

Abstract
The innate immune system senses the invasion of pathogenic microorganisms and tissue injury through Toll-like receptors (TLR), a mechanism thought to be limited to immune cells. We recently found that neurons express several TLRs, and that the levels of TLR2 and TLR4 are increased in neurons in response to energy deprivation. Here we report that TLR4 expression increases in neurons when exposed to amyloid β-peptide (Aβ1-42) or the lipid peroxidation product 4-hydroxynonenal (HNE). Neuronal apoptosis triggered by Aβ and HNE was mediated by jun N-terminal kinase (JNK); neurons from TLR4 mutant mice exhibited reduced JNK and caspase-3 activation and were protected against apoptosis induced by Aβ and HNE. Levels of TLR4 were decreased in inferior parietal cortex tissue specimens from end-stage AD patients compared to aged-matched control subjects, possibly as the result of loss of neurons expressing TLR4. Our findings suggest that TLR4 signaling increases the vulnerability of neurons to Aβ and oxidative stress in AD, and identify TLR4 as a potential therapeutic target for AD.
Keywords: Toll like receptors, amyloid β-peptide, 4-hydroxynonenal, neuron, cell death
Introduction
Toll-like receptors (TLRs) are evolutionarily conserved cell surface proteins originally identified in vertebrates on the basis of their homology with Toll, a protein that stimulates the production of antimicrobial proteins. The TLR-family members are pattern-recognition receptors that collectively recognize lipid, carbohydrate, peptide and nucleic-acid structures that are broadly expressed by different groups of microorganisms (Trinchieri and Sher., 2007). The signaling pathways activated by TLRs are broadly classified into MyD88-dependent and independent pathways (Takeda and Akira, 2005); MyD88 is the universal adapter protein recruited by all TLRs except TLR3. Major pathways activated by TLR engagement include IκB kinase (IKK) - NFκB, kinase cascades that activate mitogen-activated protein kinases (MAPK) and phosphatidylinositol 3-kinase (PI3K) - Akt. In addition, activation of jun N-terminal kinase (JNK) and p38 MAPK cascades by TLRs result in activation of different activator protein-1 (AP-1) subunits and transcription factors interacting with AP-1 (Chen et al., 2004; Doyle and O'Neill., 2006; Hoebe et al., 2006; Krishnan et al., 2007; Tang et al., 2007).
Although TLRs have been intensively studied in leukocytes where their functions have been established, recent findings suggest they can also be expressed in non-immune cells including hepatocytes and muscle cells (Liu et al., 2002; Yang et al., 2005). Some TLRs are present in the brain, where their expression where they are expressed in glial cells (microglia, astrocytes and oligodentrocytes) (Lehnardt et al., 2003; Olson and Miller., 2004; Jack et al., 2005; Walter et al., 2007) and neurons (Lefon et al., 2006; Ma et al., 2006; Kigerl etal., 2007; Rolls et al., 2007). We recently showed that TLR2 and TLR4 are expressed in cerebral cortical neurons, where their levels and downstream signaling via JNK are increased in response to energy deprivation in cell culture and ischemia in vivo (Tang et al., 2007). Neurons lacking functional TLR2 or TLR4 function exhibit increased resistance to death induced by energy deprivation and stroke (Tang et al., 2007).
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that is the most common cause of dementia in the elderly. Basic pathological changes include neurofibrillary tangles, consisting of filaments of hyper-phosphorylated tau protein and deposits of aggregated amyloid beta-protein (Aβ) in neuritic plaques. Aβ is a 39-42 amino acid peptide produced from amyloid β-protein precursor (APP) by proteolytic processing (Mattson, 2004). Aβ is heterogeneous at its C-terminus, resulting in peptides of 39-42 amino acids, among which Aβ42 is the most toxic towards neurons (Mattson, 2004). Aβ can also activate microglia, initiating a pro-inflammatory cascade that involves the production of proinflammatory cytokines (Li et al., 1996; Hu et al., 1998; Hashimoto et al., 2006). Activation of TLRs on microglia can promote the cellular uptake and clearance of Aβ (Chen et al., 2006; Tahara et al., 2006). In addition, Aβ enhances the action of TLR-2 and -4 agonists but antagonizes TLR-9-mediated inflammation in primary mouse microglial cell cultures (Lotz et al., 2005). Data from studies of AD patients and experimental models of AD suggest that Aβ damages neurons by causing membrane-associated oxidative stress and the production of the lipid peroxidation product 4-hydroxynonenal (HNE) (Kruman et al., 1997; Mark et al. 1997; Sayre et al. 1997; Lovell and Markesbery, 2006). In the present study we employed primary neuronal cultures from TLR4 mutant mice and wild-type control mice to identify key roles for neuronal TLR4 signaling in neuronal apoptosis induced by Aβ and HNE.
Materials and Methods
Animals and chemicals
TLR4 mutant mice (C.C3H-Tlr4lps-d) and wild-type mice of the same genetic background were purchased from Jackson Laboratories and maintained in our animal facility under pathogen-free conditions on a 12 h light/12 h dark cycle with continuous access to food and water. All animal procedures were approved by the National Institute on Aging Animal Care and Use Committee. Aβ1–42 was purchased from Bachem (Torrance, CA) and HNE was purchased from Cayman Chemicals (Ann Arbor, USA).
