

Abstract
Gene therapy has emerged as a promising strategy for treatment of various diseases. However, widespread implementation is hampered by difficulties in assessing the success of transfection, in particular, the spatial extent of expression in the target tissue and the longevity of expression. Thus, the development of non-invasive reporter techniques based on appropriate molecules and imaging modalities may help to assay gene expression. We have previously demonstrated the ability to detect β-gal activity based on 19F NMR chemical shift associated with release of fluorophenyl aglycones from galactopyranoside conjugates. Use of fluoropyridoxol as the aglycone provides a potential less toxic alternative and we now report the design, synthesis and structural analysis of a series of novel polyglycosylated fluorinated vitamin B6 derivatives as 19F NMR sensitive aglycones for detection of lacZ gene expression. In particular, we report the activity of 3, α4, α5-tri-O-(β-D-galactopyranosyl)-6-fluoropyridoxol 4, 3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol 12 and 3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(α-D-mannopyranosyl)-6-fluoropyridoxol 13. 4, 12, and 13 all show promising characteristics including highly sensitive 19F NMR response to β-gal activity (Δδ = 9.0 ~ 9.4 ppm), minimal toxicity for substrate or aglycone, and good water solubility. However, the differential glycosylation of 12 and 13 appears more advantageous for assessing lacZ gene expression in vivo.
Keywords: β-galactosidase, 19F NMR, lacZ gene reporter, 6-fluoropyridoxol, pH
INTRODUCTION
Gene therapy holds great promise for the treatment of diverse diseases, but widespread implementation is hindered by difficulties in assessing the success of transfection. The development of non-invasive in vivo reporter techniques based on appropriate molecules and imaging modalities would be of considerable value for assessing the location, magnitude, and persistence of expression.
The lacZ gene encoding β-galactosidase (β-gal) is widely used in molecular biology as a reporter gene to assay clonal insertion, transcriptional activation, protein expression, and protein interaction. Many colorimetric reporter molecules have been described to detect β-gal activity and these for the basis of highly effective spectrophotometric assays in vitro.1–3 However, optical methods are less practical for applications in animals in vivo or ultimately in man in the clinic due to extensive light scattering and absorption by tissues. Towards such applications new reporter molecules are being developed. Recently, Tung et al.4 presented a near infrared approach in vivo based on 9H-(1, 3-dichloro-9, 9-dimethylacridin-2-one-7-yl) β-D-galactopyranoside to detect β-gal activity in transfected tumors in live mice. Lee et al.5 described use of a radiolabeled competitive inhibitor 2-(4-[125I/123I]iodophenyl)ethyl-1-thio-β-D-galactopyranoside to detect β-gal activity in mice. Louie et al.6 introduced an NMR approach using 1-[2-(β-D-galactopyranosyloxy)propyl]-4, 7, 10-tris(carboxymethyl)-1, 4, 7, 10-tetraazacyclododecane) gadolinium (III) based on proton MRI contrast to detect β-gal activity in developing frog embryos following direct injection of substrate into eggs. We have been developing in vivo reporter molecules based on 19F NMR with structures exploiting fluorophenol, trifluorophenol, and fluoropyridoxol aglycones.7–11 To date our published investigations have focused on development of reporter molecules and we have demonstrated detection of β-gal activity in cultured tumor cells with preliminary examples of detectability in tumors in living mice 12. In a continuing effort to develop enhanced approaches for in vivo detection of β-gal, we now report the synthesis, and evaluation of polyglycosylated fluorinated vitamin B6 reporter molecules, designed to enhance water solubility, cellular penetration and enzyme response.
DESIGN
The diversity of substrates and reporter molecules for β-gal activity is indicative of broad substrate specificity. Agents have been tailored for specific imaging modalities or with particular characteristics, such as thermal stability suitable for autoclaving. However, some substrates suffer from poor aqueous solubility and inability to reach targets in vivo and some aglycone products are toxic. Our initial investigations used fluorophenyl β-D-galactopyranosides.7, 9 This approach was particularly facile, being a simple analogy of the classic “yellow” agent o-nitrophenyl β-D-galactopyranoside (ONPG). However, the product aglycone appears somewhat toxic, being closely similar to the uncoupler dinitrophenol. We were able to reduce the requisite concentration of reporter molecule by introducing a trifluoromethyl reporter moiety in place of a single fluorine atom, but this is characterized by a much smaller chemical shift response.11 Toxicity could be largely avoided by using 6-fluoropyridoxol (1, FPOL) as the aglycone and we recently demonstrated proof of principle. Introduction of a D-galactose at the 3 phenolic group of FPOL, 3-O-(β-D-galactopyranosyl)-6-fluoropyridoxol (GFPOL) yielded a 19F NMR gene expression reporter exhibiting a large chemical shift response to β-gal cleavage, but having only moderate kinetic sensitivity to β-gal.8 GFPOL was also modestly water soluble.
