

Abstract
Metallo β-lactamases (MβL's) are enzymes naturally evolved by bacterial strains under the evolutionary pressure of β-lactam antibiotic clinical use. They have a broad substrate spectrum and are resistant to all the clinically useful inhibitors, representing a potential risk of infection if massively disseminated. MβL's scaffold is designed to accommodate one or two zinc ions able to activate a nucleophilic hydroxide for the hydrolysis of the β-lactam ring. The role of zinc content on binding and reactive mechanism of action has been the subject of debate and still remains an open issue despite the large amount of data acquired. We report herein a study of the reaction pathway for binuclear CcrA from Bacteroides fragilis using density functional theory based quantum mechanics-molecular mechanics dynamical modeling. CcrA is the prototypical binuclear enzyme belonging to B1 MβL family, which includes several harmful chromosomally-encoded and transferable enzymes. The involvement of a second zinc ion in the catalytic mechanism lowers the energetic barrier for β-lactam hydrolysis, preserving the essential binding features found in mononuclear B1 enzymes (BcII from Bacillus cereus) while providing a more efficient single-step mechanism. Overall, this study suggests that uptake of a second equivalent zinc ion is evolutionary favored.
Keywords: metallo β-lactamases, β-lactams, dizinc, monozinc, metalloproteins, metalloenzymes, QM-MM, molecular dynamics
Introduction
The clinical use of β-lactams, the most widespread antibiotics on the market today, has exerted a large evolutionary pressure on bacteria, triggering several resistance mechanisms.1,2 The most effective means to counteract the action of these antibiotics is the biosynthesis of β-lactamases, a group of hydrolases, which uses different protein scaffolds and catalytic architectures to inactivate β-lactam drugs.1 β-Lactamases have been grouped into four classes based on their sequence homology.3 Classes A, C and D enzymes feature a serine residue as nucleophilic agent in the reaction, whereas class B lactamases are characterized by the presence of one or two Zn2+ ions essential for the function and feature larger substrate diversity. They are generically termed as metallo β-lactamases (MβL's), and, despite not being as ubiquitous as serine β-lactamases, they are increasingly spreading among pathogenic bacteria in the clinical settings and resistant to all the clinically useful inhibitors.4-7 Thus, there is an urgent need to understand their function at the molecular level.
MβL's are classified by sequence homology into three subclasses: B1, B2, and B3.5,8 The B1 subclass includes several chromosomally-encoded enzymes, such as the ones from Bacillus cereus (BcII).9-14Bacteroides fragilis (CcrA),15-17Elizabethkingia meningoseptica (BlaB),18,19 as well as the transferable VIM, IMP, SPM and GIM-type enzymes.20-26 Subclass B2 includes the CphA and imiS lactamases from Aeromonas species.27-29 Subclass B3, along with the extensively characterized enzyme L1 from Stenotrophomonas maltophilia,30-33 includes enzymes from environmental bacteria, such as CAU-1 from Caulobacter crescentus34 and THIN-B from Janthinobacterium lividum,35 and from opportunistic pathogens like FEZ-1 from Legionella gormanii,36-38 and GOB from E. meningoseptica.39
Atomistic structures of MβL's spanning all the three subclasses have been solved by X-ray crystallography (B1: BcII,40,41 BlaB,18 CcrA,42 IMP-1,43 SPM-1,44 (B1); B2: CphA;27 and B3: L131 and FEZ-137). The folding frame is characterized in all cases by a compact αβ/βα sandwich (two core β-sheets surrounded by α-helices), with an active site that can allocate one or two Zn2+ metal ions.4,5 This motif tolerates insertions and deletions in the sequence resulting in strikingly different loop topologies and Zn2+ content. At the active pocket, Zn1 in the first metal site is tetrahedrally coordinated by three histidine ligands (His116, His118 and His196), and a nucleophilic hydroxide45-50 in B1 and B3 enzymes. The coordination polyhedron of Zn2 in B1 enzymes is provided by the nucleophile, one water molecule, and the Asp120-Cys221-His263 triad. A similar site (with a tetrahedral geometry) is also found in mononuclear B2 enzymes as the active species. Instead, in B3 MβL's, two mutations (Cys221-Ser and Arg121-His) affect the Zn2 coordination geometry, which is bound to Asp120, His121, His263 and one or two water molecules.
Due to the MβL structural diversity, the role of Zn2+ content for the enzymatic function is far from being fully understood, despite the large amount of experimental and theoretical works on the subject. A recent, very comprehensive review51 regarding existing computational proposals on MβL's underlines the necessity of further theoretical investigations to uncover the catalytic mechanism of MβL, in particular of the influence of a second zinc ion in dizinc species. Here, we further address this issue by performing a modeling study on a prototypical B1 dizinc MβL, CcrA, one of the first MβL for which the structure has been determined by X-ray crystallography.42 Quantum mechanics-molecular mechanics (QM-MM) reaction modeling of dizinc β-lactamase CcrA from Bacteroides fragilis in complex with an antibiotic in clinical use (the cephalosporin cefotaxime) are complemented by a molecular dynamics investigation of the binding mode for a series of common β-lactams. Comparison is then made with our previous studies on a monozinc B1 MβL, BcII from Bacillus cereus,47,48 and previous proposals on MβL's.51 Our calculations add important pieces of information to those obtained by previous calculations.51 Specifically, they suggest that the insertion of a second equivalent Zn2+ ion at the active pocket elicits an elegant, efficient, and highly concerted water-aided single-step enzymatic mechanism. The monozinc B1 enzyme follows instead a two-step path, with higher activation free energy, according to consistent computations48 to those reported here. Thus, the evolutionary pressure favored – through local mutations – the uptake of a second equivalent zinc at the active site on certain bacterial strains to increase their catalytic efficiency.
Methods and Materials
CcrA-substrate adducts
Benzylpenicillin (BPC, a penicillin), cefotaxime (CEF, a cephalosporin), and imipenem (IMI, a carbapenem), three common antibiotics from different β-lactam families are docked at the active site of the dizinc B1 MβL CcrA from Bacteroides fragilis (complexes CcrA-BPC, CcrA-CEF, and CcrA-IMI, respectively). The X-ray structure of the latter has been solved at 1.85 Å resolution (PDB entry 1znb42). The structural information about inhibitors solved at the MβL's active sites along with previous docking studies47 of the same substrates on the B1 monozinc MβL BcII from Bacillus cereus are used as guidance for the manual docking of β-lactams at the metal site, further optimized trough MD simulations. The protonation states of the active site groups have been determined by large-size DFT calculations.46 As for the rest, Glu and Asp are assumed ionized whilst the histidines are protonated according to their putative H-bond patterns in the 1znb structure (His78 at Nδ, His105 at Nδ, His172 at Nε, and His286 at Nε).
The three adducts – CcrA-BPC, CcrA-CEF, and CcrA-IMI (which are immersed in a water edge 74·69·77 Å3 neutralized by 4-5 Na+ counter-ions, with a total of 38,000 atoms) undergo MD simulations. The AMBER force field (parm9452) is used for the protein and the counter-ions. The TIP3P model is used for water.53 The force field parameters for the substrates are taken from previous works47,49,54; point charges for the Zn2+-ligand center are calculated using the RESP approach, and bond and bend force constants are adapted from previous works, and have shown to consistently describe experimental distances and coordination geometry of a variety of zinc enzymes47,49,54 (see SI). A time step set of 1.5 fs is used. All bonds involving hydrogen atoms are constrained with the SHAKE algorithm following the protocol reported in previous works.47 Simulations are performed at constant temperature and pressure by coupling the systems to a Berendsen bath at 298 K and at 1 atm. Periodic boundary conditions are applied to the system. The PME method is used to evaluate long-range electrostatic interactions, and a cutoff of 12 Å is used to account for the van der Waals interactions and the short range, real part contribution to the electrostatics. A 4 ns MD trajectory is performed for each complex. RMSDs time series (see Supporting Information, SI) suggest that the systems are well equilibrated within the first ns (see SI).