Neuronal cultures
Cortical tissues were removed from 15 day-old TLR4 mutant mouse embryos (C.C3H-Tlr4lps-d) and wild-type control embryos and incubated for 15 min in a solution of 2 mg/mL trypsin in-Ca2+/Mg2+-free Hank's balanced salt solution (HBSS) buffered with 10 mmol/L HEPES (Invitrogen, Carlsbad, CA, USA). The cortical tissue was then rinsed once in HBSS, followed by 5 min incubation in HBSS containing 1 mg/mL trypsin inhibitor (Sigma), and a final rinse in HBSS. Tissues were then dissociated by trituration through the narrowed bore of a fire-polished Pasteur pipette and were distributed to polylysine-coated plastic culture dishes (Corning, Lowell, MA, USA) containing 2 mL of neurobasal medium with B27 supplements (Invitrogen). The culture density was 80–120 cells/mm of culture surface. Cultures were maintained at 37°C in a 5% CO2/94% room air, humidified incubator. All experiments were performed with neurons that had been in culture for 8 days.
Human subjects and brain specimens
Parietal cortex specimens from the brains of 10 patients with AD and 10 control subjects that had been enrolled in the University of Kentucky Alzheimer's Disease Center Autopsy Program were used for this study. The postmortem brain tissue samples were acquired under an IRB protocol approved by the University of Kentucky School of Medicine. All patients with AD met both clinical diagnostic criteria and neuropathological diagnostic criteria of AD (National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease, 1997). The control subjects had no history or neuropathological signs of a brain disorder. The sex, age, postmortem interval, Braak stage and plaque density for each subject is shown in Table 1. At autopsy, tissue specimens were rapidly removed and frozen, and were stored at -80°C.
Table1.
Clinical data on AD patients and non-demented control subjects. The numbers of neuritic plaques per 2.35 mm2 microscopic field were counted in Aβ anti body stained sections of inferior parietal lobule (the value for each subject is the mean of counts of the 5 most involved fields in each section). PMI, postmortem interval.
| Patients | Age (years) | Gender | PMI (h) | Neuritic plaques | Cause of death | Braak stage |
|---|---|---|---|---|---|---|
| Control | ||||||
| 1 | 85 | Male | 2.00 | 5.6 | Unknown | 3 |
| 2 | 86 | Female | 2.25 | 7.6 | Unknown | 2 |
| 3 | 91 | Female | 4.00 | 10.4 | Unknown | 1 |
| 4 | 86 | Female | 3.75 | 7.8 | Cardiovascular disease | 1 |
| 5 | 81 | Male | 2.00 | 13.4 | Pulmonary embolism | 2 |
| 6 | 87 | Male | 2.40 | 0.2 | Prostate cancer | 2 |
| 7 | 82 | Male | 2.10 | 1.2 | Congestive heart failure, pneumonia | 1 |
| 8 | 74 | Male | 4.00 | 0.0 | Congestive heart failure | 1 |
| 9 | 76 | Female | 2.00 | 0.0 | Chronic obstructive pulmonary disease | 1 |
| 10 | 79 | Male | 1.75 | 16.2 | Bladder cancer | 2 |
| Mean±S.D. | 82.7±5.3 | Male | 2.62±0.91 | 6.24±5.89 | ||
| Alzheimer's disease | ||||||
| 1 | 83 | Female | 4.00 | 24.6 | Aspiration pneumonia | 6 |
| 2 | 86 | Male | 4.25 | 23.4 | Bowel obstruction | 6 |
| 3 | 78 | Female | 3.75 | 34.2 | Unknown | 6 |
| 4 | 90 | Female | 2.60 | 30.4 | Unknown | 6 |
| 5 | 75 | Male | 2.33 | 19.0 | Congestive heart failure | 6 |
| 6 | 81 | Female | 3.00 | 17.4 | Unknown | 6 |
| 7 | 86 | Male | 3.25 | 19.4 | Respiratory infection | 6 |
| 8 | 74 | Male | 3.00 | 27.2 | Fall | 6 |
| 9 | 84 | Male | 4.50 | 34.8 | Unknown | 6 |
| 10 | 84 | 2.75 | 31.4 | Aspiration pneumonia | 6 | |
| Mean±S.D. | 82.1±4.8 | 3.34±0.74 | 26.18±6.41 |
RT-PCR and microarray analysis
Total RNA from cultured neurons was prepared using a standard Trizol protocol (Invitrogen). RNA purity was assessed by spectrophotometry. First-strand cDNAs were generated using the cDNA synthesis protocol of the SuperScript RT-PCR system (Invitrogen). The PCR reactions were performed using the Supermix (Invitrogen) according to the manufacturer's protocol. The sequences of the primers are: TLR2 (5V- GAGCGAGCTGGGTAAAGTAGAAA-3V, 5V- AGCCGAGGCAAGAACAAAGA-3V); TLR3 (5V- TCTCTGGGCTGAAGTGGACAA -3V, 5V- AGCAAGGGAGAATGAGCAAGTGAC-3V); TLR4 (5V-CAGTGGGTCAAGGAACAGAAGC-3V, 5V- GACAATGAAGATGATGCCAGAGC).
Each primer pair spanned at least one intron to distinguish between products amplified from cDNA and genomic DNA. The test cDNAs were always run in parallel with at least one non-cDNA negative control. The final reaction volume was 15 μl, and amplification was 30 (RT-PCR) cycles. PCR products were separated by electrophoresis in 1.75-2% agarose gels and were visualized by ethidium bromide staining.