We considered that introduction of additional sugar moieties could enhance water solubility and potentially improve enzyme sensitivity. Pertinent to this approach were reports that modification the α4- and α5-position hydroxymethyl moieties of FPOL produces modification of its pKa with relatively minor changes in chemical shift and chemical shift range.13 Further, Escherichia coli (lacZ) β-gal catalyses the hydrolysis of galactopyranosides by cleavage of the C-O bond between D-galactose and the aglycone with a double-displacement mechanism involving the formation (‘glycosylation’ step) and breakdown (‘deglycosylation’ step) of a glycosyl-enzyme intermediate via oxocarbonium-ion-like transition states. It has been observed that hydrogen bonding interaction between the enzyme and the glycosidic substrate is important in the formation of the enzyme-substrate complex and to the hydrolysis rate.14, 15 The involvement of fluorine atoms in hydrogen bonding is well documented and exemplified by some of the strongest known hydrogen bonds.16 Considerable evidence suggests that a C-F moiety can act as a weak proton acceptor and may form hydrogen bonds between the enzyme and the substrate.17–21
We have found evidence of intramolecular hydrogen bonding between α5-OH and 6-F in 1H-NMR spectra of FPOL and its derivatives. For example, the signal of α5-OH in α4-OH and α5-OH unprotected analogues, such as FPOL or 3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-6-fluoropyridoxol always appears downfield and is coupled with 5-CH2 as triplet due to the α5-OH exchange-limitation by the α5-OH and 6-F hydrogen bonding. Meanwhile, α4-OH occurs as an upfield singlet. Introduction of two additional carbohydrate residues at α4 and α5-hydroxymethyl positions of GFPOL would inhibit α5-OH and 6-F hydrogen bonding and facilitate hydrogen bonding between the enzyme and the new substrates. Thus, substrate affinity should increase and both water solubility and enzyme sensitivity could be improved, while retaining the virtues of GFPOL. 8
RESULTS AND DISCUSSION
Syntheses
Our initial approach used a one-pot technique to introduce three D-galactose moieties at the 3 phenolic and α4-, α5-hydroxymethylic sites, simultaneously. Reaction of 1 with 3.3 equivalents of 2, 3, 4, 6-tetra-O-acetyl-α-D-galactopyranosyl bromide 2 in anhydrous dichloromethane catalyzed by Hg(CN)2 afforded the fully galactopyranosylated 6-fluoropyridoxol (3) in 89% yield, which was deacetylated with NH3/MeOH giving the free galactopyranoside 3, α4, α5-tri-O-(β-D-galactopyranosyl)-6-fluoropyridoxol 4 in quantitative yield (Figure 1). The ESI-MS of 3 showed the expected molecular ion at m/z 1178 and quasimolecular ion at m/z 1179 [M+H], corresponding to the fully adorned derivative with three fully acetylated galactosides. The identity of 3 was established using 1H and 13C NMR. The anomeric protons H-1′, H-1″ and H-1‴ of D-galactoses linked to 3, α4- and α5-positions of FPOL at 5.24, 4.66 and 4.52 ppm, respectively, with three well resolved doublets (J1,2 = 8.0 Hz), as well as J2,3 ~ 10 Hz confirming that all D-galactoses are in the β-configuration with the 4C1 chair conformation, whereas in the 13C NMR spectrum, the anomeric carbons C-1′, C-1″ and C-1‴ occurred at 103.34 and 100.22 ppm.
Figure 1. Reagents and conditions.

(a) 2, 3, 4, 6-tetra-O-acetyl-α-D-galactopyranosyl bromide 2, Hg(CN)2, 4Å M.S., CH2Cl2, r.t., 12 h, 89%; (b) NH3-MeOH, 0 °C→r.t., 24 h, quantitative yields.
4 was stable in buffer and gave a single sharp 19F NMR signal. Exposure of 4 to β-gal indicated that all three β-D-galactopyranosyl C1(gal)-O linkages are sensitive resulting in multiple 19F signals around 3 ppm and 12 ~ 20 ppm (Figure 2). These results demonstrate the principle of polyglycosylation to enhance water solubility, while retaining sensitivity to β-gal, but the complex spectra suggested a need for a more sophisticated approach. We therefore designed two further molecules 3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol 12 and 3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(α-D-mannopyranosyl)-6-fluoropyridoxol 13, featuring differential glycosylation: galactosylation at the 3 phenolic group being sensitive to β-gal, and glucopyranosylation or mannopyranosylation at the α4, α5-hydroxymethyl groups to aid water solubility, but resist β-gal activity. Retro-synthetic analysis suggested two approaches through differentially protected intermediates as key synthons.
Figure 2.

19F-NMR spectra of 3, α4, α5-tri-O-(β-D-galactopyranosyl)-6-fluoropyridoxol 4 (10.1 mg, 15 mmol, lower) and its products resulting from addition of β-gal (E801A, 15 units) in PBS (pH=7.4) at 37 °C (upper). Spectra acquired in 51 s and enhanced with an exponential line broadening 40 Hz; β-D-Galp = β-D-galactopyranosyl.
6-Fluoro-α4, α5-isopropylidenepyridoxol 5 was previously prepared as part of the synthesis of 6-fluoro-3, α4-isopropylidenepyridoxol.8, 13 Testing various acids as catalysts showed 2% H2SO4 acetone solution to provide the best yield of 5 (26%). The regioselectivity of the acetonation reaction was confirmed by comparing 1H-NMR of 5 and 6-fluoro-3, α4-isopropylidenepyridoxol, in which the 5-CH2 signal of 5 appeared at 5.03 ppm as a singlet, while in 6-fluoro-3, α4-isopropylidenepyridoxol it appeared at 4.97 ppm as a doublet (JH-5,HO-5 = 1.2 Hz) due to the coupling of 5-OH. 13 Treatment of 5 with 2 using the Koenigs-Knorr glycosylation gave 3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-isopropylidene-6-fluoropyrodoxol 6 in 85% yield. The δH-1′ at 4.64 ppm is a well-resolved doublet (J1,2 = 8.0 Hz) and δC-1′ at 100.03 ppm demonstrated that the D-galactose was in the β-configuration. The correlation between 2-CH3 and H-1′ of sugar ring from the NOSEY spectrum of 6 verified that 2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl residue connected at 3 phenolic site providing further evidence that the acetonation had occurred regioselectively on 4, 5 hydroxymethyl groups.
3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-6-fluoropyridoxol 7 was obtained by cleavage of acetonide 6, but the yields were quite low (≤15%), based on several hydrolysis conditions, such as 80% AcOH, 1% HCl or 90% CF3CO2H in MeOH, CH2Cl2 or 1,4-dioxane at various temperatures (60 ~ 100 °C). A moderate amount of 1 was recoverable indicating that the β-D-galactopyranosyl C1′(gal)-O3 bond became weak and sensitive to acid hydrolysis, presumably due to the presence of the 6-fluorine atom. Condensation of 7 with 2, 3, 4, 6-tetra-O-acetyl-α-D-glucopyranosyl bromide 8 or 2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl bromide 9 in dry CH2Cl2 with Hg(CN)2 as a promoter gave 3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 10 or 3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 11 in yields of 80% or 78%, respectively. Deacetylation of 10 or 11 in NH3/MeOH from 0°C to room temperature gave the target molecules 12 and 13 in quantitative yields (Figure 3). However, the overall yields for 12 and 13 through the five-step reactions were only of 3% with limiting steps in the α4, α5-isopropylidene group formation and hydrolysis procedures.
