 95% confidence intervals for the means of the two separately treated groups. Results are reported as mean ± SEM. For all analyses the comparison was considered significant if P<0.05.
RESULTS
Histamine induces COX-2, but not COX-1, mRNA expression
In order to examine whether histamine regulates the expression of COX-1 or COX-2 gene expression in HCAEC, a time course of its effect on the mRNA expression was monitored by real-time PCR. In previous studies, 10 μM concentration of histamine was found to be sufficient to induce near maximum stimulation of inflammatory responses in endothelial cells (9, 16). Therefore, HCAEC were incubated with 10 μM histamine for 1, 2, 4 and 8 h to evaluate the kinetic profile of COX-1 and COX-2 mRNA expression. As shown in Fig. 1A, incubation of HCAEC with histamine resulted in 11-fold increase in COX-2 mRNA expression within the first hour of treatment. Histamine-stimulated COX-2 mRNA expression was transient, decreased progressively during continued incubation, and reached the basal level by 8 hours. In contrast, the expression of COX-1 mRNA was not altered by treatment of HCAEC with histamine for 1 to 8 hours. Histamine-induced COX-2 mRNA expression was concentration-dependent between 0.1 to 100 μM and an increase was evident at a concentration as low as 0.1 μM (Fig 1B). The concentration-dependent response of HCAEC to histamine did not attain a plateau even at a concentration of 100 μM. However, considering the relatively low levels of histamine in normal tissues and in circulation (1 to 3 nM) (17), concentrations greater than 100 μM were not tested in this experiment.
Figure 1.
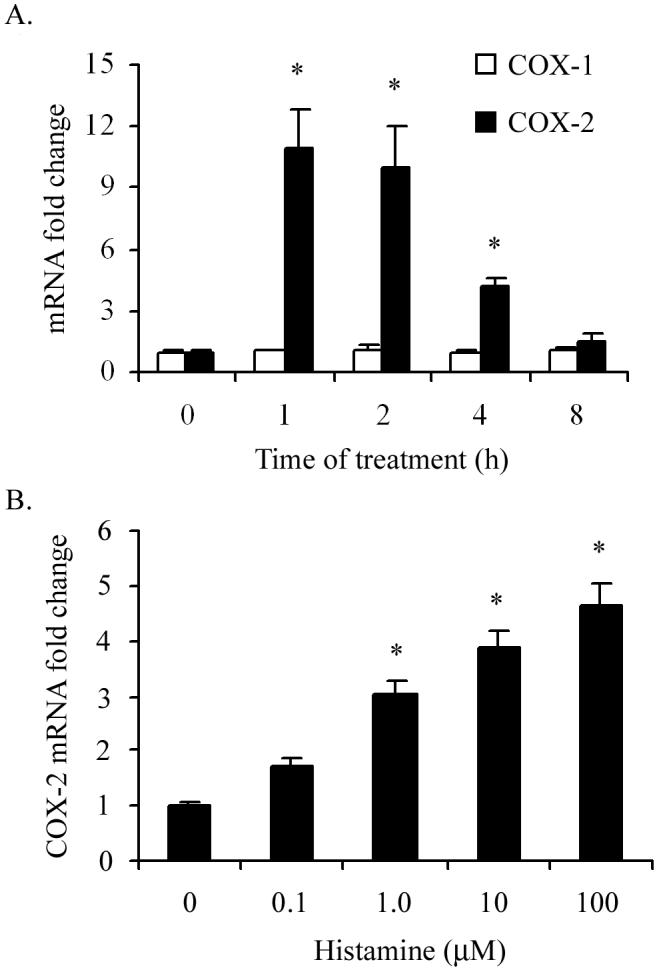
Histamine induces COX-2 but not COX-1 mRNA expression in HCAEC. HCAEC monolayers were incubated with histamine (10 μM) for 1 to 8 h (A) or with 0.1 to 100 μM histamine for 2 h (B). After the incubation, total RNA was extracted, reverse transcribed and analyzed by real-time PCR using specific primers as described in ‘Materials and Methods.’ The amplicons were normalized for β-actin and assigned a value of 1 for medium-treated cells. The magnitude of changes of mRNA expression in histamine-treated cells was calculated by comparing to the message in medium-treated HCAEC. The results presented are representative of three experiments with similar results. Each value presented is the mean ± SEM of triplicate determinations. * indicates p<0.05 when compared to medium-treated cells.
Histamine induces COX-2 protein expression
In order to determine whether histamine-induced COX-2 mRNA expression is associated with increased expression of COX-2 protein, Western blot analyses were carried out after incubating HCAEC with histamine for different time periods. As shown in Fig. 2A and 2B, only low-level expression of COX-2 protein was noted in resting HCAEC (medium-treated controls).
Figure 2.

Histamine induces COX-2 but not COX-1 protein expression in HCAEC. HCAEC monolayers were incubated with histamine (10 μM) for 2 to 24 h (A) or with 0 to 10 μM histamine for 4 h (B). After washing with phosphate-buffered saline, total cell lysates were prepared and subjected to Western blot analyses as described in ‘Materials and Methods’. The presented are representative of three experiments with similar results. Data presented in (C) and (D) are mean ± SEM of fold increases in protein expression as determined by chemiluminiscence image quantifications of three Western blot analyses * indicates p<0.05 when compared to medium-treated cells.
Histamine-induced expression of COX-2 protein was detectable within 2 h after initiating the incubation, stayed at steady state between 4 and 8 h, and returned to baseline by 24 h (Fig. 2A). Incubation of HCAEC with histamine (0.1 to 10 μM) led to a concentration-dependent increase in the expression of COX-2 protein (Fig. 2B). The level of expression of COX-1 protein in HCAEC was minimally affected by the concentration of histamine or by the duration of incubation (Fig. 2 C and D).