Immunoblot analysis
Lysates of cultured cells were obtained by washing the cells in ice-cold PBS and resuspending them in cell lysis buffer. Proteins were extracted from postmortem human brain tissue specimens using T-PER tissue protein extraction buffer containing a protease inhibitor cocktail (Sigma). Protein concentration was determined using a BCA protein assay kit (Pierce, USA) and 40 μg of protein were separated by SDS-PAGE (8-12%) and transferred to a nitrocellulose membrane. The membrane was blocked in 5% nonfat milk for 1 h at room temperature, followed by an overnight incubation at 4 °C with primary antibodies against: actin (Sigma); TLR4, TLR2, JNK and caspase-3 (Cell Signaling); GAPDH (Santa Cruz); synaptophysin, abcam and BA-1 (Wako). The membrane was then washed and incubated with a secondary antibody for 1 h at room temperature. Protein bands were visualized using a chemiluminescence detection kit (Amersham Biosciences, UK).
Immunocytochemistry
Neurons grown on 24-chamber microscope slides were fixed in 4% paraformaldehyde and incubated at 4 °C with primary TLR4 antibody (Santa Cruz) overnight, followed by a 2 h incubation with FITC-conjugated secondary antibody. Images of cells were acquired using a Zeiss Axiophot microscope (Oberkochen, Germany).
Statistical analysis
Statistical comparisons were made using unpaired t tests.
Results
Levels of TLR4 are Increased in Neurons Exposed to Aβ and HNE
Previous studies have suggested that neurons express a subset of TLRs that includes TLR2, TLR3 and TLR4 (Ma et al., 2006; Tang et al., 2007). To determine whether TLR signaling is involved in neuronal responses to Aβ and HNE, we first evaluated the expression of TLRs 2, 3 and 4 in cultured cortical neurons exposed to Aβ or HNE. Compared to control cultures, levels of TLR4 mRNA were increased within 18 h of exposure to Aβ1-42, whereas levels of TLR2 and TLR3 mRNAs were unchanged (Fig. 1A). There was a corresponding increase in the level of TLR4 protein in response to Aβ1-42 treatment which occurred in a concentration-dependent manner with Aβ1-42 concentrations of 1-10 μM (Fig. 1B).
Figure 1.
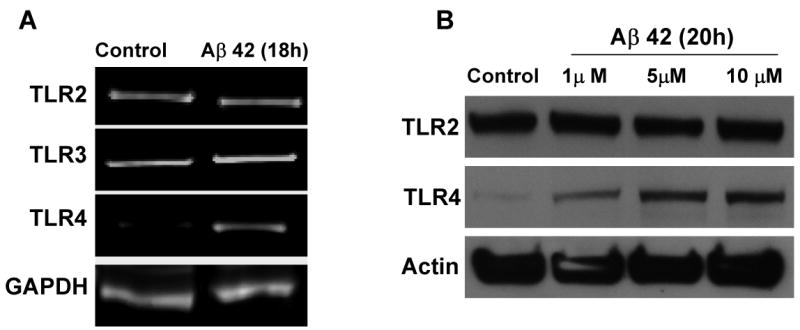
Cultured cortical neuronal cells up-regulate expression of TLR4 when exposed to Aβ1-42. A. Levels of TLR 2, 3 and 4 mRNAs in neurons treated with Aβ (10 μM) for the indicated time periods. TLR4 mRNA levels increased in response to Aβ treatment, whereas TLR2 and TLR3 mRNA levels did not change. B. Immunoblot showing relative levels of TLR2 (95 KDa) and TLR4 (90 KDa) in lysates of cultured neurons that had been treated for 20 hours with vehicle (Control) or Aβ (1, 5 or 10 μM). Levels of TLR4, but not TLR2 or TLR3, were increased in response to Aβ treatment. Analyses were performed on pooled samples from 4-8 cultures.
Membrane lipid peroxidation has been implicated in the pathogenesis of AD and the aldehyde HNE has been suggested to mediate neurotoxic effects of Aβ (Keller et al., 1997; Kruman et al., 1997; Lovell et al., 1997; Mark et al., 1997a, 1997b; Sayre et al., 1997). We therefore determined whether exposure of neurons to HNE, at concentrations (5 – 10 μM) reported to be present in neurons exposed to Aβ and in AD (Mark et al., 1997; Lovell et al., 1997), would affect TLR expression. Levels of TLR2 and TLR4 mRNAs were increased in neurons that had been exposed to 10 μM HNE for 6 or 18 hours, whereas levels of TLR3 mRNA were unchanged (Fig. 2A). Levels of TLR2 and TLR4 proteins, but not TLR3 protein, were increased within 3 h of exposure to HNE and remained elevated through 20 h of exposure (Fig. 2B, C). Because TLR4 levels increased in response to both Aβ and HNE, we immunostained cultured neurons with the TLR4 antibody to evaluate its subcellular localization. Neurons in control cultures exhibited a low level of TLR4 immunoreactivity, while neurons that had been exposed to Aβ or HNE exhibited more intense TLR4 immunoreactivity which was concentrated at the cell surface and in cytosolic compartments, but not in the nucleus (Fig. 3).
Figure 2.

Expression of TLR2 and TLR4 increase in cortical neuronal cultures in response to exposure to HNE. A. The levels of TLR 2, 3 and 4 mRNAs in neurons treated with HNE (10 μM). B and C. Immunoblots showing protein levels of TLR2 (95 KDa) and TLR4 (90 KDa) in cultured cortical neuronal cells that had been exposed to the indicated concentrations of HNE for the indicated time periods. Levels of both TLR2 and TLR4 were increased within 3 hours and remained elevated through 20 hours of exposure to HNE. Analyses were performed on pooled samples from 4-8 cultures.