Figure 3. Reagents and conditions.
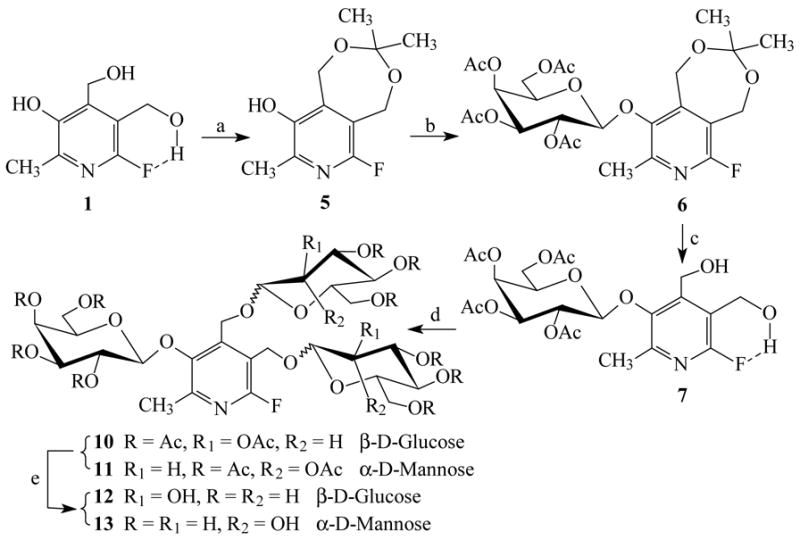
(a) 2% H2SO4, acetone, r.t. 4~5 h, 26%; (b) 2, 3, 4, 6-tetra-O-acetyl-α-D-galactopyranosyl bromide 2, Hg(CN)2, 4Å M.S., CH2Cl2, r.t., 12 h, 85%; (c) 80% AcOH, 80°C, 4~5 h, 15%; (d) 2, 3, 4, 6-tetra-O-acetyl-α-D-glucopyranosyl bromide 8 or 2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl bromide 9, Hg(CN)2, 4Å M.S., CH2Cl2, r.t., 12 h, 80%(→10) or 78%(→11), respectively; (e) NH3-MeOH, 0°C→r.t., 24 h, quantitative yields.
The acidic 3 phenolic group para to 6-fluorine atom in FPOL should be easily converted into the monoanion under mild base conditions, 8, 13, 22 suggesting an alternate approach to selectively benzylate the 3-OH under carefully controlled conditions. Benzyl bromide (1.1 equiv.) was added dropwise over a period of 4 ~ 5 h to the well-stirred reaction mixture of 1 in a dichloromethane-aqueous biphasic system (pH 10~ 11) using tetrabutylammonium bromide (TBAB) as the phase-transfer catalyst yielding 3-O-benzyl-6-fluoropyridoxol 14 in 76% yield. The structure was established on the basis of the coupling characteristics of α4, α5-CH2 as doublets (JH-4,HO-4 = 6.0 Hz, JH-5,HO-5 = 5.4 Hz) and α4, α5-OH as triplets in the 1H-NMR spectrum. Condensation of 14 with 8 or 9 gave 3-O-benzyl-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 15 or 3-O-benzyl-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 16 in satisfactory yields. Removal of the benzyl-protecting group afforded acceptors α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 17 or α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 18 in quantitative yields, which were subjected to a procedure similar to that described above for the preparation of galactosides giving 10 or 11 in high yields (88% or 85%, respectively). After work up and deacetylation, the target compounds 12 and 13 were obtained in 57% and 52% overall yields over five-step reactions (Figure 4).
Figure 4. Reagents and conditions.
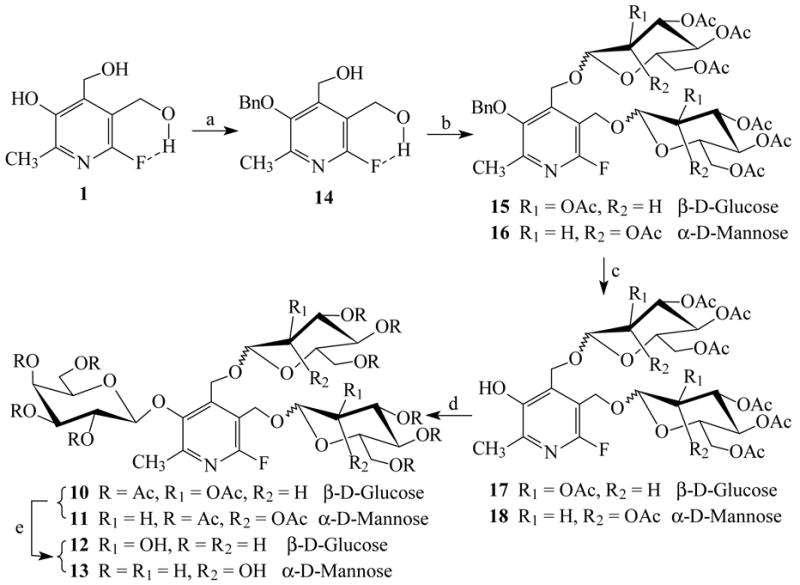
(a) benzyl bromide (1.1 equiv.), CH2Cl2-H2O, pH 10~11, 50°C, TBAB, 4~5 h, 76%; (b) 2, 3, 4, 6-tetra-O-acetyl-α-D-glucopyranosyl bromide 8 or 2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl bromide 9, Hg(CN)2, 4Å M.S., CH2Cl2, r.t., 12 h, 90%(→15) or 85%(→16), respectively; (c) 25psi H2, Pd/C, r.t., 12 h, quantitative yields; (d) 2, 3, 4, 6-tetra-O-acetyl-α-D-galactopyranosyl bromide 2, Hg(CN)2, 4Å M.S., CH2Cl2, r.t., 12 h, 88%(→10) or 85%(→11), respectively; (e) NH3-MeOH, 0°C→r.t., 24 h, 95%(→12) or 94%(→13), respectively.