Histamine stimulates production of PGI2 and PGE2
In view of our finding that histamine stimulated the expression of COX-2 mRNA and protein in HCAEC, we asked whether the newly expressed protein is functionally active. It is well established that COX-2 catalyzes the conversion of arachidonic acid to PGH2, which in turn is rapidly converted to PGE2, PGD2, PGF2α, PGI2 and TXA2 by the terminal synthases. Here we determined the production of PGI2 because of its well-recognized vasodilatory and immunomodulatory functions. Accordingly, HCAEC monolayers were incubated with selected concentrations of histamine (1, 10 and 100 μM) for 4, 8, and 12 h and the production of PGI2 was monitored by quantifying the levels of 6-keto PGF1α in culture supernatants. As shown in Fig. 3A, incubation of HCAEC with histamine for 8 or 12 h led to a statistically significant increase in the production of PGI2 which was concentration- and time-dependent. To determine whether histamine-stimulated PGI2 production is indeed associated with the increased function of COX-2, HCAEC were pre-treated with COX-2-specific inhibitor NS-398 for 30 min prior to stimulation with histamine, and PGI2 production was measured after 12 h. As shown in Fig 3B, NS-398 caused an approximately 50% reduction of the basal PGI2 production and complete inhibition of histamine-induced PGI2 production. In order to further confirm the association between histamine-induced COX-2 expression and PGI2 production, HCAEC were transfected with COX-2-specific siRNA or negative control siRNA, and COX-2 mRNA expression and PGI2 production were determined. As evident from Fig 3C, transfection of HCAEC with active siRNA resulted in the inhibition of constitutive and histamine-induced COX-2 mRNA expression by approximately 70% and 75%, respectively. In agreement with the siRNA-induced decrease in COX-2 mRNA expression, both the basal and histamine-stimulated production of PGI2 were decreased proportionately (Fig 3D).
Figure 3.

Histamine-induced COX-2 expression is functionally active and contributes to the increased production of PGI2. HCAEC monolayers were incubated with medium or histamine (1.0 to 100 μM) for 4, 8, and 12 h (A) or with 10 μM of histamine in the presence or absence of 1.0 μM COX-2 inhibitor NS-398 (B), and the production PGI2 was determined by quantifying 6-keto PGF1α by EIA. To determine the association of COX-2 mRNA expression and PGI2 production, HCAEC monolayers were transfected with COX-2 specific siRNA or negative control siRNA for 24 h. The cells were then treated with 10 μM histamine for 2 h for the determination of COX-2 mRNA expression by real-time PCR (C), or 12 h for determination of PGI2 production (D). Data presented in (A) and (B) are mean ± SEM of quadruplicate determinations of a representative experiment out of three. Each value presented in (C) and (D) is the mean ± SEM of triplicate determinations of a representative experiment out of two. * indicates p<0.05 when compared to values for medium-treated cells (A), cells treated without NS-398 (B), and reagent control treated cells (C) and (D).
Histamine-induced COX-2 gene expression and PGI2 production in HCAEC is mediated via H1R
Previous studies from our laboratory have shown that histamine-induced production of IL-6 and IL-8 (9) as well as the expression of TLRs (16) in human endothelial cells is mediated via H1 receptors. To determine whether histamine-induced COX-2 expression and PGI2 production are mediated through H1R, HCAEC monolayers were incubated with histamine in the presence of H1R antagonist fexofenidine or the H2R antagonist famotidine, and mRNA expression and PGI2 production were monitored. As shown in Figure 4A, incubation of HCAEC with 10 μM histamine for 2 h resulted in a seven-fold increase in COX-2 mRNA expression, which was completely abrogated by the H1R antagonist fexofenidine. In contrast, the H2R antagonist famotidine did not affect histamine-stimulated COX-2 gene expression. The involvement of H1R in histamine-stimulated COX-2 expression is further confirmed by the finding that an H1R-specific agonist was able to mimic the effect of histamine and was inhibited by fexofinidine but not by famotidine (data not shown). In addition, the H2R agonist dimaprit failed to stimulate COX-2 expression (data not shown). Neither fexofenidine nor famotidine had any effect on the basal expression of COX-2.
Figure 4.
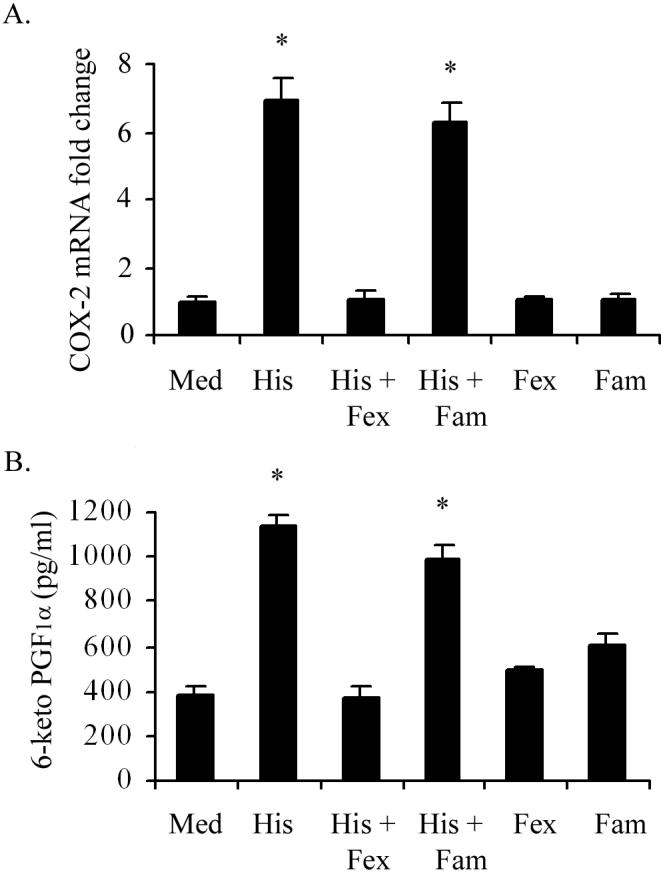
H1R antagonist blocks histamine-induced COX-2 mRNA expression and associated PGI2 production in HCAEC. HCAEC monolayers were preincubated for 30 min with the H1R antagonist, fexofenadine (Fex, 10 μM) or the H2R antagonist famotidine (Fam, 10 μM) followed by incubation with medium or histamine (His, 10 μM) for 2 h for the analyses of COX-2 mRNA expression by real-time PCR (A) or for 24 h for of the production PGI2 (B). Each value presented in (A) is the mean ± SEM for triplicate determinations of a representative experiment out of three. Each value presented in (B) is the mean ± SEM for quadruplicate determinations of a representative experiment out of three. * indicates p<0.05 when compared to values for medium-treated cells.
Next we determined whether histamine-stimulated PGI2 production is also mediated via H1R activation. Once again, as in the case of COX-2 expression, histamine-induced production of PGI2 was completely inhibited by the H1R antagonist, fexofenidine and not by the H2R antagonist, famotidine (Fig 4B).