Figure 3.
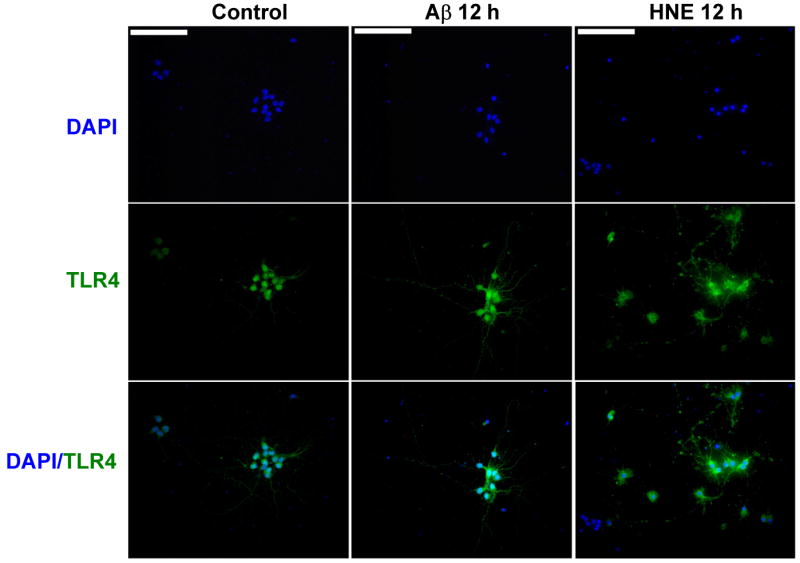
Aβ and HNE induce increases in TLR4 immunoreactivity in cultured cortical neurons. Images of TLR4 immunofluorescence (green) in cultured neurons in a control culture, and cultures that had been exposed for 12 hours to either 10 μM Aβ1-42 or 5 μM HNE. Scale bar = 100μM.
Neurons deficient in TLR4 receptors are resistant to Aβ - and HNE -induced JNK activation and apoptosis
Previous studies have shown that Aβ and HNE can induce neuronal apoptosis by a mechanism involving activation of JNK and caspase-3 (Loo et al., 1993; Kruman et al., 1997; Mattson et al., 1998; Camandola et al., 2000; Troy et al., 2001). Because the expression of TLR4 increased in response to Aβ and HNE, we performed experiments with cortical cultures established from TLR4 mutant mice to establish the role of TLR4 in neuronal vulnerability to Aβ and HNE. In contrast to the robust increase in JNK activation in wild-type neurons in response to Aβ (Fig. 4 A) and HNE (Fig. 4C), neurons deficient in TLR4 did not exhibit a JNK response to either Aβ or HNE. These results suggested an essential role for TLR4 in neuronal JNK activation induced by Aβ and HNE.
Figure 4.
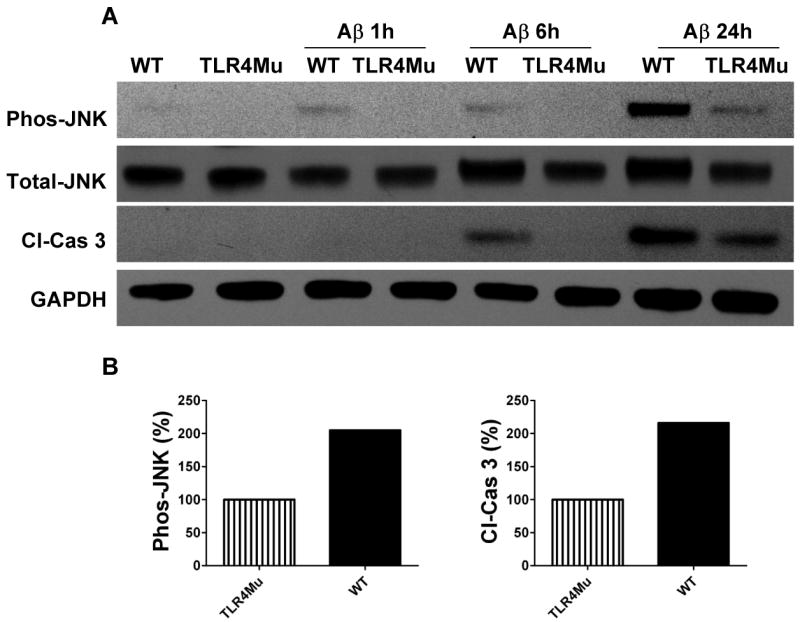
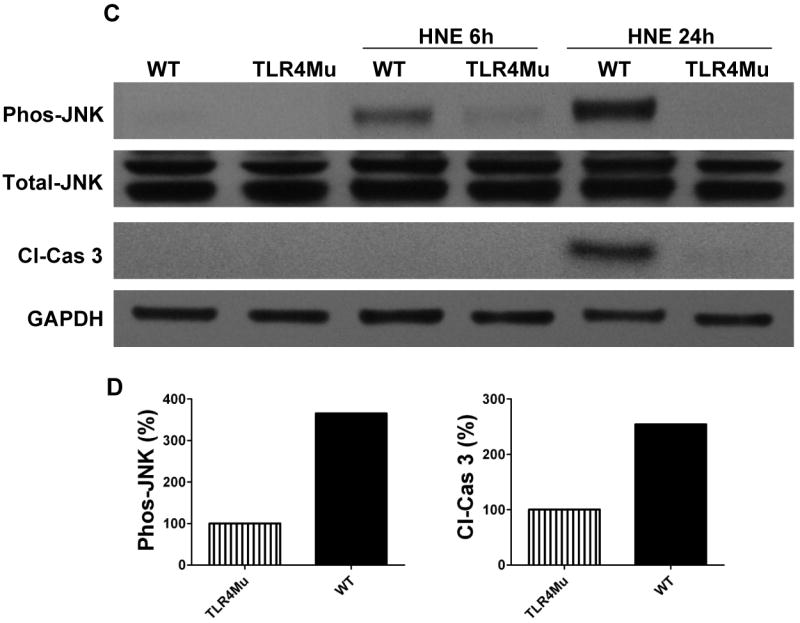
Evidence that TLR4 mediates activation of JNK and caspase-3 in response to Aβ and HNE in cortical neurons. Immunoblot analysis of phospho-JNK (46 kDa), total JNK (46 kDa) and cleaved caspase-3 (19 kDa) in cell lysates of neurons in cortical cultures established from either wild-type (WT) or TLR4 deficient (TLR4mu) mice. The neurons were left untreated or were exposed to 5 μM Aβ1-42 (panel A) or 5 μM HNE (panel C) for the indicated time points. Results of densitometric analysis of phospho-JNK and cleaved caspase-3 protein levels in WT compared with TLR4 mutant cells from Aβ treated (panel B) and HNE treated (panel D) samples at 24 hours. Analyses were performed on pooled samples from at least 3 separate cultures that had been established from 10-14 embryos in each group.