Recognizing the differential reactivity of the 3 phenolic group over the hydroxymethyl groups, most recently, we have successfully specifically galactopyranosylated 1 on the 3 phenolic group directly with 2 using the above phase-transfer catalysis technique yielding 7.8 Figure 5 depicts a very efficient route to synthesize the target compounds 12 and 13 using just three-steps with higher overall yields (67% and 65%, respectively).
Figure 5. Reagents and conditions.

(a) 2, 3, 4, 6-tetra-O-acetyl-α-D-galactopyranosyl bromide 2, CH2Cl2-H2O, pH 10~11, r.t., TBAB, 4~5 h, 88%; (b) 2, 3, 4, 6-tetra-O-acetyl-α-D-glucopyranosyl bromide 8 or 2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl bromide 9, Hg(CN)2, 4Å M.S., CH2Cl2, r.t., 12 h, 80%(→10) or 78%(→11), respectively; (c) NH3-MeOH, 0°C→r.t., 24 h, 95%(→12) or 94%(→13), respectively.
Characteristics
4, 12 and 13 each gave a single narrow 19F NMR signal between δ −2.0 ~ −3.3 ppm essentially invariant (Δδ ≤ 0.06 ppm) with pH in the range 3 to 12 and temperatures from 25 to 37 °C in whole rabbit blood, 0.9% saline or PBS. Addition of β-gal (E801A) in PBS at 37 °C to 4, 12 and 13 caused rapid hydrolysis releasing the aglycones α4, α5-di-O-(β-D-galactopyranosyl)-6-fluoropyridoxol, α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol and α4, α5-di-O-(α-D-mannopyranosyl)-6-fluoropyridoxol, which also appeared as single narrow 19F signals between δ −11.20 ~ −12.40 ppm (Δδ = 9.0 ~ 9.4 ppm) (Table 1). Action of β-gal on 4 was complicated by action on each of the galactose residue apparently randomly to generate multiple signals representing 1 together with partially galactosylated products (Figure 2). The β-gal hydrolysis of 4, 12, and 13 proceeded in a smooth manner indicating that the liberated aglycones have no inhibitory effects on β-gal (Figure 6). The kinetic curves suggest straightforward first-order kinetics, which were much more rapid for all substrates than for GFPOL. 12 and 13 gave single products upon exposure to β-gal (Figure 7). Addition of 12 and 13 to stably transfected human breast MCF-7-lacZ tumor cells showed cleavage of 12 or 13 (Figure 8) and this proceeded in an initially smooth monotonic manner at rates of 18.6 or 19.6 μmol/min per million MCF7-lacZ cells, respectively. 12 and 13 have much higher aqueous solubility than GFPOL (75 mM, vs. 12, 196 mM and 13, 173 mM in PBS).
Table 1.
| Reporters | 4 | 12 | 13 | GFPOL |
|---|---|---|---|---|
| δF (substrate) | −3.02 | −2.85 | −2.14 | −3.22 |
| δF (product) | −12.37 | −12.16 | −11.22 | −11.21 |
| ΔδF | 9.35 | 9.31 | 9.08 | 7.99 |
| ν(μmol/min/unit) | 34.0 | 35.0 | 38.0 | 4.3 |
ppm with respect to sodium trifluoroacetate.
β-gal (E801A) added at 37°C in PBS (0.1 M, pH=7.4).
Figure 6.

The kinetic hydrolysis time courses of 4 (◆), 12 ( ), 13 (□) (15.0 mmol each) and GFPOL (○) (10.0 mmol) by β-gal (E801A, 15 units) hydrolysis in PBS (0.1 M, pH=7.4, 600 μL) at 37 °C.
Figure 7.

19F-NMR spectra of 3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(β-Dglucopyranosyl)-6-fluoropyridoxol 12 (10.1 mg, 15 mmol, lower) and its hydrolysis by β-gal (E801A, 15 units) in PBS (0.1 M, pH=7.4, 600 μL) at 37 °C (upper). Spectra acquired in 205 s and enhanced with exponential line broadening = 40 Hz.
Figure 8.

19F-NMR spectra of 3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol 12 (5.1 mg, 7.5 mmol) with stably transfected MCF7-lacZ cells (5×106) in PBS (0.1M, pH=7.4, 600 μL) at 37 °C. Spectra acquired in 51 s and enhanced with an exponential line broadening = 100 Hz. (DUFPOL: α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol).
The products α4, α5-di-O-(β-D-galactopyranosyl)-6-fluoropyridoxol (DGFPOL), α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol (DUFPOL) and α4, α5-di-O-(α-D-mannopyranosyl)-6-fluoropyridoxol (DMFPOL) of the action of β-gal on 4, 12 and 13 also exhibit large 19F NMR chemical shift response to pH (Δδ = ~ 11.0 ppm) in the range of pH 1 ~ 12 (Figure 9, Table 2), but there is no spectral overlap with the substrates.
Figure 9.
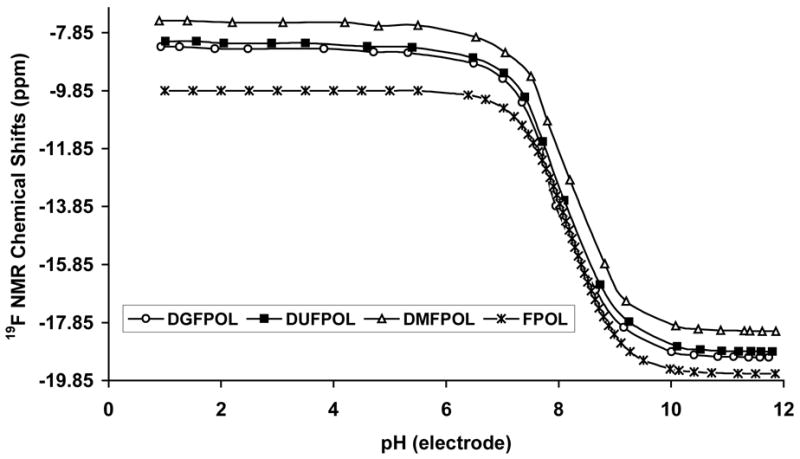
19F NMR chemical shifts pH titration curve of DGFPOL, DUFPOL and DMFPOL in 0.9% saline at 37°C. (DGFPOL: α4, α5-di-O-(β-D-galactopyranosyl)-6-fluoropyridoxol; DUFPOL: α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol; DMFPOL: α4, α5-di-O-(α-D-mannopyranosyl)-6-fluoropyridoxol).