Histamine amplifies LPS-induced expression of COX-2 mRNA and protein
Having documented the direct effect of histamine on COX-2 expression, we examined whether histamine is capable of modulating the expression of COX-2 that was initiated by other inflammatory agents. To determine the potential synergy between histamine and LPS on expression of COX-2, HCAEC monolayers were incubated for 2 h with LPS (100 ng/ml) in the presence and absence of 10 μM histamine and the expression of COX-2 mRNA and protein were determined (Fig. 5). The results presented in Fig. 5A demonstrate that histamine and LPS independently stimulated COX-2 mRNA expression by 6 fold and 12 fold, respectively. Interestingly, in addition to its direct effect, histamine was found to amplify LPS-stimulated COX-2 gene expression, as indicated by the 26-fold increase over the constitutive level of expression-a significantly synergistic effect. The Western blot analyses of COX-1 and COX-2 proteins confirm that the amplifying effect of histamine on LPS-stimulated COX-2 expression is not only limited to the transcriptional level but is also evident at the translational level (Fig 5B). The quantification of COX-2 signals shows that incubation of HCAEC with histamine, LPS, and LPS + histamine, resulted in 6, 8, and 16 fold increases in COX-2 expression, respectively (Fig 5C). On the other hand, changes in the level of expression of COX-1 protein in HCAEC was minimal after incubation for the same duration of time with histamine (1.5 fold), LPS (1.9 fold), or LPS + histamine (1.9 fold).
Figure 5.
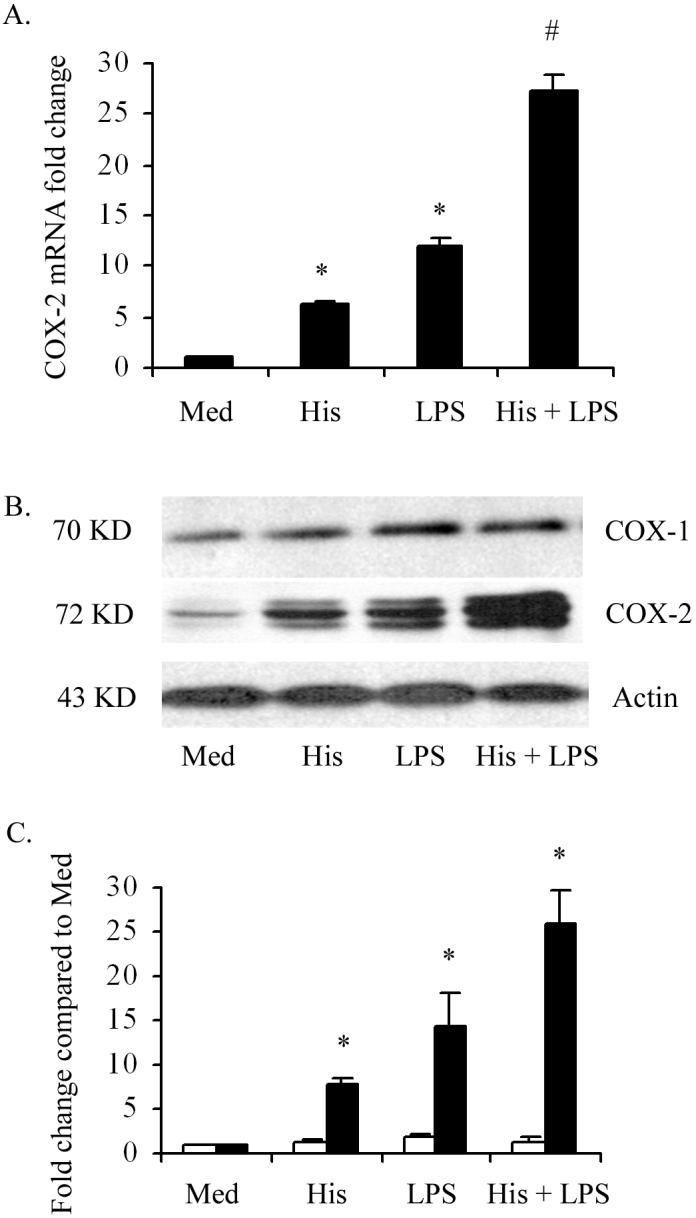
Histamine (His) and LPS synergistically stimulate COX-2 mRNA and protein expression in HCAEC. HCAEC monolayers were incubated with histamine (10 μM), LPS (100 ng/ml), or histamine + LPS for 2 h for the analyses of COX-2 mRNA expression by real-time PCR (A), or for 4 h for the Western blot analyses of COX-1 and COX-2 proteins (B). The results presented are representive of three experiments with similar results. Each value presented in (A) is the mean ± SEM of triplicate determinations. The data presented in (C) are mean ± SEM of fold increases in protein expression as determined by chemiluminiscence image quantifications of three Western blot analyses. * indicates p<0.05 when compared to medium-treated cells. # indicates p<0.05 when compared to the sum of the means of histamine-treated and LPS-treated cells.
Histamine amplifies LPS-induced endothelial production of PGI2 and PGE2, but does not affect TXA2 production
In order to examine whether histamine-induced COX-2 expression as well as its amplification by LPS are translated into functional activities, we monitored the kinetics of the production of PGI2, PGE2 and TXA2 by HCAEC. HCAEC monolayers were incubated with histamine (10 μM), LPS (100 ng/ml) or a combination of histamine (10 μM) and LPS (100 ng/ml) for 4 to 48 h and the production of 6-keto PG-F1α, PGE2 and TXB2 (stable product of TXA2) were monitored. Incubation of HCAEC with histamine or LPS caused stimulation of PGI2 (Fig. 6A) and PGE2 production (Fig. 6B) in a time-dependent fashion. As in the case of COX-2 mRNA and protein expression, histamine markedly enhanced LPS-induced production of PGI2 and PGE2 at all time points tested. The ability of histamine to amplify the LPS effect was evident even at 48 h after stimulation of HCAEC which indicates the existence of a substantial synergy between histamine and LPS in COX-2-mediated PGI2 and PGE2 production. As shown in Fig 6C (solid bars), histamine did not stimulate TXA2 production at any of the time periods tested. Although incubation of HCAEC with LPS for 24 h and 48 h resulted in significant increases in TXA2 synthesis, unlike in the case of PGI2 and PGE2, histamine failed to modulate TXA2 synthesis either in the presence or absence of LPS (Fig. 6C).
Figure 6.