CcrA-CEF enzymatic reaction
The reaction path of CcrA-CEF adduct is investigated by first principles MD in a fully Hamiltonian QM-MM scheme, which combines Car-Parrinello DFT MD and classical MD. The QM-MM interface takes into account electrostatic and mechanical coupling of the entire protein frame and solvation bath with the QM cell. This scheme has been shown to describe accurately a variety of enzymatic systems, including the monozinc BcII metallo β-lactamase48 and other zinc-dependent enzymes.45 First principles calculations for the QM are here used. This contrasts to several other studies of MβL, which use semi-empirical and SCC-DFTB methods. Although first principles calculations access shorter timescale (in this work ∼150 ps), they offer the advantage of being transferable. Within the Car-Parrinello scheme, in addition, statistical mechanics approaches to simulate rare events may be used to investigate reactions in biological systems.55,56
In the present QM-MM approach, the reactive portion of the complex, namely the zinc first ligand shell along with the CEF bi-cyclic portion (Figure 2), is treated at the quantum level (DFT-BLYP57,58) and the remaining part at the classical level (AMBER force field52). The valence electrons are described by a plane wave basis set up to a cutoff of 70 Ry. Plane waves are not affected by basis set superposition errors, which might otherwise influence the quality of the simulation. The interactions between valence electrons and ionic cores are described with norm-conserving Martins-Troullier pseudopotentials. The BLYP functional, along with the plane-waves basis set, has been used for a variety of reactions in aqueous solution and in enzymes. Notice that the use of popular B3LYP functional along with in plane-wave based calculations is computationally, prohibitively expensive and so is not used here. Car-Parrinello molecular dynamics59 simulations are carried out with a time step of 0.14 fs (totally ∼70 ps for each reaction path, with initial equilibration of reactants of 20 ps, and 5 ps for each step of constrained MD48,55) and a fictitious electron mass of 500 a.u.; constant temperature simulations are achieved by coupling the system with a Nosé-Hoover thermostat at 500 cm-1 frequency. The non-bonded interactions between the MM and QM regions are treated as in ref. 60. The free energy of activation for the hydrolytic mechanism is evaluated with the method of thermodynamics integration along the putative reaction coordinate (RC), namely dRC=O(OH-)-C8(β-lactam ring) (following the β-lactam standard numbering scheme), using the same protocol used for monozinc BcII reaction.48 The free energy of activation can be related to kcat through transition state theory for comparison with the experiment in case the system is in saturation conditions. Estimation of ΔF provides an approximate value of the catalytic rate due to problems related with DFT accuracy and sampling and because of the inherent limitations of rate constants definition. Two reaction paths are taken into account differing by the H-bond between Asn233 and the substrate (C8 carbonyl oxygen) as different configurations produced by the MD simulations.
Gromacs v3.261 is used as the engine for the classical MD simulations, CPMD v3.762 for the investigation of the reaction path, and VMD software for analysis and rendering.63
Results and Discussion
Here we discuss the findings on dizinc CcrA in complex with bi-cyclic β-lactams from three different types (BPC, CEF, and IMI). First, we provide general features observed for all the MD complexes, then focus on cefotaxime, for which the reaction mechanism is described by QM-MM simulations. Next, we compare the features of the Michaelis CcrA-CEF complex with those of CcrA-BPC and CcrA-IMI. Finally, the modularity exploited in binding and reactive mechanism by B1 MβL's upon second metal uptake is discussed by comparing CcrA to mononuclear BcII enzyme (Scheme 1).
Scheme 1.
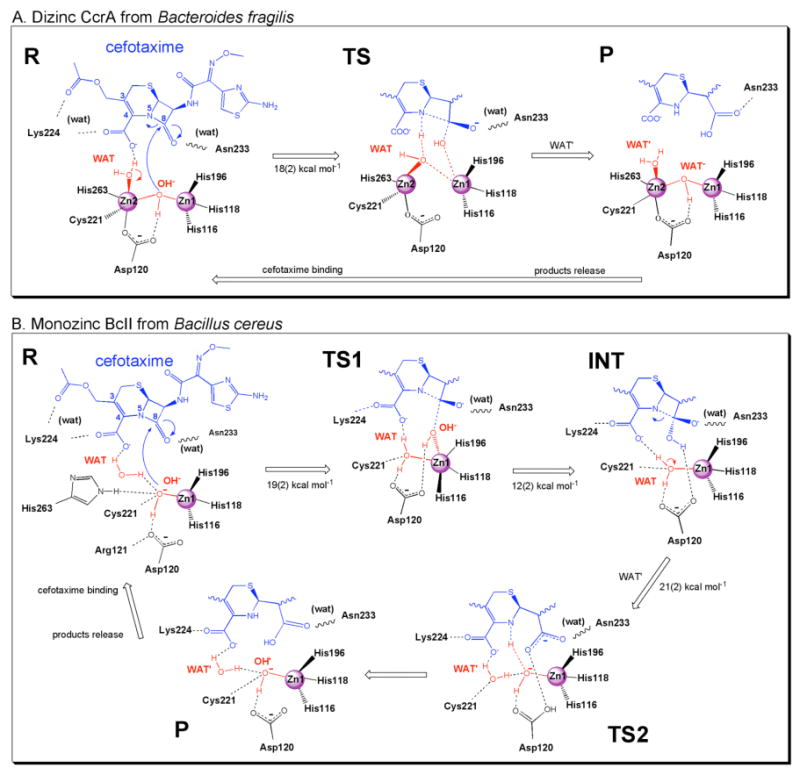
Cefotaxime hydrolysis catalyzed by (A) binuclear MβL CcrA from B.fragilis and (B) mononuclear BcII from B.cereus. The reactant state (R) corresponds to the ES complex, and the product state (P) corresponds to the EP complex.
Binding and reactivity of dizinc MβL's
General features of CcrA-substrate adducts
The overall MβL fold is maintained along the entire simulated time for all three adducts (4ns, see Figure 1 and SI). The root mean square deviations (RMSDs) with respect to the X-ray structure fluctuate around very low values (0.9(2), 1.0(3), and 1.0(2) Å for the backbone respectively, SI). In fact, RMS fluctuations are similar and slightly smaller than those derived from X-ray B–factors (see SI), possibly because of the lower mobility of the large loops involved in β–lactams binding (Figure 1), as also found for β-lactam binding at BcII enzyme. The β-lactam substrates at the active cleft are very stable as well (average RMSD: 2.0(3) Å).
Figure 1.
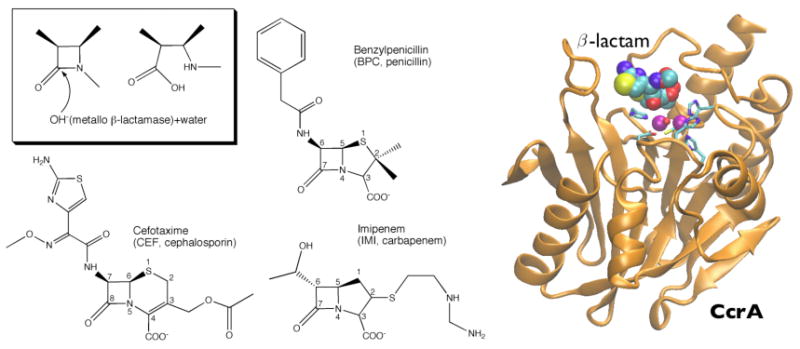
(Left) β-Lactam hydrolysis catalyzed by MβL is sketched in the inset. Also shown are substrates used in this work. (Right) Binding mode of CcrA from Bacteroides fragilis. Secondary structures are represented in orange cartoons; purple spheres indicate the Zn2+ cofactors present at the active site, whereas coordinated metal ligands are depicted in stick representation and the β-lactam in space-filled balls.