Table 2.
Acidities and 19F NMR/pH properties of DGFPOL, DUFPOL, DMFPOL and FPOL in saline at 25 °C
| pH Indicators | DGFPOL | DUFPOL | DMFPOL | FPOL24 |
|---|---|---|---|---|
| pKa | 7.95 | 8.08 | 8.18 | 8.20 |
| δF acid | −8.34 | −8.15 | −7.44 | −9.85 |
| δF base | −19.05 | −18.85 | −18.15 | −19.61 |
ppm with respect to sodium trifluoroacetate.
Conclusion
These results provide further evidence for the broad specificity of β-gal and the feasibility of modifying substrate structures to enhance enzyme sensitivity and water solubility. The additional sugar residues in 4, 12, and 13 compared with GFPOL all lead to faster cleavage kinetics with β-gal. Significantly, the differential glycosylation provides structures that respond to β-gal with generation of single products. The results with stably transfected breast cancer cells indicate the potential for future studies in vivo.
EXPERIMENTAL
General methods
NMR spectra were recorded on a Varian Inova 400 spectrometer (400 MHz for 1H, 100 MHz for 13C, 376 MHz for 19F) with CDCl3, or DMSO-d6 as solvents. 1H and 13C chemical shifts are referenced to TMS as internal standard, and 19F to a dilute solution of NaTFA in a capillary as external standard (37 °C). Compounds were characterized by acquisition of 1H, 13C, DEPT, 1H-1H COSY or NOESY experiments at 25 °C. Microanalyses were performed on a Perkin-Elmer 2400CHN microanalyser. Mass spectra were obtained by positive and negative ESI-MS using a Micromass Q-TOF hybrid quadrupole/time-of-flight instrument (Micromass UK Ltd.). Reactions requiring anhydrous conditions were performed under nitrogen or argon. Hg(CN)2 was dried before use at 50 °C for 1h, CH2Cl2 was dried over Drierite, and acetonitrile was dried on CaH2 and kept over molecular sieves under N2. Solutions in organic solvents were dried with anhydrous sodium sulfate, and concentrated in vacuo below 45 °C. 2, 3, 4, 6-tetra-O-acetyl-α-D-galactopyranosyl bromide 2 and 2, 3, 4, 6-tetra-O-acetyl-α-D-glucopyranosyl bromide 8 were purchased from the Sigma Chemical Company. 2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl bromide 9 was prepared according to the literature method.23 Column chromatography was performed on silica gel (200 ~ 300 mesh) by elution with cyclohexane-EtOAc and silica gel GF254 (Aldrich) used for analytical TLC. Detection was effected by spraying the plates with 5% ethanolic H2SO4 (followed by heating at 110 °C for ~10 min.) or by direct UV illumination of the plate.
For enzyme kinetic experiments, 4, 12 and 13 (10.1 mg, 15 mmol) were dissolved in PBS (0.1 M, pH=7.4, 600 μL), and a PBS solution of β-gal (0.1 M, pH=7.4, 15 μL, 1 unit/μL, E801A, Promega, Madison, WI, USA) was added and NMR data were acquired immediately at 37 °C.
MCF7-lacZ human breast cancer cells stably transfected to express β-gal were grown in culture under standard conditions and harvested. 12 or 13 were added to suspension of cells (5×106) in PBS and observed by NMR for 1 h.
Syntheses
3, α4, α5-tri-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-6-fluoro-pyridoxol 3
A solution of 2, 3, 4, 6-tetra-O-acetyl-α-D-galactopyranosyl bromide 2 (1.35 g, 3.3 mmol, 1.1 equiv.) in anhydrous CH2Cl2 (8 mL) was added dropwise to the solution of 6-fluoropyridoxol 1 (0.18 g, 1.0 mmol) and Hg(CN)2 (1.01 g, 4.0 mmol) in dry MeCN (10 mL) containing powdered molecular sieves (4 Å, 2.0 g) with vigorous stirring at r.t. under argon in the dark for 12h. The mixture was diluted with CH2Cl2 (30 mL), filtered through Celite, washed, dried (Na2SO4) and concentrated in vacuo. The residue was purified on a silica gel column (1:3 cyclohexane-EtOAc) to yield 3 (1.05 g, 89%) as