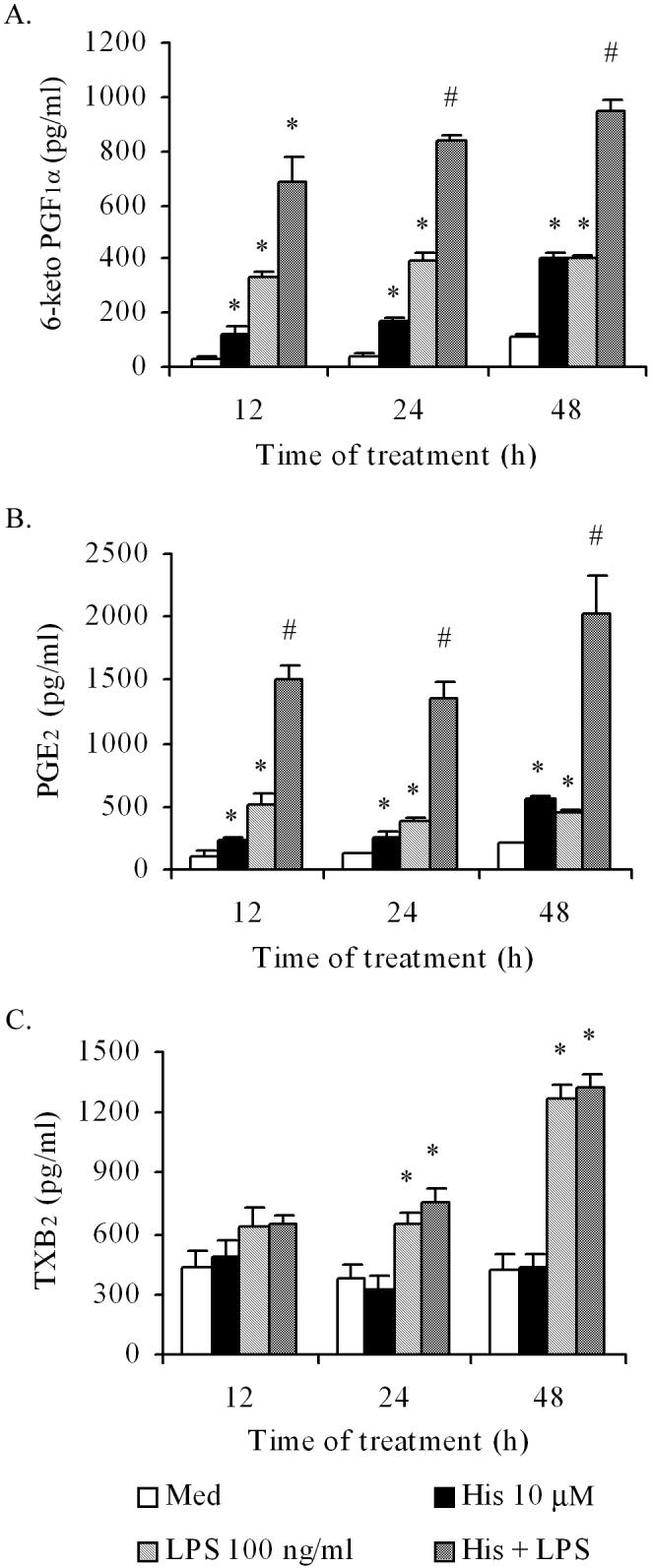
Histamine (His) amplifies LPS-induced endothelial production of PGI2 and PGE2, but does not affect TXA2 production. HCAEC monolayers were incubated with histamine (10 μM), LPS (100 ng/ml), or the combination of the two for 12, 24, or 48 hours, and the culture supernatants were analyzed for PGI2 (6-keto PGF1α), PGE2, or TXA2 (TXB2) by EIA. Data presented in (A) and (B) are mean ± SEM of quadruplicate determinations of a representative experiment of three. (C) Represents mean ± SEM of quadruplicate determinations of a representative experiment of two. * indicates p<0.05 when compared to the medium-treated cells. # indicates p<0.05 when compared to the sum of the means of histamine-treated and LPS-treated cells.
Histamine stimulates the expression of PGI2 synthase and PGE2 synthase, but not TXA2 synthase, mRNA expression
To determine whether histamine-induced increases in PGI2 and PGE2 production are associated with changes in PGI2 synthase and PGE2 synthase gene expression, HCAEC monolayers were incubated with histamine (10 μM) for 2, 8 and 12 h, and the mRNA levels were quantified by real-time RT-PCR. As shown in Fig, 7A, incubation of HCAEC with histamine resulted in time-dependent increase in PGI2 synthase mRNA expression and attained a 3-4 fold increase in 8 hours before returning to basal levels by 12 hours post-treatment. The expression of PGE2 synthase mRNA in response to histamine was of lesser magnitude and it required 8 h to attain a 40% increase (Fig. 7B). In contrast, TXA2 synthase mRNA expression in HCAEC was minimally affected by histamine treatment (Fig. 7C).
Figure 7.
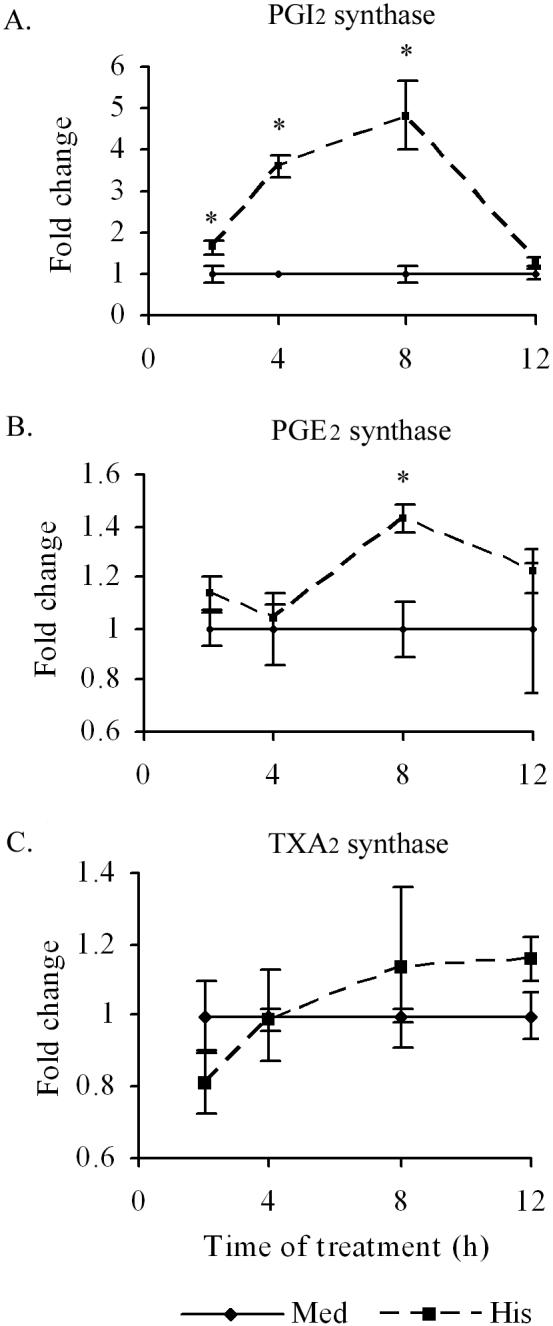
Histamine induces a time dependent PGI2 synthase and PGE2 synthase, but not TXA2 synthase, mRNA expression, in HCAEC. HCAEC monolayers were incubated with histamine (10 μM) or medium for 2 to 12 h. After the incubation, total RNA was extracted, reverse transcribed and analyzed by real-time PCR using specific primers as described in ‘Materials and Methods.’ The amplicons were normalized for β-actin and assigned a value of 1 for medium-treated cells. The magnitude of changes of mRNA expression in histamine-treated cells was calculated by comparing to the message in the medium-treated HCAEC. The results presented are representative of three experiments with similar results. Each value presented is the mean ± SEM of triplicate determinations. * indicates p<0.05 when compared to medium-treated cells.