Because JNK activation and consequent mitochondrial alterations and caspase-3 activation are implicated in stress induced neuronal death (Okuno et al., 2004), we measured levels of activated (cleaved) caspase-3 in the same cell lysates used to measure JNK levels. Aβ and HNE treatments resulted in a much greater increase in activated caspase-3 after 24 h in wild-type neurons compared to TLR4 mutant neurons (Fig. 4A, C) suggesting a pro-apoptotic role for TLR4 in neurons exposed to Aβ and HNE. In order to determine whether JNK activation was required for apoptosis induced by Aβ and HNE, we tested the effect of a JNK inhibitor on Aβ- and HNE-induced caspase-3 activation in wild-type neurons. Neurons treated with the JNK inhibitor SP600125 exhibited reduced levels of activated JNK and of activated caspase-3 in response to Aβ and only reduced the level of activated JNK in response to HNE compared to vehicle-treated control neurons (Fig. 5A, C), suggesting an essential role for JNK in Aβ and HNE-induced neuronal apoptosis. As we observed no differences in activated caspase-3 levels between control and SP600125-treated cells following 24 h HNE treatment, we performed further experiments to determine whether Aβ and HNE activate other kinases. Our data showed that indeed both Aβ and HNE activate other p38 MAPKs and Akt (Fig. 6).
Figure 5.
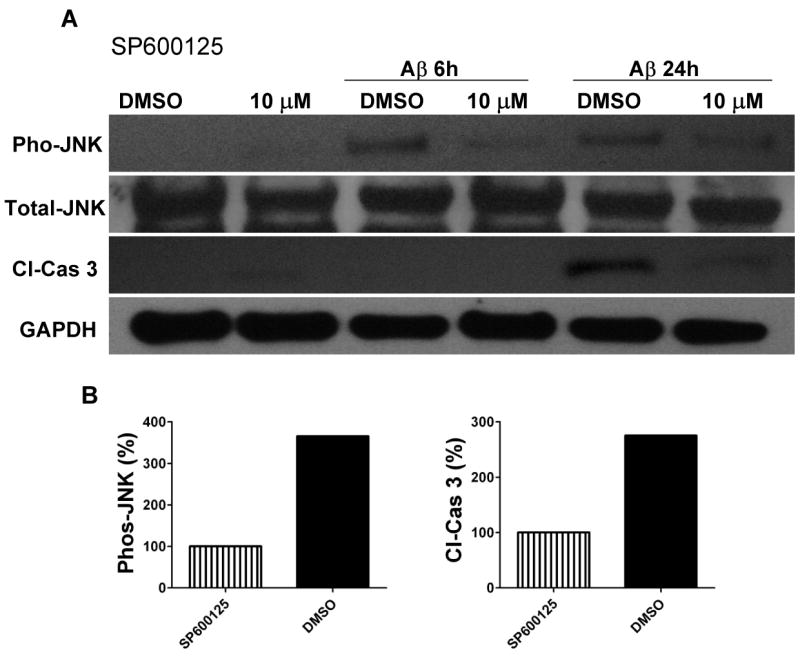
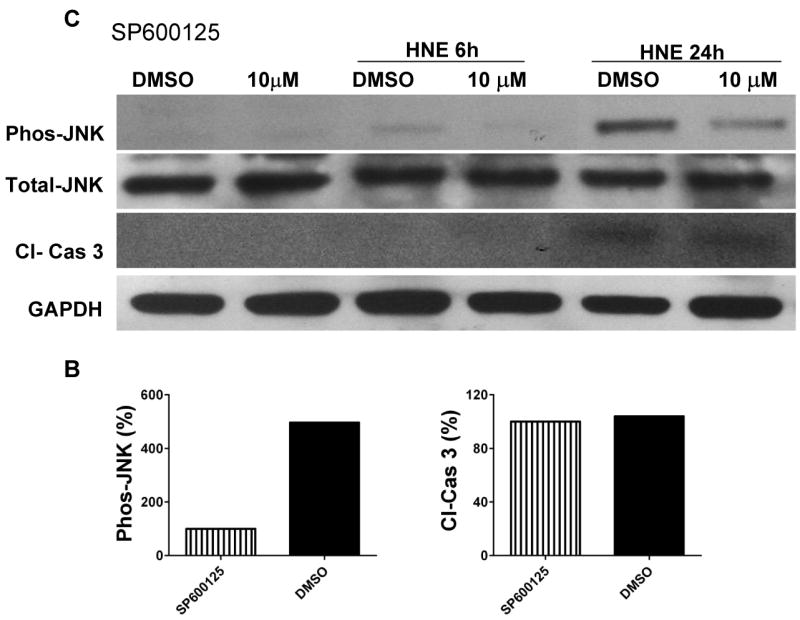
Evidence that JNK mediates activation of the apoptotic caspase-3 protein in response to Aβ and HNE in cortical neurons. Immunoblot analysis of phospho-JNK (46 kDa), total JNK (46 kDa) and cleaved caspase-3 (19 kDa) in cell lysates of neurons in cortical cultures that were pretreated for 10 minutes with the JNK inhibitor SP600125 (10 μM) or vehicle (DMSO). The cultures were then exposed for the indicated time periods to either 5 μM Aβ1-42 (panel A) or 5 μM HNE (panel C). Results of densitometric analysis of phospho-JNK and cleaved caspase-3 protein levels in DMSO treated cells compared with SP600125 (10 μM) cells from Aβ treated (panel B) and HNE treated (panel D) samples at 24 hours.
Figure 6.
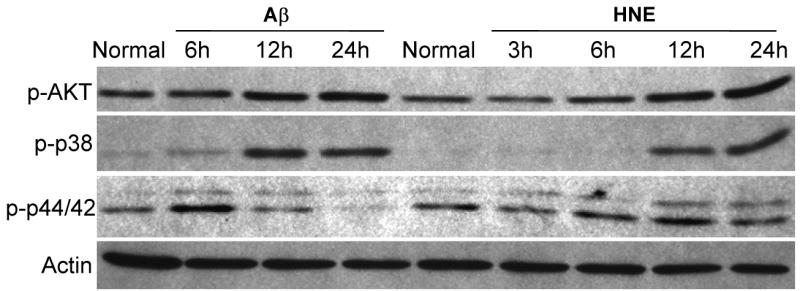
Evidence that Aβ1-42 and HNE can activate multiple kinases including p44/42, p38 and Akt kinases. Immunoblot analysis using antibodies that selectively recognize activated forms of the indicated kinases (p-p44/42, p-p38 and p-Akt) in lysates of cortical cells established from wild-type mice. The neurons were treated with 5 μM Aβ1-42 or 5 μM HNE for the indicated time periods.
TLR4 expression is reduced in the inferior parietal cortex of AD patients