syrup, Rf 0.30 (1:3 cyclohexane-EtOAc), NMR (CDCl3), δH: 5.24 (1 H, d, J1′,2′ = 8.0 Hz, H-1′), 5.04 (1 H, dd, J2′,3′ = 9.8 Hz, H-2′), 4.73 (1 H, dd, J3′,4′ = 3.4 Hz, H-3′), 3.98 (1 H, dd, J4′,5′ = 2.4 Hz, H-4′), 4.02~4.10 (3 H, m, H-5′, H-6′), 4.66 (1 H, d, J1″,2″ = 8.0 Hz, H-1″), 4.52 (1 H, d, J1‴,2‴ = 8.0 Hz, H-1‴), 5.15 (2 H, dd, J2″,3″ = J2‴,3‴ = 10.0 Hz, H-2″, H-2‴), 5.07 (2 H, dd, J3″,4″ = J3‴,4‴ = 3.6 Hz, H-3″, H-3‴), 5.52 (2 H, dd, J4″,5″ = J4‴,5‴ = 3.2 Hz, H-4″, H-4‴), 3.88 (2 H, m, H-5″, H-5‴), 4.18 (2 H, dd, J5″,6a″ = J5‴,6a‴ = 3.6 Hz, J6a″,6b″ = J6a‴,6b‴ = 9.2 Hz, H-6a″, H-6a‴), 4.11 (2 H, dd, J5″,6b″ = J5‴,6b‴ = 6.8 Hz, H-6b″, H-6b‴), 4.48 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 13.2 Hz, CH2-4a, CH2-5a), 4.12 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 13.2 Hz, CH2-4b, CH2-5b), 2.43 (3 H, s, CH3-2), 2.18, 2.17, 2.16, 2.15, 2.12, 2.11, 2.10, 2.09, 2.08, 2.07, 2.06, 2.05 (36 H, 12s, 12×CH3CO) ppm; δC: 170.84, 170.79, 170.77, 170.73, 170.68, 170.54, 170.53, 170.49, 170.45, 170.35, 170.31, 170.28 (12×CH3CO), 146.47 (d, 3JF-C = 14.5 Hz, Py-C2), 148.16 (d, 4JF-C = 3.8 Hz, Py-C3), 133.10 (s, Py-C4), 112.51 (d, 2JF-C = 31.3 Hz, Py-C5), 155.04 (d, 1JF-C = 231.2 Hz, Py-C6), 103.34 (s, C-1′), 100.22 (s, C-1″, C-1‴), 70.75 (s, C-2′), 71.14 (s, C-3′), 70.61 (s, C-4′), 71.56 (s, C-5′), 67.03 (s, C-6′), 67.45 (s, C-2″, C-2‴), 68.39 (s, C-3″, C-3‴), 66.31 (s, C-4″, C-4‴), 68.55 (s, C-5″, C-5‴), 61.99 (s, C-6″, C-6‴), 61.54 (s, CH2-4), 61.67 (s, CH2-5), 21.03, 20.94, 20.90, 20.89, 20.87, 20.85, 20.83, 20.79, 20.77, 20.76, 20.74, 20.72 (12s, 12×CH3CO), 18.77 (s, CH3-3) ppm. ESIMS: m/z 1178 [M+] (26%), 1179 [M+1] (14%). Anal. Calcd. for C50H64NO30F(%): C, 50.96, H, 5.48, N, 1.19; Found: C, 50.93, H, 5.46, N, 1.15.
3, α4, α5-tri-O-(β-D-galactopyranosyl)-6-fluoropyridoxol 4
A solution of 3 (0.9 g) in anhydrous MeOH (20 mL) containing 0.5 M NH3 was vigorously stirred from 0 °C to r.t. overnight until TLC showed complete reaction and evaporated to dryness in vacuo. Chromatography of the crude syrup on silica gel with EtOAc/MeOH (4:1) afforded 4 (0.52 g) as a syrup in quantitative yield, Rf 0.10 (1:4 MeOH-EtOAc), NMR (DMSO-d6), δH: 4.95 (1 H, d, J1′,2′ = 8.2 Hz, H-1′), 4.76 (1 H, dd, J2′,3′ = 10.0 Hz, H-2′), 4.91 (1 H, dd, J3′,4′ = 2.8 Hz, H-3′), 5.11 (1 H, dd, J4′,5′ = 2.3 Hz, H-4′), 3.77 (1 H, m, H-5′), 3.90 (1 H, dd, J5′,6a′ = 6.4 Hz, J6a′,6b′ = 12.4 Hz, H-6a′), 3.68 (1 H, dd, J5′,6b′ = 3.6 Hz, H-6b′), 4.22 (2 H, d, J1″,2″ = J1‴,2‴ = 8.0 Hz, H-1″, H-1‴), 3.29 (2 H, dd, J2″,3″ = J2‴,3‴ = 10.6 Hz, H-2″, H-2‴), 3.51 (2 H, dd, J3″,4″ = J3‴,4‴ = 3.2 Hz, H-3″, H-3‴), 3.62 (2 H, dd, J4″,5″ = J4‴,5‴ = 2.4 Hz, H-4″, H-4‴), 3.46 (2 H, m, H-5″, H-5‴), 3.66 (2 H, dd, J5″,6a″ = J5‴,6a‴ = 3.6 Hz, J6a″,6b″ = J6a‴,6b‴ = 10.4 Hz, H-6a″, H-6a‴), 3.39 (2 H, dd, J5″,6b″ = J5‴,6b‴ = 6.6 Hz, H-6b″, H-6b‴), 4.48 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 13.0 Hz, CH2-4a, CH2-5a), 4.44 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 13.0 Hz, CH2-4b, CH2-5b), 2.32 (3H, s, CH3-2) ppm; δC: 144.65 (d, 3JF-C = 14.5 Hz, Py-C2), 147.87 (d, 4JF-C = 3.9 Hz, Py-C3), 137.36 (d, 4JF-C = 3.8 Hz, Py-C4), 115.15 (d, 2JF-C = 32.3 Hz, Py-C5), 154.26 (d, 1JF-C = 226.6 Hz, Py-C6), 103.19 (s, C-1′), 101.67 (s, C-1″, C-1‴), 70.36 (s, C-2′), 73.94 (s, C-3′), 69.44 (s, C-4′), 76.08 (s, C-5′), 62.88 (s, C-6′), 72.10 (s, C-2″, C-2‴), 73.50 (s, C-3″, C-3‴), 68.26 (s, C-4″, C-4‴), 75.02 (s, C-5″, C-5‴), 60.60 (s, C-6″, C-6‴), 68.77 (s, CH2-4), 68.92 (s, CH2-5), 19.19 (s, CH3-3) ppm. ESIMS: m/z 673 [M+] (6%), 674 [M+1] (10%). Anal. Calcd. for C26H40NO18F(%): C, 46.34, H, 5.99, N, 2.08; Found: C, 46.30, H, 5.96, N, 2.05.