The aortic tissues of HDC-/- mice are deficient in COX-2 mRNA expression with no change in COX-1 mRNA
Histidine decarboxylase (HDC) is the only enzyme that generates histamine from the amino acid histidine through a single enzymatic reaction. Because the HDC-/- mouse cannot synthesize histamine, this model was used to test the in vivo role of histamine on COX-1 and COX-2 gene expression. Accordingly, the expression of COX-1 and COX-2 mRNA in aortic tissues of wild-type (WT) and HDC-/- mice was determined. The semi-quantitative RT-PCR data presented in Fig. 8 demonstrate that aortic tissues of wild-type controls (lanes 1 and 2) constitutively express significant amounts of COX-2 mRNA whereas the tissues from HDC-/- mice express markedly lower levels (Fig. 8, lanes 3 and 4). The level of expression of COX-1 mRNA in the aortic tissues of HDC-/- mice was comparable to that of wild type controls. The deficient expression of COX-2 in HDC-/- mice aortic tissues and the in vitro data showing increased expression of COX-2 mRNA and protein in histamine-stimulated HCAEC suggest that histamine plays a key role in the regulation of the COX-2 pathway.
Figure 8.

Semi-quantitative reverse transcriptase-PCR analyses of the expression of COX-1 and COX-2 mRNA in aortic tissues of two wild-type and two HDC-/- mice. After exsanguination, 1-1.5 cm of thoracic aorta was dissected and rinsed with cold phosphate buffered saline. Total RNA was extracted from the tissues using TRIzol Reagent and cDNA prepared by reverse transcription for PCR reaction. PCR amplification of cDNA was performed with Taq Polymerase and PCR products were electrophoresed on agarose gel. rpL7 mRNA expression was used for normalization.
DISCUSSION
The importance of the mast cell and its products in vascular inflammation is well recognized (18, 19) and is suggested by increased levels of histamine in the coronary circulation (11) and increased synthesis of this amine in atherosclerotic lesions (20). Increasing interest is now focused on the role of COX-2 and prostanoid homeostasis in the pathogenesis of atherosclerosis (21). Previous reports from our laboratory have documented the ability of mast cell proteases and histamine to interact with endothelial cells and enhance inflammatory responses as determined by the production of IL-6 and IL-8 (9, 22, 23) and expression of TLR2 and TLR4 (16). In the present study we demonstrate that histamine, acting through H1R, selectively induces the expression of functionally active COX-2 and stimulates the production of PGI2 and PGE2 in HCAEC. The increases in PGI2 and PGE2 production by histamine-treated HCAEC were found to be associated with increased expression of mRNA encoding PGI2 and PGE2 synthases, the terminal enzymes involved in the conversion of PGH2 to PGI2 and PGE2 (24, 25). The finding that pretreatment of HCAEC with COX-2-specific inhibitor NS-398 or transfection of the cells with gene silencing siRNA inhibited histamine-stimulated prostanoid production documents that COX-2 expression contributes to the increase in prostanoid production. Interestingly, histamine did not stimulate COX-1 and TXA2 synthase gene expression or the production of TXA2. The preferential effect of histamine on the induction of COX-2 expression with resultant production of PGE2 and PGI2 and its lack of influence on COX-1 expression and TXA2 production support the concept of a distinct coupling pattern of COX-2 with PGE2 and PGI2 synthases and that of COX-1 with TXA2 synthase (25, 26). It is intriguing that, despite the coexistence of both COX-1 and COX-2 in HCAEC, histamine is able to segregate its influence on COX-2/PGE2/PGI2 pathway and not on COX-1/TXA2 pathway. It is also of interest that the increase in the production of prostanoids is not always associated with parallel induction of the expression or activity of a distinct prostaglandin synthase (27). Since a physiological balance in the production of PGI2 and TXA2 by endothelial cells is critical for the maintenance of vascular integrity and control of thrombosis (28, 29), the histamine-induced shift of prostanoid equilibrium in favor of PGI2 production is noteworthy and supports its well-recognized vasodilatory and vasoprotective function.
It is well-recognized that the cellular responses of histamine are mediated through a family of histamine receptors (H1, H2, H3 and H4) variably expressed in different cell types (30). Human endothelial cells predominantly express H1R which are involved in histamine-mediated hypersensitivity reactions and inflammatory responses (9, 31). The present finding that histamine-induced COX-2 expression and PGI2 production are inhibited by the H1R antagonist, fexofenidine, and not by the H2R antagonist, famotidine, suggest that histamine-mediated prostanoid homeostasis in HCAEC is regulated via H1R activation. These findings are in agreement with our previous reports demonstrating a distinct role of H1R in histamine-mediated proinflammatory cytokine production and TLR expression (16) and of others on histamine-induced released of PGI2 in human umbilical vein endothelial cells (32, 33). It should be noted that the stimulatory effect of histamine on COX-2 expression is not always mediated via H1R but is rather dependent on the cell type. For instance, histamine has been shown to stimulate COX-2 expression and generation of PGE2 in colon cancer cells through H2R (34). However, the lack of involvement of H2R in histamine-mediated prostanoid synthesis in HCAEC is further confirmed by the fact that dimaprit, an H2R-specific agonist, failed to stimulate the production of PGI2 (data not shown). Therefore, H1R seems to be an important player in the regulation of inflammatory responses and prostanoid homeostasis in vascular endothelium.