CcrA-CEF: binding and reactivity
CEF binding dehydrates almost completely the active site: only one water molecule is found in the close proximity of the metal ions (Figure 4). This molecule (WAT in Figure 2) is interacting with Zn2 (d=3.4(4) Å, Figure 3), H-bonding in addition to CEF carboxylate group (C4 substituent in cephalosporins, Figure 2) and to the C3 substituent. Although not strongly bonding to Zn2 as in the X-ray structure (2.3 Å), WAT is able to maintain a trigonal bi-pyramidal geometry (common of penta-coordinated rather than tetrahedral geometry for Zn2 sphere). The C4 substituent, which is present in all bi-cyclic β-lactams, also H-bonds to backbone N(Asn233) and forms a water-mediated salt bridge with Lys224. The β-lactam carbonyl (O-C8 in Figure 1), mainly H-bonds to Nε(Asn233) (Figure 2, and SI). These minimal binding features are present in all β-lactams (basically present in BcII-CEF adduct either, see below and ref. 47) and are supposed to play a major role in substrate binding. On the other hand, the C3 substituent interacts with Lys224 in loop Cys221-Thr240, which includes several conserved residues among MβL's,4 and C7 substituent binds to Glu62 in loop Ile61-Val67 (see SI). This is by far the most flexible region of the protein, and undergoes major conformational changes upon substrate binding.
Figure 2.
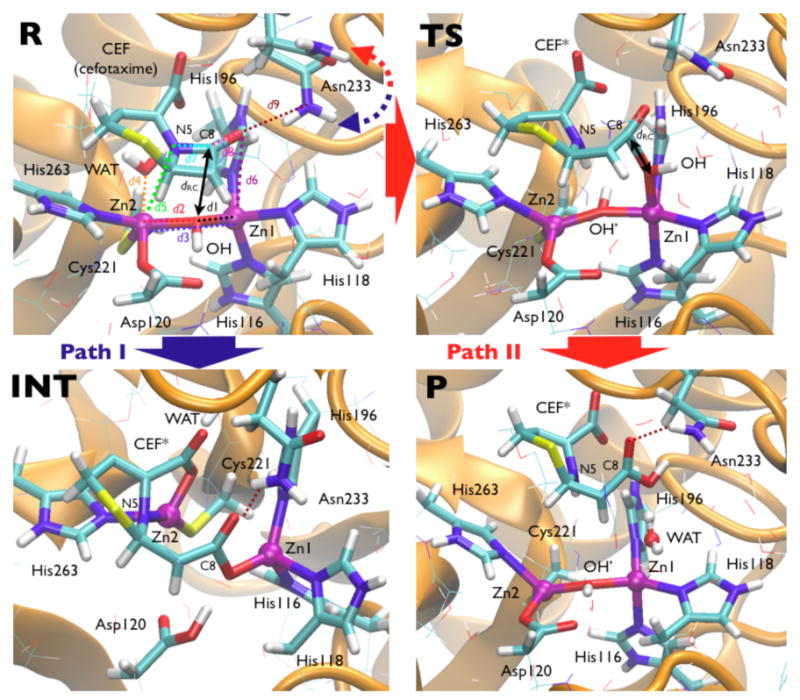
Shown are selected snapshots from the QM-MM investigation of cefotaxime hydrolysis (see Scheme 1A). In R, the reactant state, the quantum atoms are represented in sticks, distance labels refer to Figure 3, and the scaffold is pictured as orange cartoons in the background. Two pathways are reported as in the text: Path I: R→INT (in blue) indicates the unfavorable higher free energy path where Asn233 H-bonds to C8 carbonyl; and Path II: R→TS→P (in red) which gives the favorable mechanism when Asn233 does not H-bond to C8 carbonyl (Figure 3, top).
Figure 3.
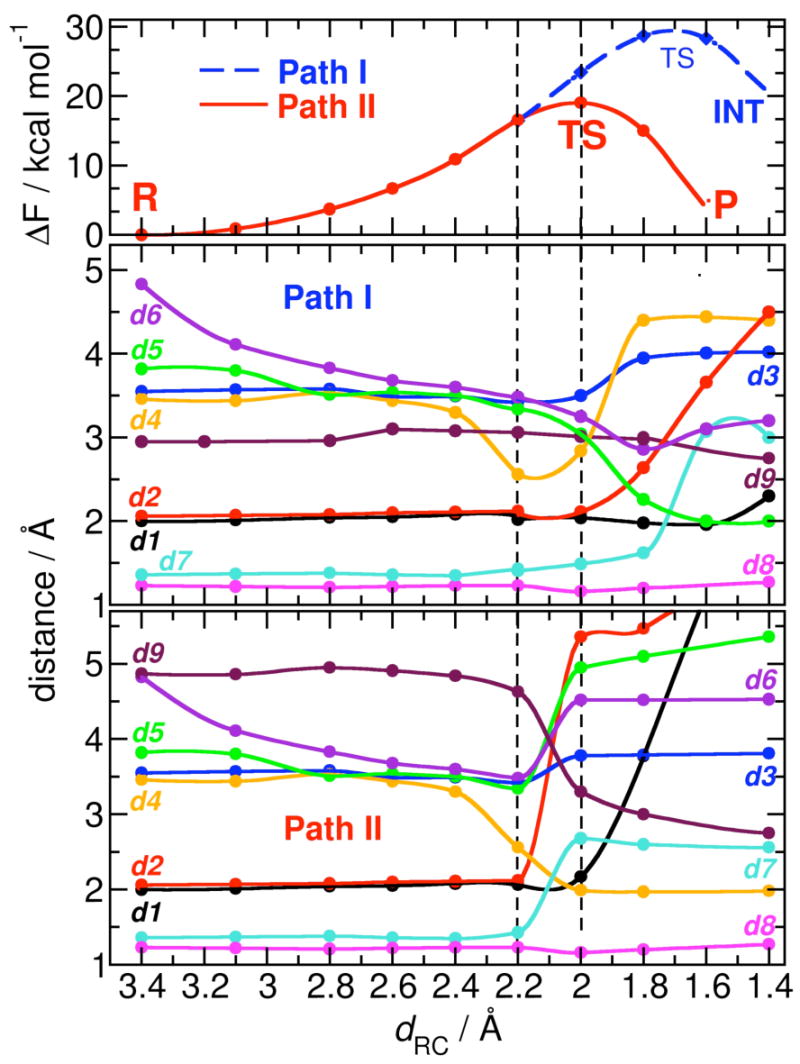
Free energy profiles (top) and relevant average distances (bottom) along the two cefotaxime hydrolysis pathways in which the O(C8)-Asn233 H-bond is either present (Path I) or not (Path II). Bond distance labels as in Figures 1 and 2.