α4, α5-O-isopropylidene-6-fluoropyridoxol 5
A suspension of 1 (0.50 g, 2.67 mmol) in anhydrous acetone (40 mL) containing 2% c. H2SO4 was stirred for 4 ~ 5h, at the end of which time TLC (4:1 cyclohexane-EtOAc) indicated complete reaction, then cold saturated Na2CO3 solution was added with vigorous stirring up to pH between 8 ~ 9. The precipitate was filtered off and concentration of the reaction mixture under reduced pressure followed by purification on flash silica gel column (4:1 cyclohexane-EtOAc) gave 5 (0.64 g, 26%) as a syrup, Rf 0.34 (4:1 cyclohexane-EtOAc), NMR (CDCl3), δH: 7.45 (1 H, s, HO-3), 5.03 (2 H, s, CH2-5), 4.57 (2 H, s, CH2-4), 2.33 (3 H, s, CH3-2), 1.55 (6 H, s, 2×CH3) ppm; δC: 146.40 (d, 3JF-C = 14.5 Hz, Py-C2), 144.32 (d, 4JF-C = 3.8 Hz, Py-C3), 130.68 (s, Py-C4), 111.21 (d, 2JF-C = 32.8 Hz, Py-C5), 152.20 (d, 1JF-C = 231.2 Hz, Py-C6), 100.15 (s, CMe2), 58.70 (d, 3JF-C = 3.0 Hz, CH2-5), 54.51 (s, CH2-4), 31.62 (s, C(CH3)2), 17.58 (s, CH3-2) ppm. Anal. Calcd. for C11H14NO3F(%): C, 58.13, H, 6.21, N, 6.17; Found: C, 58.08, H, 6.16, N, 6.11.
3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-O-isopropylidene-6-fluoropyridoxol 6
To a solution of 5 (0.62 g, 2.72 mmol) and Hg(CN)2 (0.88 g, 3.50 mmol) in dry CH2Cl2 (10 mL) containing freshly activated 4Å molecular sieves (2.0 g) was added dropwise 2 (1.23 g, 3.0 mmol, 1.1 equiv.). The mixture was stirred overnight in the dark at r.t. under N2 until TLC indicated complete reaction. Work up as for 3 gave 6 (1.29 g, 85%), Rf 0.40 (2:3 cyclohexane-EtOAc), NMR (CDCl3), δH: 4.64 (1 H, d, J1′,2′ = 8.0 Hz, H-1′), 5.25 (1 H, dd, J2′,3′ = 10.0 Hz, H-2′), 5.02 (1 H, dd, J3′,4′ = 3.6 Hz, H-3′), 5.41 (1 H, dd, J4′,5′ = 3.2 Hz, H-4′), 3.97 (1 H, m, H-5′), 4.21 (1 H, dd, J5′,6a′ = 4.4 Hz, J6a′,6b′ = 11.2 Hz, H-6a′), 4.13 (1 H, dd, J5′,6b′ = 7.2 Hz, H-6b′), 5.10 (1 H, d, JCH2-4a,CH2-4b = 8.0 Hz, CH2-4a), 4.67 (1 H, d, JCH2-4a,CH2-4b = 8.0 Hz, CH2-4b), 5.14 (1 H, d, JCH2-5a,CH2-5b = 9.6 Hz, CH2-5a), 5.12 (1 H, d, JCH2-5a,CH2-5b = 9.6 Hz, CH2-5b), 2.42 (3 H, s, CH3-2), 2.17, 2.09, 2.08, 1.99 (12 H, 4s, 4×CH3CO), 1.61, 1.59 (6 H, 2s, 2×CH3) ppm; δC: 170.78, 170.39, 170.26, 170.11 (4s, 4×CH3CO), 145.48 (d, 3JF-C = 15.2 Hz, Py-C2), 133.16 (d, 4JF-C = 4.0 Hz, Py-C3), 126.26 (s, Py-C4), 116.95 (d, 2JF-C = 32.1 Hz, Py-C5), 154.30 (d, 1JF-C = 229.0 Hz, Py-C6), 101.41 (s, CMe2), 100.03 (s, C-1′), 68.70 (s, C-2′), 70.82 (s, C-3′), 67.12 (s, C-4′), 71.53 (s, C-5′), 64.28 (s, C-6′), 55.38 (s, CH2-4), 61.58 (s, CH2-5), 31.88 (s, C(CH3)2), 20.90, 20.89, 20.82, 20.77 (4s, 4×CH3CO), 18.77 (s, CH3-2) ppm. Anal. Calcd. for C25H32NO12F(%): C, 53.84, H, 5.79, N, 2.51; Found: C, 53.79, H, 5.74, N, 2.49.
3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-6-fluoropyridoxol 7
A mixture of 6 (1.25 g, 2.50 mmol) in 80% AcOH (40 mL) was stirred at 80 °C for 4 ~ 5 h, till TLC (1:3 cyclohexane-EtOAc) showed reaction complete. The cooled mixture was neutralized with cold saturated Na2CO3 solution, extracted with EtOAc (4×30 mL), concentrated and purified by flash silica gel column with 1:4 cyclohexane-EtOAc giving 7 (0.17 g, 15%) as a syrup, Rf 0.18 (1:4 cyclohexane-EtOAc), NMR (CDCl3), δH: 4.79 (1 H, d, J1′,2′ = 8.0 Hz, H-1′), 5.55 (1 H, dd, J2′,3′ = 10.6 Hz, H-2′), 5.10 (1 H, dd, J3′,4′ = 3.6 Hz, H-3′), 5.41 (1 H, dd, J4′,5′ = 3.6 Hz, H-4′), 3.88 (1 H, m, H-5′), 4.24 (1 H, dd, J5′,6a′ = 4.4 Hz, J6a′,6b′ = 12.0 Hz, H-6a′), 4.09 (1 H, dd, J5′,6b′ = 6.0 Hz, H-6b′), 5.01 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 12.4 Hz, CH2-4a, CH2-5a), 4.62 (1 H, d, JCH2-4a,CH2-4b = 12.4 Hz, CH2-4b), 4.66 (1 H, d, JCH2-5a,CH2-5b = 12.4 Hz, CH2-5b), 3.50 (1 H, m, α4-HO, exchangeable with D2O), 3.56 (1 H, m, α5-HO, exchangeable with D2O), 2.47 (3 H, s, CH3-2), 2.23, 2.17, 2.02, 2.00 (12 H, 4s, 4×CH3CO)ppm; δC: 170.32, 170.28, 170.18, 169.48 (4×CH3CO), 150.33 (d, 3JF-C = 15.2 Hz, Py-C2), 147.62 (d, 4JF-C = 4.6 Hz, Py-C3), 146.32 (d, 3JF-C = 4.5 Hz, Py-C4), 120.17 (d, 2JF-C = 32.0 Hz, Py-C5), 157.60 (d, 1JF-C = 235.8 Hz, Py-C6), 102.39 (s, C-1′), 68.91 (s, C-2′), 70.74 (s, C-3′), 67.19 (s, C-4′), 71.93 (s, C-5′), 61.98 (s, C-6′), 55.91 (s, CH2-4), 59.60 (s, CH2-5), 20.99, 20.85, 20.70, 20.67 (4s, 4×CH3CO), 19.46 (s, CH3-2) ppm. Anal. Calcd. for C22H28NO12F(%): C, 51.05, H, 5.46, N, 2.71; Found: C, 51.00, H, 5.39, N, 2.68.