In addition to its direct effect, histamine was able to synergize the effect of LPS on the expression of COX-2 and production of PGE2 and PGI2 without modulating TXA2 production. The failure of histamine to produce TXA2 was not due to the lack of TXA2 synthase activity because LPS-treated HCAEC generated significant amounts of TXA2 after 24 h and 48 h of treatment (Fig 6C). Since the production of TXA2 is thought to be coupled to COX-1 and TXA2 synthase (25) and histamine does not stimulate the expression of COX-1 (Figure 1 and 2) or TXA2 synthase in HCAEC (Fig 7C), the lack of TXA2 production by histamine-treated HCAEC is predictable. The fact that histamine failed to modulate TXA2 production either directly or in the presence of LPS suggests a novel mechanism by which this mast cell mediator regulates prostanoid homeostasis in HCAEC. Such a selective release of PGI2 without altering TXA2 synthesis is seen in hypoxia-induced COX-2 expression in human umbilical vein endothelial cells (35). It is noteworthy that, although TXA2 is predominantly produced by mature human platelets, which express only COX-1 (36), TXA2 is produced in other cell types such as monocytes and macrophages which express both COX-1 and COX-2 (37). However, distinct contribution of COX-1 and COX-2 to the production of a particular prostanoid in response to specific inflammatory signals has not been established. Thus, the present finding that LPS, but not histamine, was able to induce TXA2 production by HCAEC suggests that the type of inflammatory agent encountering the cells influences the pattern of prostaglandin production.
The ability of histamine to synergize LPS-induced COX-2 expression and prostanoid production underscores the potential role of this mast cell mediator to amplify infection-associated inflammatory responses in vascular endothelium. It is well-recognized that TLRs are critical components of the innate immune system and each of these TLRs recognizes a distinct pathogen-associated molecule to initiate the inflammatory response (38-43). Among the TLRs identified thus far, TLR4, in association with its accessory molecules, recognizes LPS (43-47). Recently, we demonstrated that histamine has the ability to stimulate the expression of TLR4 mRNA and protein, and amplify LPS-induced production of cytokines in human umbilical vein endothelial cells (16). Therefore, although the effect of histamine on TLR4 expression in HCAEC was not examined in this study, it is reasonable to suggest that the amplification of LPS-induced COX-2 expression and enhancement in the production of PGI2 and PGE2 is due to increased expression of functionally active TLR4. The assumption is further supported by the finding that, although histamine alone is a poor inducer of NF-κB translocation in endothelial cells, it markedly enhances LPS-induced NF-κB activation, an index of amplified TLR4 activation (9). It is noteworthy that dysfunctional TLR4 polymorphism, which affects the extracellular domain of the receptor, is associated with a reduction in systemic levels of proinflammatory mediators (48) and cardiovascular events (49). Furthermore, compared to control subjects, individuals with TLR4 polymorphism who presented with significantly lower intima-media thickness in the carotid arteries had a 65% reduction of 11-dehydro-TXB2 in the urine indicating decreased systemic production of TXA2 in these individuals.
The circulating levels of PGI2 and TXA2 are pivotal for the normal functioning of the cardiovascular system (21, 50) and the syntheses of both PGI2 and TXA2 are increased in patients with atherosclerosis (14, 15). PGI2 is an antithrombotic and vasodilator molecule that can decrease vascular remodeling and cholesterol uptake (51, 52). This is particularly evident from the fact that disruption of the prostacyclin receptor (IPR) gene leads to increased intima/media ratio in response to vascular injury and promote initiation and progression of atherosclerosis in hyperlipidemic mouse (53, 54) suggesting a protective role for prostacyclin in vascular remodeling. TXA2, on the other hand, is a prothrombotic and vasoconstricting agent, and enhances vascular remodeling (51, 52). Since PGI2 and TXA2 exert opposing influences in the cardiovascular system, the upregulation of the expression of COX-2, PGE2 synthase, and PGI2 synthase in HCAEC by histamine emphasizes its importance in the maintenance of vascular integrity.
In conclusion, the present study demonstrates that incubation of HCAEC with histamine leads to increased expression of COX-2 with resultant enhancement in the production of PGE2 and PGI2. We also present evidence that histamine-induced production of PGE2 and PGI2 can be attributed to increased gene expression of PGE2 and PGI2 synthases. Interestingly, histamine did not affect TXA2 synthase gene expression or TXA2 production. In addition to the direct effect, histamine is capable of amplifying LPS-stimulated expression of COX-2 and production of PGE2 and PGI2 potentially via histamine-stimulated expression of TLR4 (16). Both the direct and synergizing effects of histamine on endothelial cell activation are found to be mediated via H1R activation. The role of histamine in the regulation of COX-2 expression in the vasculature is further supported by the finding that the aortic tissue of HDC-/- mouse, which is deficient in histamine, has significantly reduced expression of COX-2 mRNA without a change in the levels of COX-1 mRNA. These results underscore the important role of histamine in prostanoid homeostasis in the vasculature.
Acknowledgment
The authors are grateful to Dr. John Belmont, Professor Emeritus in Pediatrics at the University of Kansas Medical Center for contributing to the statistical analyses; and to Mr. Donald D. Smith for his technical assistance.
Abbreviations
- COX
cyclooxygenase
- EIA
enzyme immunoassay
- H1R
histamine H1 receptor
- H2R
histamine H2 receptor
- HCAEC
human coronary artery endothelial cells
- TX
thromboxane
Footnotes
This work was supported by National Institutes of Health Grant R01-HL070101 and Joseph and Elizabeth Carey Arthritis Fund from the Kansas University Endowment Association.