The β-lactam C8 carbonyl oxygen strongly H-bonds to Asn233 along this reaction pathway (d9 in Figure 3), whereas it does not bind Zn1 due to the steric orientation of CEF at the catalytic pocket (Zn1 keeps a tetrahedral geometry in R). Furthermore, cefotaxime is found to H-bond to Asn233 for about 90% of the time during the MD trajectory. This interaction is not predicted to be essential in cefotaxime hydrolysis by monozinc BcII,47,48 and mutagenesis studies indicated that Asn233 is not essential for catalysis in CcrA.15 Asn233 fluctuations randomly occur along the MD trajectory when its side-chain flips out of the active site losing the H-bond with the β-lactam, eventually replaced by water molecules coming from the bulk solvent (R in Figure 3). The timescale of such an event is estimated to be few tenths of nanoseconds, and appears to be well sampled within ∼4 ns trajectories. However, it is very unlikely to occur spontaneously during a first principles QM-MM dynamics (∼70 ps timescale). Based on these observations, this is the only large conformational change not accessible within QM-MM timescale, so that two hydrolytic pathways are explored with the same QM-MM protocol: Path I, where the β-lactam carbonyl does H-bond directly to Asn233, and Path II, in which Asn233 is flipped out, solvated by bulk water molecules (Figure 3). Initial QM-MM equilibrations (∼20 ps) are performed to check the stability of starting MD configurations for Path I and II. Although, as already stated, this conformation is present for about 10% of the MD simulated time, it points to a non-crucial role of Asn233 for the function, which in turn is consistent with an increase of KM in the N233A, N233L and N233E,15,64 without largely affecting kcat/KM. In this respect, it is worth noting that so far Asn233-Tyr is the only mutation which significant improves the catalytic power in another MβL (BcII) (unpublished results), and it is the only alternative residue found in MβL's at position 233 (namely, in B1 MβL blaB, from E. meningoseptica39), that can perform a similar role. Moreover, a Tyr residue is found in glyoxalase II (a dizinc enzyme belonging to the metallo β-lactamase superfamily65) at the same topological position, and might have a similar binding function.
The putative reaction coordinate dRC, namely the distance between the nucleophilic oxygen O(OH-) and β-lactam carbonyl carbon C8 (Figure 2), is very short during MD simulations (3.4(2) Å) suggesting that this model provides a good picture of the Michaelis complex (R, Figure 3). The catalytic pathway of CEF hydrolysis is studied using hybrid QM-MM simulations, starting from an average-dRC MD snapshot equilibrated by ∼20 ps QM-MM dynamics on the reactant state, R. The free simulation suggests that the coordination sphere of the metal ion is preserved as in the X-ray structure (Figure 3, Scheme 1A) and dRC is similar to the MD value (dRC=3.3(3) Å). dRC is then adiabatically shortened to force the hydrolysis of the β-lactam CEF (products state, P).
In Path I, the nucleophile OH- approaches the β-lactam C8 maintaining its coordination with metal ions. The whole adduct is not relevantly affected by any other structural changes until dRC=2.0 Å. At dRC=1.8 Å, the Zn2-OH- bond weakens (d2=2.6 Å, Figure 3), while the Zn1-Zn2 distance increases simultaneously (d3=4.0 Å). The metal-ligand bond lengths do not experience significant structural modifications, but WAT molecule definitively detaches from Zn2, being scattered away from the active site (d4=4.4 Å). At the same time, Zn2 weakly binds to N5 (d5=2.3 Å), consequently lengthening the C8-N5 bond in CEF (d7=1.6 Å). At dRC=1.6 Å, the force on the constrained O(OH-)-C8 distance is negative indicating that the transition state has been reached, and can be located reconstructing the free energy profile at dRC∼1.7 Å. When the constraint on the O(OH-)-C8 distance is released, the system falls freely on a stable intermediate state (INT, Figure 3), where the C8-N5 bond is broken (d7=3.1 Å), and the development of partial negative charge on N5 is stabilized by a stronger N5-Zn2 bond (d5=1.6 Å). The C4-carboxylate group from the substrate becomes a Zn2 ligand at this point. Reorganization of Zn2 coordination sphere induces detachment of WAT from the site. C8 fully acquires sp2 hybridization, and its newly formed carboxyl moiety binds Zn1 in a bi-dentate manner. This is accompanied by the protonation of Asp120, likely promoted to stabilize Zn1 coordination sphere.
The formation of INT requires a large activation free energy (ΔF=30(3) kcal mol-1), which is clearly inconsistent with the fact that cefotaxime is readily hydrolyzed by CcrA with an estimated free energy of ∼17 kcal mol-1. In addition, the presence of an intermediate is not consistent with the fact that the process is likely supposed to be a single step process, as proposed by experimental evidences on similar adducts.13 Finally, in INT the strong N5-Zn2 and O(C8)-Zn1 bonds seem to prevent, due to steric hindrance, the approach of a water molecule from the bulk solvent. This is required to protonate the β-lactam N5 and to release the products. A possible proton donor candidate in INT conformation can be the protonated Asp120, as previously proposed, although this would require a significant rearrangement of the active site. For cefotaxime hydrolysis however this path is already largely energetically unfavorable, but for other substrates it cannot be definitively excluded.
Interestingly, this state closely resembles the crystal structure of L1 MβL from B3 subclass recently solved in complex with hydrolyzed moxalactam.66 3′-Elimination of the substrate bypasses N5 protonation producing a stable intermediate by product inhibition. In this conformation, as well as in the INT state, N5 and the β-lactam carboxylate are bound to Zn2, and the hydrolyzed C8 moiety is bound to Zn1. The only difference with the X-ray structure is the presence of a hydroxide-water bridging the Zn2+ ions. This moiety can eventually occupy this position giving a longer sampling time in the QM-MM simulation. It is worth noticing that QM cell including only CEF bi-cyclic core does not allow a possible 3′-elimination of C3 acetate as in L1 case, which may make N5 protonation unnecessary. Since many cephalosporins equally do not possess a good 3′ leaving group this might suggest their presence could favor the formation of stable INT-like intermediate state. Moreover, PM3-MD computational structures reported for the intermediate of hydrolysis of nitrocefin67 have similar features in comparison with INT: the N5 atom is bound to Zn2, and the hydrolyzed C8 carboxylate is bound to Zn1. However, the C4 carboxylate is not interacting with Zn2. So, the INT species is the outcome of a poorly efficient first step of cefotaxime hydrolysis, but based on these independent evidences it further indicates the feasibility of such an intermediate state in MβL-mediated hydrolysis of other substrates.
In Path II, the reaction proceeds similarly to Path I until dRC=2.2 Å (the only noticeable events up to that stage are the increase of the C8-N5 bond (d7=1.4 Å, Figure 3, Scheme 1A), and the stronger bond between Zn2 and WAT, d5=2.5 Å). At dRC=2.0 Å, the force on RC nullifies, indicating formation of the TS through a cascade of almost simultaneous events (Figure 3).† (i) OH- moves out from the Zn1-Zn2 plane attacking C8 in a apical position with respect to Zn1, while the Zn1-OH- bond is being weakened (d1=2.2 Å). (ii) The OH--Zn2 bond is lost upon nucleophilic attack, and the Zn1-Zn2 distance increases to 3.8 Å. A hydroxide simultaneously bonded to Zn1 and Zn2 is in fact expected to be a poor nucleophile, so it is reasonable that OH- attacks while detaching from at least one Zn2+ ion. As a consequence, the Zn2 coordination number changes from 5 to 4. (iii) WAT ligand, previously weakly bonded at the apical position of the Zn2 bi-pyramidal polyhedron, moves towards Zn1 to give rise to a tetrahedral geometry. The WAT-Zn2 distance (d5) gradually decreases thus lowering the WAT pKa. (iv) The Zn2-bound WAT comes closer to the β-lactam ring, consequently increasing the C8-N5 distance. The partial negative charge on N5 together with the enhanced nucleophilicity of WAT produces a proton shuttle from WAT to the N5 atom that finally triggers the C8-N5 bond cleavage (d7=2.6 Å). (v) Deprotonated water (Figure 3, WAT → OH-′) binds Zn1 completing the tetrahedral coordination sphere of Zn2, and perfectly replacing the position of the OH- nucleophile in R state. Zn1 at this point switches to a penta-coordinated bi-pyramidal coordination from the initial tetrahedral one. (vi) Upon nucleophilic attack and C8-N5 bond breaking, C8 acquires an sp2 hybridization. (vii) The O(C8)-Asn233 H-bond is formed again: the partially hydrolyzed substrate gets farther from the metal center upon TS formation (mainly due to OH- switching on Zn1 apical position), being able to H-bond to Asn233.