If the expression of TLR4 in neurons renders them vulnerable to death in AD, then a reduced level of TLR4 would be expected as a result of the death of TLR4-expressing neurons in brain regions affected in AD. We therefore measured relative levels of TLR4 protein in specimens of inferior parietal lobule obtained postmortem from end-stage AD patients and age-matched control subjects (see Table 1 for information on individual AD and control subjects).
Immunoblot analysis showed that the level of TLR4 was significantly lower in samples from AD patients compared to control subjects (Fig. 7A, B). We also measured relative levels of the neuronal synaptic protein synaptophysin, and the microglial cell marker IBA-1, in the same protein samples used to measure TLR4 levels. Samples from AD patients exhibited lower levels of synaptophysin and higher levels of IBA-1 compared to samples from control subjects (Fig. 7A).
Figure 7.


Levels of TLR4 protein are decreased in association with neuronal degeneration in postmortem brain tissue samples from AD patients compared to control subjects. A. Immunoblots showing protein levels of TLR4 (90 kDa), synaptophysin (38 kDa) and IBA-1 (17 kDa) in samples of inferior parietal cortex of AD and control human subjects. B. Results of densitometric analysis of TLR4 protein levels in samples from AD and control subjects. Values are the mean and SEM. *p = 0.038 (unpaired t test).
Discussion
Our previous findings showed that TLR2 and TLR4 are expressed in cerebral cortical neurons, where their levels and downstream signaling via JNK are increased in response to energy deprivation in cell culture and ischemia in vivo (Tang et al., 2007). In the present study we show that TLR4 expression increases during exposures to Aβ1-42 and the lipid peroxidation product HNE. JNK and caspase-3 activity levels were increased in neurons exposed to Aβ and HNE, and selective elimination of TLR4 function significantly suppressed the abilities of Aβ and HNE to induce activation of JNK and caspase-3. These findings suggest that neurons expressing TLR4 are vulnerable to degeneration in AD. Consistent with the latter possibility, we found that levels of TLR4 are decreased in AD brain tissue samples compared to samples from control subjects. The latter result suggests that neurons expressing TLR4 are vulnerable to degeneration in AD because TLR4 is expressed primarily in neurons (Tang et al., 2007). Moreover, TLR4 expression is increased in glial cells in response to activation (inflammation); because glial cell activation is increased in AD brain (Mrak and Griffin, 2005), it is unlikely that the decrease in TLR4 in AD is due to a decrease in glial cells. Collectively, our findings from cell culture studies and analysis of AD and control subjects suggest a possible contribution of TLR4-mediated signaling in the neuronal degeneration that may be triggered by Aβ and membrane lipid peroxidation in AD.