REFERENCES
- 1.Barnes PJ. Histamine receptors in the lung. Agents Action. 1991;33:103–122. doi: 10.1007/978-3-0348-7309-3_9. [DOI] [PubMed] [Google Scholar]
- 2.Hill SJ. Multiple histamine receptors: Properties and functional characteristics. Biochem. Soc. Trans. 1992;20:122–125. doi: 10.1042/bst0200122. [DOI] [PubMed] [Google Scholar]
- 3.Elenkov IJ, Webster E, Papanicolaou DA, Fleisher TA, Chrousos GP, Wilder RL. Histamine potently suppresses human IL-12 and stimulates IL-10 production via H2 receptors. J. Immunol. 1998;161:2586–2593. [PubMed] [Google Scholar]
- 4.Dileepan KN, Lorsbach RB, Stechschulte DJ. Mast cell granules inhibit macrophage-mediated lysis of mastocytoma cells (P815) and nitric oxide production. J. Leukoc. Biol. 1993;53:446–453. doi: 10.1002/jlb.53.4.446. [DOI] [PubMed] [Google Scholar]
- 5.Clark RA, Gallin JI, Kaplan AP. The selective chemotactic activity of histamine. J. Exp. Med. 1975;142:1462–1476. doi: 10.1084/jem.142.6.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dileepan KN, Simpson KM, Lynch SR, Stechschulte DJ. Dismutation of eosinophil superoxide by mast cell granule superoxide dismutase. Biochem. Arch. 1989;5:153–160. [Google Scholar]
- 7.Ogden BE, Hill HR. Histamine regulates lymphocyte mitogenic responses through activation of specific H1 and H2 histamine receptors. Immunol. 1980;41:107–114. [PMC free article] [PubMed] [Google Scholar]
- 8.Beer DJ, Matloff SM, Rocklin RE. The influence of histamine on immune and inflammatory responses. Adv. Immunol. 1984;35:209–268. doi: 10.1016/s0065-2776(08)60577-5. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Chi Y, Stechschulte DJ, Dileepan KN. Histamine-induced production of IL-6 and IL-8 by human coronary artery endothelial cells is enhanced by endotoxin and TNF-α. Microvasc. Res. 2001;61:253–262. doi: 10.1006/mvre.2001.2304. [DOI] [PubMed] [Google Scholar]
- 10.Kalsner S, Richards R. Coronary arteries of cardiac patients are hyper reactive and contain stores of amines: a mechanism for coronary spasm. Science. l984;223:l435–1437. doi: 10.1126/science.6701530. [DOI] [PubMed] [Google Scholar]
- 11.Sakata YK, Komamura K, Hirayama A, Nato S, Kitakaze M, Hori M, Kodama K. Elevation of the plasma histamine concentration in the coronary circulation in patients with variant angina. Am. J. Cardiol. 1996;77:1121–1126. doi: 10.1016/s0002-9149(96)00147-6. [DOI] [PubMed] [Google Scholar]
- 12.Delneste Y, Lassalle P, Jeannin P. Histamine induces IL-6 production by human endothelial cells. Clin. Exp. Immunol. 1994;98:344–349. doi: 10.1111/j.1365-2249.1994.tb06148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeannin P, Delneste Y, Gossette P, Molet S, Lassalle P, Hamid Q, Tsicopoulos A, Tonnel AB. Histamine induces IL-8 secretion by endothelial cells. Blood. 1994;84:2229–2233. [PubMed] [Google Scholar]
- 14.FitzGerald GA, Smith B, Pedersen AK, Brash AR. Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N. Engl J. Med. 1984;310:1065–1068. doi: 10.1056/NEJM198404263101701. [DOI] [PubMed] [Google Scholar]
- 15.Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;102:840–845. doi: 10.1161/01.cir.102.8.840. [DOI] [PubMed] [Google Scholar]
- 16.Talreja J, Kabir MH, Filla M, Stechschulte DJ, Dileepan KN. Histamine induces Toll-like receptors 2 and 4 expression in endothelial cells and enhances sensitivity to Gram-positive and Gram-negative bacterial cell wall components. Immunol. 2004;113:224–233. doi: 10.1111/j.1365-2567.2004.01946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato Y, Nagai A, Saito M, Ito T, Koga M, Tsuboi R. Food-dependent exercise-induced anaphylaxis with a high level of plasma noradrenaline. J. Dermatol. 2007;34:110–113. doi: 10.1111/j.1346-8138.2006.00227.x. [DOI] [PubMed] [Google Scholar]
- 18.Kaartinen M, Penttila A, Kovanen PT. Mast cells in rupture-prone areas of human coronary atheromas produce and store TNF-alpha. Circulation. 1996;94:2787–2792. doi: 10.1161/01.cir.94.11.2787. [DOI] [PubMed] [Google Scholar]
- 19.Kaartinen M, Penttila A, Kovanen PT. Mast cells accompany microvessels in human coronary atheromas: implications for intimal neovascularization and hemorrhage. Atherosclerosis. 1996;123:123–131. doi: 10.1016/0021-9150(95)05794-3. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi S, Tanimoto A, Arima N, Xu H, Murata Y, Hamada T, Makishima K, Sasaguri Y. Effects of histamine and interleukin-4 synthesized in arterial intima on phagocytosis by monocytes/macrophages in relation to atherosclerosis. FEBS Lett. 2001;505:217–222. doi: 10.1016/s0014-5793(01)02823-x. [DOI] [PubMed] [Google Scholar]
- 21.Grosser T, Fries S, Fitzgerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jehle AB, Li Y, Stechschulte AC, Stechschulte DJ, Dileepan KN. Endotoxin and mast cell granule proteases synergistically activate human coronary artery endothelial cells to generate interleukin-6 and interleukin-8. J. Interferon Cytokine Res. 2000;20:361–368. doi: 10.1089/107999000312298. [DOI] [PubMed] [Google Scholar]
- 23.Chi L, Li Y, Stehno-Bittel L, Gao J, Morrison DC, Stechschulte DJ, Dileepan KN. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J. Interferon Cytokine Res. 2001;21:231–240. doi: 10.1089/107999001750169871. [DOI] [PubMed] [Google Scholar]
- 24.Smith WL. Prostanoid biosynthesis and mechanisms of action. Am. J. Physiol. 1992;263:F181–191. doi: 10.1152/ajprenal.1992.263.2.F181. [DOI] [PubMed] [Google Scholar]
- 25.Ueno N, Takegoshi Y, Kamei D, Kudo I, Murakami M. Coupling between cyclooxygenases and terminal prostanoid synthases. Biochem. Biophys. Res.Commun. 2005;338:70–76. doi: 10.1016/j.bbrc.2005.08.152. [DOI] [PubMed] [Google Scholar]