When the constraint of RC is released, the system evolves onto the products state (P, Figure 3), where OH-′ reorients H-bonding to Asp120 and bridging the Zn1 and Zn2 ions as in R state. The CEF substrate is completely hydrolyzed, the β-lactam ring is open and C8 acquires planar sp2 hybridization, and it is finally detached from the metal center losing the OH--Zn1 bond. The O(C8)-Asn233 H-bond is stronger than in TS, indicating that the flexibility and electrostatic properties of this residue play a role for binding as well as for the product release of degraded cephalosporins. As soon as the product is going to be expelled from the active site, water molecules start to solvate the metal site first interacting with Zn1 (meanwhile returned to a tetrahedral geometry), and eventually coordinating Zn2 as in the unbound structure.42 The calculated estimated ΔF=18(2) kcal mol-1 is consistent with (i) experimental evidences that suggest a single-step, efficient reaction for cephalosporin hydrolysis68 with (ii) a free energy of activation of about 17 kcal mol-1 for similar reactions,49 and with (iii) the fact the uncatalyzed reaction in water solution has an estimated barrier of 48(3) kcal mol-1.48
This also suggests that Asn233 has been selected by evolution as the best compromise for its involvement in tuning the binding, catalysis, and product release processes. Asn233 is in fact able to interact with the β-lactam carbonyl moiety accommodating the substrate in an optimal position for the nucleophilic attack, but at the same time it can temporarily lose this interaction allowing the attack on the carbonyl along with the reorganization of the metal center, finally facilitating release of the inactivated β-lactam from the active site.
Binding of other β-lactams
IMI and BPC are accommodated at the active pocket in a productive complex: dRC is in fact relatively short (3.2(2) Å for IMI binding, and 3.5(2) Å for BPC). This may be consistent with the fact that both are efficiently hydrolyzed by CcrA (Figures 1,4 and SI). The key interactions of bi-cyclic core at the CcrA catalytic cleft are preserved: (i) the H-bond between the carbonyl group and Nε(Asn233), although allowing fluctuations on its persistency (80-90% with 3.5 Å H-bond cutoff); (ii) the C3-C4 carboxylate forms a H-bond with N(Asn233), and (iii) a water-mediated salt bridge with Lys224 (see SI). (iv) A water molecule (WAT) is buried at the cleavage site, in an equivalent position to that in the CcrA-CEF adduct (and BcII complexes previously reported),48 providing an identical degree of solvation for the metal center and bridging Zn2 with C3-C4 carboxylate. This WAT molecule plays an important role in binding and catalysis, at least for B1 MβL's, which share the same metal architecture, i.e. WAT can be displayed as fifth apical ligand of Zn2 metal, readily activated upon nucleophilic attack as proton shuttle for N5-C7(C8) bond breaking (Figure 4). Thus, despite the slightly different relative orientations of the β-lactam ring and the carboxylate moiety in the three adducts, these interactions are preserved, confirming that these common elements are flexible enough to accommodate the different substrates and elicit a broad spectrum activity in MβL's. The main differences of binding among the three substrates consist indeed in the nonspecific interactions of C2-C3 substituents with loop Ile61-Val67 that may be responsible for the slightly different catalytic activity (see SI).
Figure 4.
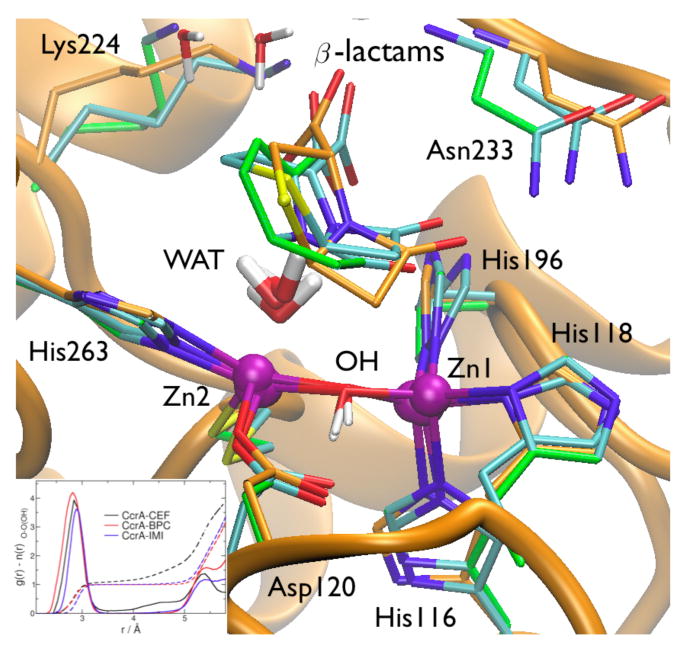
Superimposition of selected MD snapshots of dizinc CcrA in complex with cefotaxime (CEF, orange carbon skeleton), benzylpenicillin (BPC, green skeleton) and imipenem (IMI, cyan skeleton). The CcrA fold is pictured in the background. In the inset shows the O-Owat radial distribution function for the OH- nucleophile upon binding of the different substrates.
Moreover, the conservation of these key interactions suggests that minimal, common determinants play an important role for substrate binding7 (Figure 4, and SI). Thus, a similar water-assisted hydrolysis mechanism might be generally plausible for penicillins, cephalosporins and carbapenems (with still the possibility of a two-steps pathway as in the case of nitrocefin). They might also provide a rationale on why monobactams such as aztreonam, lacking the bi-cyclic core and C3-C4 carboxylate, are not efficiently hydrolyzed by MβL's.68
Other models of substrate-enzyme complexes for binuclear B1 MβL's have been reported in literature.54,69 In particular, in an MD-based model of the complex of CcrA with imipenem (IMI), the authors assumed a protonated Asp120 bound to Zn2 in the active site. This assumption, while not altering the protein conformation, leads to a different H-bonding and solvation patterns at the active site, producing a complex in which the reaction coordinate is substantially longer than in our model (5.5(3) Å). This fact most probably stems from the different protonation state of Asp120, which induces a flip in the Zn1-bound OH- group and a seemingly non-productive enzyme-substrate complex. The breaking of the Zn2-OH- bridge, which has been modeled to provide a more productive Michaelis complex in that model (dRC=4.3(4) Å), is instead spontaneously produced in our work upon the nucleophilic attack driving the completion of the reaction.
Similarities between monozinc and dizinc B1 MβL's
Substrate binding
This and previous work47 suggest that in B1 MβL subclass CcrA and BcII accommodate a variety of β-lactams, such as benzylpenicillin, impenem, and cefotaxime. Comparing the MD structures of CcrA and BcII proteins in complex with these ligands provides insights on the common determinants of substrate binding for mono and binuclear metal sites. Few interactions are conserved (Figure 3, and SI): (i) the volatile H-bond between C7-C8 carbonyl and Asn233 side chain. This is stronger and more persistent in CcrA possibly because of the presence of Ser235 (not present in BcII), that largely stabilizes the fluctuations of Asn233 side chain at the metal site; (ii) the H-bond between the C3-C4 carboxylate and the Asn233 backbone. The fact that such key interaction is formed with the backbone and not with the side chain is consistent with the catalytic activity of BcII and CcrA Asn233 mutants;15 (iii) the water-mediated salt-bridge between the C3-C4 carboxylate and Lys224; (iii) the water mediated electrostatic interactions between the C3-C4 carboxylate and the metal ion(s). The water molecule (WAT) is highly persistent, and surprisingly, is spatially conserved in all systems (Figure 5), although WAT acts as a weak ligand of Zn2 in CcrA, whereas in BcII it reorients for H-bonding to the attacking OH-. It is actually striking that these common minimal features are sufficient to accommodate different β-lactams producing productive conformation for the enzymatic reaction in all complexes: indeed, dRC ranges from 3.2 to 3.5 Å. In all complexes moreover the β-lactam ring docks parallel to the Zn1-OH--(Zn2) plane, with O(C7-C8) on top of Zn1 (but not directly bonded as reported by other models49,54,67), C7-C8 on top of O(OH-), and N5 on top of Zn2 when present, all with similar and short distances ranging from 3.6 to 4.5 Å (RMSD= 0.4 Å for bi-cyclic core, Figures 4, 5, and Tables in SI). This configuration is particularly favorable for the reaction, as the HOMO of the nucleophilc agent (the OH- group) and the LUMO localized on the β-lactam ring are perfectly aligned for nucleophilic attack.45,46
Figure 5.