TLRs are widely conserved in organisms ranging from sponges to humans, and are believed to have evolved as a mechanism to protect against invading pathogens including bacteria, viruses and parasites (Beutler, 2005; Barton, 2007; Wiens et al., 2007). However, TLRs are also activated in immune cells (T cells, dendritic cells and macrophages) in response to tissue injury, including trauma, hemorrhagic shock and ischemia (Mollen et al., 2006; Frink et al., 2007). We found that TLR2 and TLR4 are up-regulated and activated in neurons in response to an ischemic stroke (Tang et al., 2007). Previous studies have shown that other molecules involved in innate immunity, including components of the complement cascade, are activated in neurons and glial cells in AD (Nataf et al., 1999; Mack et al., 2006; Shen and Meri., 2003; Zanjani et al., 2005). Our findings suggest that TLR4 signaling may also play a role in AD pathogenesis, possibly being activated by Aβ and membrane-associated oxidative stress. While activation of TLR4 in neurons might contribute to their death, the activation of TLR2 in microglia has been reported to enhance their phagocytic uptake of Aβ, a potentially beneficial process (Chen et al., 2006). Another recent study by Tahara and colleagues demonstrated that activation of a microglial cell line (BV-2 cells) with TLR2, TLR4 or TLR9 ligands enhances their uptake of Aβ (Tahara et al., 2006). Therefore, the roles of TLRs in AD are likely to be complex with activation of different TLRs resulting in different types of responses in neurons and glial cells.
Molecules produced by pathogens have been shown to act as TLR ligands (Trinchieri and Sher., 2007). However, an unresolved issue in the field of TLRs concerns the endogenous ligands that activate them during tissue injury and inflammatory disease states. It has been proposed that heat-shock protein 70 (HSP70) is a ligand for TLR2 and TLR4 (Asea et al., 2002; Vabulas et al., 2002). Levels of HSP70 are increased in AD (Yoo et al., 1999) and we have observed that levels of HSP70 are increased in association with TLR2 and 4 activation in neurons subjected to energy deprivation (Tang et al., 2007) and in neurons exposed to HNE (unpublished data). Another candidate for the endogenous ligand that activates TLR4 in neurons in response to Aβ or HNE mediated toxic conditions is hyaluronan fragments. Levels of hyaluronate were reported to be increased in the temporal cortex of AD patients compared to age-matched control subjects (Jenkins and Bachelard, 1988). Hyaluronases are activated in injured neural tissue resulting in the generation of hyaluronan fragments (Al'Qteishat et al., 2006) that have been reported capable of activating TLR2 and TLR4 in immune cells (Termeer et al., 2002; Jiang et al., 2005). It is therefore possible that hyaluronan fragments contribute to the activation of TLRs in neurons and glial cells in AD.
Activation of one or more TLRs in neurons and glial cells may occur in response to injury to the nervous system. Our findings suggest that membrane lipid peroxidation is one injury-associated factor that can result in TLR4 activation in neurons. Lipid peroxidation and HNE are implicated in the degeneration of neurons that occurs in ischemic stroke (Keller et al., 1998) and several neurodegenerative disorders including AD (Keller and Mattson, 1998), ALS (Pedersen et al., 1998) and Parkinson's disease (Yoritaka et al., 1996). It will therefore be of considerable interest to elucidate the relationships between oxidative stress, TLR activation and the pathogenesis of these different disorders. Our findings therefore suggest that agents that target TLR expression, endogenous TLR ligands or TLR signal transduction molecules might be of therapeutic value for such disorders.
Acknowledgments
We wish to thank Hiayang Zhu and Ouyang Xin for their excellent technical support. This work was supported by Intramural Research Program of the National Institute on Aging, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al Qteishat A, Gaffney JJ, Krupinski J, Slevin M. Hyaluronan expression following middle cerebral artery occlusion in the rat. Neuroreport. 2006;17:1111–1114. doi: 10.1097/01.wnr.0000227986.69680.20. [DOI] [PubMed] [Google Scholar]
- Al'Qteishat A, Gaffney J, Krupinski J, Rubio F, West D, Kumar S, Kumar P, Mitsios N, Slevin M. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain. 2006;129:2158–2176. doi: 10.1093/brain/awl139. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Barton GM. Viral recognition by Toll-like receptors. Semin Immunol. 2007;19:33–40. doi: 10.1016/j.smim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Beutler B. The Toll-like receptors: analysis by forward genetic methods. Immunogenetics. 2005;57:385–392. doi: 10.1007/s00251-005-0011-3. [DOI] [PubMed] [Google Scholar]
- Camandola S, Poli G, Mattson MP. The lipid peroxidation product 4-hydroxy-2,3-nonenal increases AP-1-binding activity through caspase activation in neurons. J Neurochem. 2000;74:159–168. doi: 10.1046/j.1471-4159.2000.0740159.x. [DOI] [PubMed] [Google Scholar]
- Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- Chen R, Lim JH, Jono H, Gu XX, Kim YS, Basbaum CB, Murphy TF, Li JD. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem Biophys Res Commun. 2004;324:1087–1094. doi: 10.1016/j.bbrc.2004.09.157. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SL, O'Neill LA. Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol. 2006;72:1102–1113. doi: 10.1016/j.bcp.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Frink M, Hsieh YC, Thobe BM, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. TLR4 regulates Kupffer cell chemokine production, systemic inflammation and lung neutrophil infiltration following trauma-hemorrhage. Mol Immunol. 2007;44:2625–2630. doi: 10.1016/j.molimm.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Nawa M, Chiba T, Aiso S, Nishimoto I, Matsuoka M. Transforming growth factor beta2 autocrinally mediates neuronal cell death induced by amyloid-beta. J Neurosci Res. 2006;83:1039–1047. doi: 10.1002/jnr.20804. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Jiang Z, Georgel P, Tabeta K, Janssen E, Du X, Beutler B. TLR signaling pathways: opportunities for activation and blockade in pursuit of therapy. Curr Pharm Des. 2006;12:4123–4134. doi: 10.2174/138161206778743466. [DOI] [PubMed] [Google Scholar]
- Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ. Amyloid-beta peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998;785:195–206. doi: 10.1016/s0006-8993(97)01318-8. [DOI] [PubMed] [Google Scholar]
- Ii M, Sunamoto M, Ohnishi K, Ichimori Y. beta-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996;720:93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Jenkins HG, Bachelard HS. Glycosaminoglycans in cortical autopsy samples from Alzheimer brain. J Neurochem. 1988;51:1641–1645. doi: 10.1111/j.1471-4159.1988.tb01135.x. [DOI] [PubMed] [Google Scholar]
- Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Mattson MP. Roles of lipid peroxidation in modulation of cellular signaling pathways, cell dysfunction, and death in the nervous system. Rev Neurosci. 1998;9:105–116. doi: 10.1515/revneuro.1998.9.2.105. [DOI] [PubMed] [Google Scholar]
- Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer S, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial MnSOD prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- Krishnan J, Selvarajoo K, Tsuchiya M, Lee G, Choi S. Toll-like receptor signal transduction. Exp Mol Med. 2007;39:421–438. doi: 10.1038/emm.2007.47. [DOI] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon M, Megret F, Lafage M, Prehaud C. The innate immune facet of brain: human neurons express TLR-3 and sense viral dsRNA. J Mol Neurosci. 2006;29:185–194. doi: 10.1385/JMN:29:3:185. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, Gambotto AA, Humphris EL, Vodovotz Y, Billiar TR. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect Immun. 2002;70:3433–3442. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N, Wiltfang J, Gerber J, Nau R. Amyloid beta peptide 1-40 enhances the action of Toll-like receptor-2 and - 4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem. 2005;94:289–298. doi: 10.1111/j.1471-4159.2005.03188.x. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lu J, Kosaras B, Sidman RL, Volpe JJ, Vartanian T. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175:209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack WJ, Sughrue ME, Ducruet AF, Mocco J, Sosunov SA, Hassid BG, Silverberg JZ, Ten VS, Pinsky DJ, Connolly ES., Jr Temporal pattern of C1q deposition after transient focal cerebral ischemia. J Neurosci Res. 2006;83:883–889. doi: 10.1002/jnr.20775. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997a;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997b;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Partin J, Begley JG. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–437. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- Montine KS, Olson SJ, Amarnath V, Whetsell WO, Jr, Graham DG, Montine TJ. Immunohistochemical detection of 4-hydroxy-2-nonenal adducts in Alzheimer's disease is associated with inheritance of APOE4. Am J Pathol. 1997;150:437–443. [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Nataf S, Stahel PF, Davoust N, Barnum SR. Complement anaphylatoxin receptors on neurons: new tricks for old receptors? Trends Neurosci. 1999;22:397–402. doi: 10.1016/s0166-2236(98)01390-3. [DOI] [PubMed] [Google Scholar]
- Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel SH, Smith RG, Kasarskis E, Mattson MP. Protein modification by the lipid peroxidation product 4-hydroxynonenal in spinal cord tissue of ALS patients. Ann Neurol. 1998;44:819–824. doi: 10.1002/ana.410440518. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- Shen Y, Meri S. Yin and Yang: complement activation and regulation in Alzheimer's disease. Prog Neurobiol. 2003;70:463–472. doi: 10.1016/j.pneurobio.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal Role for Neuronal Toll-Like Receptors in Ischemic Brain Injury and Functional Deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- Wiens M, Korzhev M, Perovic-Ottstadt S, Luthringer B, Brandt D, Klein S, Muller WE. Toll-like receptors are part of the innate immune defense system of sponges (demospongiae: porifera) Mol Biol Evol. 2007;24:792–804. doi: 10.1093/molbev/msl208. [DOI] [PubMed] [Google Scholar]
- Yang X, Coriolan D, Murthy V, Schultz K, Golenbock DT, Beasley D. Proinflammatory phenotype of vascular smooth muscle cells: role of efficient Toll-like receptor 4 signaling. Am J Physiol Heart Circ Physiol. 2005;289:H1069–1076. doi: 10.1152/ajpheart.00143.2005. [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BC, Seidl R, Cairns N, Lubec G. Heat-shock protein 70 levels in brain of patients with Down syndrome and Alzheimer's disease. J Neural Transm Suppl. 1999;57:315–322. doi: 10.1007/978-3-7091-6380-1_22. [DOI] [PubMed] [Google Scholar]
- Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- Zanjani H, Finch CE, Kemper C, Atkinson J, McKeel D, Morris JC, Price JL. Complement activation in very early Alzheimer disease. Alzheimer Dis Assoc Disord. 2005;19:55–66. doi: 10.1097/01.wad.0000165506.60370.94. [DOI] [PubMed] [Google Scholar]