- 26.Yu Y, Fan J, Hui Y, Rouzer CA, Marnett LJ, Klein-Szanto AJ, FitzGerald GA, Funk CD. Targeted cyclooxygenase gene (ptgs) exchange reveals discriminant isoform functionality. J. Biol. Chem. 2007;282:1498–1506. doi: 10.1074/jbc.M609930200. [DOI] [PubMed] [Google Scholar]
- 27.Camacho M, López-Belmonte J, Vila L. Rate of vasoconstrictor prostanoids released by endothelial cells depends on cyclooxygenase-2 expression and prostaglandin I synthase activity. Circ. Res. 1998;83:353–365. doi: 10.1161/01.res.83.4.353. [DOI] [PubMed] [Google Scholar]
- 28.Bunting S, Moncada S, Vane JR. The prostacyclin--thromboxane A2 balance: pathophysiological and therapeutic implications. Br. Med. Bull. 1983;39:271–276. doi: 10.1093/oxfordjournals.bmb.a071832. [DOI] [PubMed] [Google Scholar]
- 29.Wu KK, Thiagarajan P. Role of endothelium in thrombosis and hemostasis. Annu. Rev. Med. 1996;47:315–331. doi: 10.1146/annurev.med.47.1.315. [DOI] [PubMed] [Google Scholar]
- 30.Hough LB. Genomics meets histamine receptors: new subtypes, new receptors. Mol. Pharmacol. 2001;59:415–419. [PubMed] [Google Scholar]
- 31.Walsh GM, Annunziato L, Frossard N, Knol K, Levander S, Nicolas JM, Taglialatela M, Tharp MD, Tillement JP, Timmerman H. New insights into the second generation antihistamines. Drugs. 2001;61:207–236. doi: 10.2165/00003495-200161020-00006. [DOI] [PubMed] [Google Scholar]
- 32.Baenziger NL, Force LE, Becherer PR. Histamine stimulates prostacyclin synthesis in cultured human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 1980;92:1435–1440. doi: 10.1016/0006-291x(80)90447-7. [DOI] [PubMed] [Google Scholar]
- 33.McIntyre TM, Zimmerman GA, Satoh K, Prescott SM. Cultured endothelial cells synthesize both platelet-activating factor and prostacyclin in response to histamine, bradykinin, and adenosine triphosphate. J. Clin. Invest. 1985;76:271–280. doi: 10.1172/JCI111957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cianchi F, Cortesini C, Schiavone N, Perna F, Magnelli L, Fanti E, Bani D, Messerini L, Fabbroni V, Perigli G, Capaccioli S, Masini E. The role of cyclooxygenase-2 in mediating the effects of histamine on cell proliferation and vascular endothelial growth factor production in colorectal cancer. Clin. Cancer. Res. 2005;11:6807–6815. doi: 10.1158/1078-0432.CCR-05-0675. [DOI] [PubMed] [Google Scholar]
- 35.Cook-Johson RJ, Demasi M, Cleland LG, Gamble JR, Saint DA, James MJ. Endothelial cell COX-2 expression and activity in hypoxia. Biochim. Biophys. Acta. 2006;1761:1443–1449. doi: 10.1016/j.bbalip.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 36.FitzGerald GA. Mechanisms of platelet activation: thromboxane A2 as an amplifying signal for other agonists. Am. J. Cardiol. 1991;68:11B–15B. doi: 10.1016/0002-9149(91)90379-y. [DOI] [PubMed] [Google Scholar]
- 37.Fu YX, Watson G, Jimenez JJ, Wang Y, Lopez DM. Expansion of immunoregulatory macrophages by granulocyte-macrophage colony-stimulating factor derived from a murine mammary tumor. Cancer Res. 1990;50:227–234. [PubMed] [Google Scholar]
- 38.Lehner MD, Morath S, Michelsen KS, Schumann RR, Hartung T. Induction of cross-tolerance by lipopolysaccharide and highly purified lipoteichoic acid via different Toll-like receptors independent of paracrine mediators. J. Immunol. 2001;166:5161–5167. doi: 10.4049/jimmunol.166.8.5161. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi O, Akira S. MyD88 as a bottle neck in Toll/IL-1 signaling. Curr. Top. Microbiol. Immunol. 2002;270:155–167. doi: 10.1007/978-3-642-59430-4_10. [DOI] [PubMed] [Google Scholar]
- 41.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 42.Krutzik SR, Sieling PA, Modlin RL. The role of Toll-like receptors in host defense against microbial infection. Curr. Opin. Immunol. 2001;13:104–108. doi: 10.1016/s0952-7915(00)00189-8. [DOI] [PubMed] [Google Scholar]
- 43.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 44.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 45.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 46.Kennedy MN, Mullen GE, Leifer CA, Lee CW, Mazzoni A, Dileepan KN, Segal DM. A complex of soluble MD-2 and lipopolysaccharide serves as an activating ligand for Toll-like receptor 4. J. Biol. Chem. 2004;279:34698–34704. doi: 10.1074/jbc.M405444200. [DOI] [PubMed] [Google Scholar]
- 47.Talreja J, Dileepan K, Puri S, Kabir MH, Segal DM, Stechschulte DJ, Dileepan KN. Human conjunctival epithelial cells lack lipopolysaccharide responsiveness due to deficient expression of MD2 but respond after interferon-gamma priming or soluble MD2 supplementation. Inflammation. 2005;29:170–181. doi: 10.1007/s10753-006-9014-y. [DOI] [PubMed] [Google Scholar]
- 48.Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 49.Ameziane N, Beillat T, Verpillat P, Chollet-Martin S, Aumont MC, Seknadji P, Lamotte M, Lebret D, Ollivier V, de Prost D. Association of the Toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscler. Thromb. Vasc. Biol. 2003;23:e61–64. doi: 10.1161/01.ATV.0000101191.92392.1D. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat. Rev. Drug Discov. 2006;5:75–86. doi: 10.1038/nrd1929. [DOI] [PubMed] [Google Scholar]
- 51.Davidge ST. Prostaglandin H synthase and vascular function. Circ. Res. 2001;89:650–660. doi: 10.1161/hh2001.098351. [DOI] [PubMed] [Google Scholar]
- 52.Dogne JM, Hanson J, Pratico D. Thromboxane, prostacyclin and isoprostanes: therapeutic targets in atherogenesis. Trends Pharmacol. Sci. 2005;26:639–644. doi: 10.1016/j.tips.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yakode M, Kita T, Narumiya S. Roles of thromboxane A2 and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J. Clin. Invest. 2004;114:784–794. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Egan KM, Lawson JA, Fries S, Koller B, Rader DA, Smyth EM, FitzGerald GA. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]