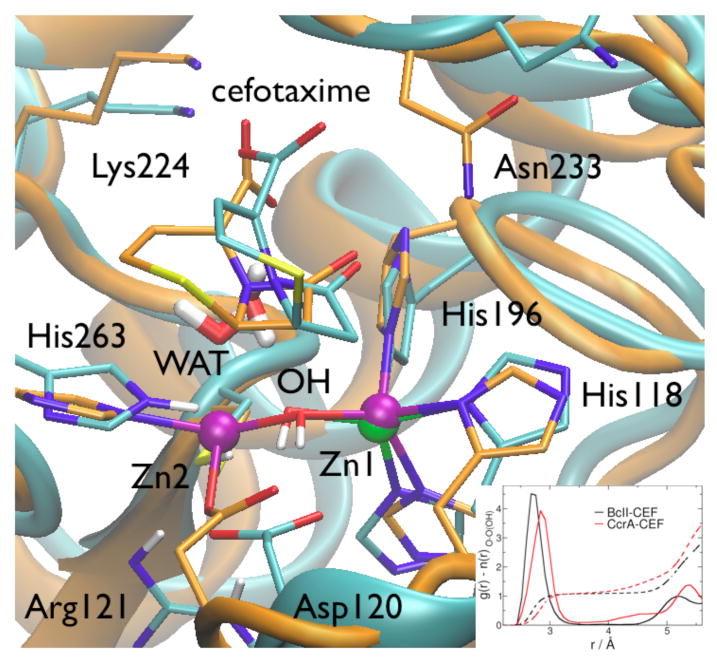
Superimposition of selected MD snapshots of monozinc BcII (cyan skeleton and cartoons) and dizinc CcrA (orange) in complex with cefotaxime (CEF). The inset shows the O-Owat radial distribution function for the nucleophile in both the conformations.
Catalytic hydrolysis
The architecture of the metal center is able to preserve key interactions for the reaction on passing from the monozinc to the dizinc species: (i) in CcrA, the Asp120-Cys221-His263 triad binds the second Zn2+, which helps to orient and stabilize the nucleophilic OH- and WAT moieties; (ii) in BcII the same triad along with Arg121 (residue that is only present in the monozinc species) forms a highly organized H-bond network that appears to be functionally equivalent, as it similarly orients the nucleophile and the catalytic water for the reaction (Figure 5, Scheme 1). Thus, these residues may be modularly exploited according to the zinc content, to preserve the structural key features of the reaction cycle. In addition, the overall effective charge at the active site is preserved through punctual mutations: the charge of the second zinc ion in dizinc species is replaced in monozinc by the presence of a not ionized Cys221, and Arg121, directly and strongly H-bonded to Asp120.
Regardless to the metal content, our calculations suggest similar features of the mechanism for cefotaxime substrate: both the monozinc and dizinc enzymes are able to promote (i) the nucleophilic attack by a Zn1-bound hydroxide, and (ii) to catalytically activate a water (WAT) for a proton shuttle on N5 that cleaves the β-lactam ring. The way these two processes are modulated by MβL's strongly depends on zinc content and flexibility. In CcrA, the coordination numbers of Zn1 and Zn2 change between 4 and 5 (Figures 1,3): in particular at TS, Zn1 accepts a fifth ligand (OH-′, deprotonated WAT) upon OH- nucleophilic attack, distorting the tetrahedral polyhedron to a bi-pyramidal sphere, and vice versa Zn2 simultaneously switches from 5 to 4 ligands, from bi-pyramidal to tetrahedral once it loses the OH-, and promoting the activation of a water molecule (WAT) to protonate the bridging N5 atom. Then, after product release, Zn1 re-acquires the tetrahedral coordination, whereas Zn2 (still bound to 4 ligands) can eventually switch back to the initial coordination once water molecules have completely diffused at the free active pocket (Figure 3, Scheme 1). In BcII, Zn1 exactly undergoes the same rearrangement during the first step of the reaction (R → TS → INT) grabbing a water as fifth ligand in a metastable state.48 Experimentally, characterizing a change of coordination number at the TS is difficult because Zn2+ is silent to most spectroscopic methods; instead, computational methods may provide, like in this case, a direct computational evidence of the metal polyhedron reorganization.48
Thus, in monozinc BcII, the only Zn2+ ion forcedly aids the two steps, so that WAT first replaces the nucleophile during the first step entering the Zn1 coordination shell, and then is activated in the second step as proton donor for β-lactam N5 (the latter being the rate limiting step of the reaction). In CcrA, the presence of Zn2 merges these two movements in a concerted unique step: Zn2 already activates WAT as soon as the OH--Zn1 bond is lost upon nucleophilic attack. This might contribute, at least in part, to its observed larger catalytic efficiency (ΔF=18 kcal mol-1vs. 21 kcal mol-1 for BcII).
Because the zinc ligands and most active site residues are highly conserved among B1 MβL subclass, binding and mechanism found for CcrA and BcII may be likely for the entire B1 subclass, where β-lactams might follow similar catalytic pathways as all the groups involved in the catalysis do not depend on the substrate chemical diversity. Of course slightly different activation free energies are expected, because of the substrate binding modes generated by different β-lactam substituents.
Implications for B2 and B3 MβL's
At a rather speculative level other MβL subclasses might share similar features. Members of B2 MβL subclass, such as CphA, are known to be active only in the mononuclear form, with Zn2+ in the Zn2 site.19,27 Recent studies70 suggest that the B3 β-lactamase GOB could also be active as a mono-Zn2 enzyme. Even if these cases may hold a different mechanism, these data, altogether with the present work, strongly suggest a central role of Zn2 in MβL-mediated catalysis, and call for more detailed studies in this direction. In B3 MβL's, where a His group usually substitutes Cys221, preliminary MD modeling of FEZ1 binuclear37 adducts instead indicates a similar binding mode and the presence of water molecule equivalent to the catalytic water in CcrA and BcII, suggesting a water-assisted pathway may be also shared with binuclear B3 enzymes.
Conclusion
The evolutionary insertion of the second equivalent zinc passing from BcII to CcrA species in B1 MβL subclass basically preserves the binding ability of the enzyme, but relevantly affects the mechanism of reaction as emerged from consistent quantum molecular modeling of both the enzymes. The role of Zn2+ in the second site is crucial for better stabilizing the negative charge developed at β-lactam N5 atom upon nucleophilic attack and its detachment from the second zinc. This leads in fact to a simultaneous protonation of N5 by the Zn2-N5 bridging water and the consequent breaking of β-lactam ring, clearly favoring a functional advantage in binuclear B1 MβL's over the mononuclear species. Nevertheless, the flexible and modular conformation of the second zinc site is able to mimic the electrostatic environment of the second ion also in mononuclear BcII, preserving the binding and catalytic properties (i.e. catalytic water position).
The catalytic water is the common, principal feature in B1 MβL catalysis. The C3-C4 β-lactam carboxylate itself paradoxically stabilizes the catalytic water at the active site upon binding, so that the water/β-lactam entity should be considered as (i) the active substrate in the Michaelis complex and (ii) the favorite template for the design of new inhibitors. The flexibility of the zinc site is of paramount importance in the mechanism: regardless the zinc content, the hydrolysis is promoted only by conformational changes at the coordination metal sphere. In the binuclear site then it is particularly elegant the concerted way the zinc ions rearrange their ligands to activate the reactive moieties – the nucleophilic hydroxide and the catalytic water – for a single step β-lactam hydrolysis.
All these ingredients turned out to be important and need to be considered for the design of new kind of inhibitors. As seen, the broad-spectrum activity of MβL's (B1 and B3 subclasses) is mainly developed by few key interactions. Hence, transition state analogues should mimic these common minimal determinants developed at the active site during the reaction. Importantly, new drugs should also favor and promote a rearrangement of the metal sphere as seen during the transition state, inducing a similar shifting of zinc coordination number upon binding.
Supplementary Material
Supporting Information Available: analysis of MD trajectories of CcrA adducts with cefotaxime, benzypenicillin and imipenem. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
MURST-COFIN and NIH are acknowledged for financial support, and INFM and PSC for CPU time. AJV thanks ANPCyT and HHMI for financial support. AJV is a staff member from CONICET and an HHMI International Research Scholar.
Footnotes
Clearly, the statistical errors of the method and the metastable nature of this configuration can lead to an approximate structural estimation of TS.
References
- 1.Fisher JF, Meroueh SO, Mobashery S. Chemical Reviews. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 2.Wilke MS, Lovering AL, Strynadka NCJ. Current Opinion in Microbiology. 2005;8:525–533. doi: 10.1016/j.mib.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 3.Hall BG, Barlow M. Journal of Antimicrobial Chemotherapy. 2005;55:1050–1051. doi: 10.1093/jac/dki130. [DOI] [PubMed] [Google Scholar]
- 4.Galleni M, Lamotte-Brasseur J, Rossolini GM, Spencer J, Dideberg O, Frere JM, Grp MBLW. Antimicrobial Agents and Chemotherapy. 2001;45:660–663. doi: 10.1128/AAC.45.3.660-663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garau G, Di Guilmi AM, Hall BG. Antimicrobial Agents and Chemotherapy. 2005;49:2778–2784. doi: 10.1128/AAC.49.7.2778-2784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh TR, Toleman MA, Poirel L, Nordmann P. Clinical Microbiology Reviews. 2005;18:306-+. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowder MW, Spencer J, Vila AJ. Accounts Of Chemical Research. 2006;39:721–28. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 8.Hall BG, Barlow M. Journal of Molecular Evolution. 2003;57:255–260. doi: 10.1007/s00239-003-2473-y. [DOI] [PubMed] [Google Scholar]
- 9.Orellano EG, Girardini JE, Cricco JA, Ceccarelli EA, Vila AJ. Biochemistry. 1998;37:10173–10180. doi: 10.1021/bi980309j. [DOI] [PubMed] [Google Scholar]
- 10.Prosperi-Meys C, Wouters J, Galleni M, Lamotte-Brasseur J. Cellular and Molecular Life Sciences. 2001;58:2136–2143. doi: 10.1007/PL00000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bounaga S, Laws AP, Galleni M, Page MI. Biochemical Journal. 1998;331:703–711. doi: 10.1042/bj3310703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasia RM, Ceolin M, Vila AJ. Protein Science. 2003;12:1538–1546. doi: 10.1110/ps.0301603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasia RM, Vila AJ. Journal of Biological Chemistry. 2004;279:26046–26051. doi: 10.1074/jbc.M311373200. [DOI] [PubMed] [Google Scholar]
- 14.Rasia RM, Vila AJ. Biochemistry. 2002;41:1853–1860. doi: 10.1021/bi010933n. [DOI] [PubMed] [Google Scholar]
- 15.Yanchak MP, Taylor RA, Crowder MW. Biochemistry. 2000;39:11330–11339. doi: 10.1021/bi0010524. [DOI] [PubMed] [Google Scholar]
- 16.Wang ZG, Fast W, Benkovic SJ. Biochemistry. 1999;38:10013–10023. doi: 10.1021/bi990356r. [DOI] [PubMed] [Google Scholar]
- 17.Wang ZG, Fast W, Benkovic SJ. Journal of the American Chemical Society. 1998;120:10788–10789. [Google Scholar]
- 18.Garcia-Saez I, Hopkins J, Papamicael C, Franceschini N, Amicosante G, Rossolini GM, Galleni M, Frere JM, Dideberg O. Journal of Biological Chemistry. 2003;278:23868–23873. doi: 10.1074/jbc.M301062200. [DOI] [PubMed] [Google Scholar]
- 19.Rossolini GM, Franceschini N, Riccio ML, Mercuri PS, Perilli M, Galleni M, Frere JM, Amicosante G. Biochemical Journal. 1998;332:145–152. doi: 10.1042/bj3320145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Docquier JD, Lamotte-Brasseur J, Galleni M, Amicosante G, Frere JM, Rossolini GM. Journal of Antimicrobial Chemotherapy. 2003;51:257–266. doi: 10.1093/jac/dkg067. [DOI] [PubMed] [Google Scholar]
- 21.Oelschlaeger P, Mayo SL, Pleiss J. Protein Science. 2005;14:765–774. doi: 10.1110/ps.041093405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamaguchi Y, Kuroki T, Yasuzawa H, Higashi T, Jin WC, Kawanami A, Yamagata Y, Arakawa Y, Goto M, Kurosaki H. Journal of Biological Chemistry. 2005;280:20824–20832. doi: 10.1074/jbc.M414314200. [DOI] [PubMed] [Google Scholar]
- 23.Materon IC, Beharry Z, Huang WZ, Perez C, Palzkill T. Journal of Molecular Biology. 2004;344:653–663. doi: 10.1016/j.jmb.2004.09.074. [DOI] [PubMed] [Google Scholar]
- 24.Oelschlaeger P, Schmid RD, Pleiss J. Protein Engineering. 2003;16:341–350. doi: 10.1093/protein/gzg049. [DOI] [PubMed] [Google Scholar]
- 25.Moali C, Anne C, Lamotte-Brasseur J, Groslambert S, Devreese B, Van Beeumen J, Galleni M, Frere JM. Chemistry & Biology. 2003;10:319–329. doi: 10.1016/s1074-5521(03)00070-x. [DOI] [PubMed] [Google Scholar]
- 26.Toleman Ma, Simm AM, Murphy TA, Gales AC, Biedenbach DJ, Jones RN, Walsh TR. Journal of Antimicrobial Chemotherapy. 2002;50:673–679. doi: 10.1093/jac/dkf210. [DOI] [PubMed] [Google Scholar]
- 27.Garau G, Bebrone C, Anne C, Galleni M, Frere JM, Dideberg O. Journal of Molecular Biology. 2005;345:785–795. doi: 10.1016/j.jmb.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 28.Valladares MH, Felici A, Weber G, Adolph HW, Zeppezauer M, Rossolini GM, Amicosante G, Frere JM, Galleni M. Biochemistry. 1997;36:11534–11541. doi: 10.1021/bi971056h. [DOI] [PubMed] [Google Scholar]
- 29.Crawford PA, Yang KW, Sharma N, Bennett B, Crowder MW. Biochemistry. 2005;44:5168–5176. doi: 10.1021/bi047463s. [DOI] [PubMed] [Google Scholar]
- 30.Walsh TR, Hall L, Assinder SJ, Nichols WW, Cartwright SJ, Macgowan AP, Bennett PM. Biochimica Et Biophysica Acta-Gene Structure and Expression. 1994;1218:199–201. doi: 10.1016/0167-4781(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 31.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. Journal of Molecular Biology. 1998;284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 32.Spencer J, Clarke AR, Walsh TR. Journal of Biological Chemistry. 2001;276:33638–33644. doi: 10.1074/jbc.M105550200. [DOI] [PubMed] [Google Scholar]
- 33.Garrity JD, Carenbauer AL, Herron LR, Crowder MW. Journal of Biological Chemistry. 2004;279:920–927. doi: 10.1074/jbc.M309852200. [DOI] [PubMed] [Google Scholar]
- 34.Docquier JD, Pantanella F, Giuliani F, Thaller MC, Amicosante G, Galleni M, Frere JM, Bush K, Rossolini GM. Antimicrobial Agents and Chemotherapy. 2002;46:1823–1830. doi: 10.1128/AAC.46.6.1823-1830.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Docquier JD, Lopizzo T, Liberatori S, Prenna M, Thaller MC, Frere JM, Rossolini GM. Antimicrobial Agents and Chemotherapy. 2004;48:4778–4783. doi: 10.1128/AAC.48.12.4778-4783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mercuri PS, Bouillenne F, Boschi L, Lamote-Brasseur J, Amicosante G, Devreese B, Van Beeumen J, Frere JM, Rossolini GM, Galleni M. Antimicrobial Agents and Chemotherapy. 2001;45:1254–1262. doi: 10.1128/AAC.45.4.1254-1262.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Saez I, Mercuri PS, Papamicael C, Kahn R, Frere JM, Galleni M, Rossolini GM, Dideberg O. Journal of Molecular Biology. 2003;325:651–660. doi: 10.1016/s0022-2836(02)01271-8. [DOI] [PubMed] [Google Scholar]
- 38.Mercuri PS, Garcia-Saez I, De Vriendt K, Thamm I, Devreese B, Van Beeumen J, Dideberg O, Rossolini GM, Frere JM, Galleni M. Journal of Biological Chemistry. 2004;279:33630–33638. doi: 10.1074/jbc.M403671200. [DOI] [PubMed] [Google Scholar]
- 39.Bellais S, Aubert D, Naas T, Nordmann P. Antimicrobial Agents and Chemotherapy. 2000;44:1878–1886. doi: 10.1128/aac.44.7.1878-1886.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies AM, Rasia RM, Vila AJ, Sutton BJ, Fabiane SM. Biochemistry. 2005;44:4841–4849. doi: 10.1021/bi047709t. [DOI] [PubMed] [Google Scholar]
- 41.Fabiane SM, Sohi MK, Wan T, Payne DJ, Bateson JH, Mitchell T, Sutton BJ. Biochemistry. 1998;37:12404–12411. doi: 10.1021/bi980506i. [DOI] [PubMed] [Google Scholar]
- 42.Concha NO, Rasmussen BA, Bush K, Herzberg O. Structure. 1996;4:823–836. doi: 10.1016/s0969-2126(96)00089-5. [DOI] [PubMed] [Google Scholar]
- 43.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, Abdel-Meguid SS. Biochemistry. 2000;39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 44.Murphy TA, Catto LE, Halford SE, Hadfield AT, Minor W, Walsh TR, Spencer J. Journal of Molecular Biology. 2006;357:890–903. doi: 10.1016/j.jmb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Dal Peraro M, Vila AJ, Carloni P. Journal of Biological Inorganic Chemistry. 2002;7:704–712. doi: 10.1007/s00775-002-0346-2. [DOI] [PubMed] [Google Scholar]
- 46.Dal Peraro M, Vila AJ, Carloni P. Inorganic Chemistry. 2003;42:4245–4247. doi: 10.1021/ic026059j. [DOI] [PubMed] [Google Scholar]
- 47.Dal Peraro M, Vila AJ, Carloni P. Proteins-Structure Function and Genetics. 2004;54:412–423. doi: 10.1002/prot.10554. [DOI] [PubMed] [Google Scholar]
- 48.Dal Peraro M, Llarrull LI, Rothlisberger U, Vila AJ, Carloni P. Journal of the American Chemical Society. 2004;126:12661–12668. doi: 10.1021/ja048071b. [DOI] [PubMed] [Google Scholar]
- 49.Diaz N, Suarez D, Merz KM. Journal of the American Chemical Society. 2001;123:9867–9879. doi: 10.1021/ja0113246. [DOI] [PubMed] [Google Scholar]
- 50.Diaz N, Suarez D, Merz KMM. Journal of the American Chemical Society. 2000;122:4197–4208. [Google Scholar]
- 51.Estiu G, Suarez D, Merz KM. J Comput Chem. 2006;27:1240–62. doi: 10.1002/jcc.20411. [DOI] [PubMed] [Google Scholar]
- 52.Cornell W, Cieplak P, Bayly C, Gould I, Merz K, Ferguson D, Spellmeyer D, Fox T, Caldwell J, Kollman PA. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 53.Joergensen W, Chandrasekhar J, Madura J, Impey R, Klein M. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 54.Suarez D, Diaz N, Merz KM. Journal of Computational Chemistry. 2002;23:1587–1600. doi: 10.1002/jcc.10157. [DOI] [PubMed] [Google Scholar]
- 55.Sprik M, Ciccotti G. Journal of Chemical Physics. 1998;109:7737–7744. [Google Scholar]
- 56.Iannuzzi M, Laio A, Parrinello M. Physical Review Letters. 2003;90:238302. doi: 10.1103/PhysRevLett.90.238302. [DOI] [PubMed] [Google Scholar]
- 57.Becke AD. Phys Rev A. 1998;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 58.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 59.Car R, Parrinello M. Physical Review Letters. 1985;55:2471–2474. doi: 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- 60.Laio A, VandeVondele J, Rothlisberger U. J Chem Phys. 2002;116:6941–6947. [Google Scholar]
- 61.Lindahl E, Hess B, van der Spoel D. J Mol Mod. 2001;7:306–317. [Google Scholar]
- 62.Hutter J, Alavi A, Deutsch T, Ballone P, Bernasconi M, Focher P, Goedecker S, editors. Max Planck Institut fuer Festkoerperforschung. IBM Research; Stuttgart: pp. 1995–1999. 3.9. [Google Scholar]
- 63.Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics. 1996;14:33-&. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 64.Haruta S, Yamamoto ET, Eriguchi Y, Sawai T. Fems Microbiology Letters. 2001;197:85–89. doi: 10.1111/j.1574-6968.2001.tb10587.x. [DOI] [PubMed] [Google Scholar]
- 65.Cameron AD, Ridderstrom M, Olin B, Mannervik B. Structure. 1999;7:1067–1078. doi: 10.1016/s0969-2126(99)80174-9. [DOI] [PubMed] [Google Scholar]
- 66.Spencer J, Read J, Sessions RB, Howell S, Blackburn GM, Gamblin SJ. Journal of the American Chemical Society. 2005;127:14439–14444. doi: 10.1021/ja0536062. [DOI] [PubMed] [Google Scholar]
- 67.Park H, Brothers EN, Merz KM. Journal of the American Chemical Society. 2005;127:4232–4241. doi: 10.1021/ja042607b. [DOI] [PubMed] [Google Scholar]
- 68.Felici A, Amicosante G, Oratore A, Strom R, Ledent P, Joris B, Fanuel L, Frere JM. Biochemical Journal. 1993;291:151–155. doi: 10.1042/bj2910151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suarez D, Brothers EN, Merz KM. Biochemistry. 2002;41:6615–6630. doi: 10.1021/bi0121860. [DOI] [PubMed] [Google Scholar]
- 70.Morán Barrio J., González J.M., Lisa M.N., Costello A.L., Dal Peraro M., Carloni P., Bennett B., Tierney D.L., Limansky A.S., Viale A.M. and Vila A.J., submitted
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: analysis of MD trajectories of CcrA adducts with cefotaxime, benzypenicillin and imipenem. This material is available free of charge via the Internet at http://pubs.acs.org.