Alternately 7 was synthesized from 1 directly by phase transfer catalysis: to a well stirred CH2Cl2 (10 mL)-H2O (10 mL) biphasic mixture (pH 10 ~ 11) of 1 (0.5 g, 2.67 mmol) and TBAB (0.1 g, 0.31 mmol), a solution of 2 (1.21 g, 2.94 mmol, 1.1 equiv.) in CH2Cl2 (10 mL) was added dropwise over a period of 4 ~ 5h at r.t., and the stirring continued for an additional hour. The products were extracted (EtOAc; 4×20 mL), washed free of alkali, dried (Na2SO4), and concentrated. The residue was purified by column chromatography on silica gel (1:4 cyclohexane-EtOAc) to afford 7 (1.08 g, 88%) as syrup, which is identical in all respects to the product obtained above.
3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 10 and 3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-manno-pyranosyl)-6-fluoropyridoxol 11
Condensation of 7 (0.5 g, 1.1 mmol) with 2, 3, 4, 6-tetra-O-acetyl-α-D-glucopyranosyl bromide 8 or 2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl bromide 9 (1.0 g, 2.40 mmol, 1.1 equiv.) in dry CH2Cl2 (10 mL) with Hg(CN)2 (0.63 g, 2.50 mmol) as a promoter, according to the procedures described for the preparation of 3 and 6, furnished 3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 10 and 3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 11, respectively.
3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 10 (1.04 g, 80%), syrup, Rf 0.30 (1:3 cyclohexane-EtOAc), NMR (CDCl3), δH: 5.06 (1 H, d, J1′,2′ = 7.8 Hz, H-1′), 5.28 (1 H, dd, J2′,3′ = 8.8 Hz, H-2′), 4.98 (1 H, dd, J3′,4′ = 4.8 Hz, H-3′), 4.73 (1 H, dd, J4′,5′ = 2.8 Hz, H-4′), 3.95 (1 H, m, H-5′), 4.19 (1 H, dd, J5′,6a′ = 3.6 Hz, J6a′,6b′ = 10.8 Hz, H-6a′), 4.02 (1 H, dd, J5′,6b′ = 5.2 Hz, H-6b′), 5.36 (1 H, d, J1″,2″ = 8.0 Hz, H-1″), 5.39 (1 H, d, J1‴,2‴ = 8.0 Hz, H-1‴), 5.12 (1 H, dd, J2″,3″ = 7.2 Hz, H-2″), 5.15 (1 H, dd, J2‴,3‴ = 6.8 Hz, H-2‴), 5.04 (1 H, dd, J3″,4″ = 3.2 Hz, H-3″), 5.07 (1 H, dd, J3‴,4‴ = 3.6 Hz, H-3‴), 4.76 (1 H, dd, J4″,5″ = 2.8 Hz, H-4″), 4.78 (1 H, dd, J4‴,5‴ = 2.8 Hz, H-4‴), 3.91 (1 H, m, H-5″), 3.93 (1 H, m, H-5‴), 4.08 (1 H, dd, J5″,6a″ = 3.2 Hz, J6a″,6b″ = 9.4 Hz, H-6a″), 4.10 (1 H, dd, J5‴,6a‴ = 3.0 Hz, J6a″,6b″ = 10.0 Hz, H-6a‴), 4.04 (1 H, dd, J5″,6b″ = 7.6 Hz, H-6b″), 4.07 (1 H, dd, J5‴,6b‴ = 6.8 Hz, H-6b‴), 4.55 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 11.2 Hz, CH2-4a, CH2-5b), 4.49 (1 H, d, JCH2-4a,CH2-4b = 11.2 Hz, CH2-4b), 4.91 (1 H, d, JCH2-5a,CH2-5b = 11.2 Hz, CH2-5a), 2.34 (3 H, s, CH3-2), 2.05, 1.98, 1.97, 1.96, 1.95, 1.94, 1.93, 1.92, 1.91, 1.90, 1.89, 1.88 (36 H, 12s, 12×CH3CO) ppm; δC: 170.83, 170.80, 170.76, 170.72, 170.70, 170.56, 170.28, 170.20, 170.17, 170.00, 169.82, 169.75 (12×CH3CO), 152.14 (d, 3JF-C = 16.0 Hz, Py-C2), 149.81 (s, Py-C3), 138.42 (d, 3JF-C = 11.4 Hz, Py-C4), 117.48 (d, 2JF-C = 32.0 Hz, Py-C5), 157.56 (d, 1JF-C = 233.5 Hz, Py-C6), 102.66 (s, C-1′), 98.00 (s, C-1″), 98.06 (s, C-1‴), 71.09 (s, C-2′), 68.65 (s, C-2″), 68.95 (s, C-2‴), 74.46 (s, C-3′), 70.84 (s, C-3″), 71.51 (s, C-3‴), 70.05 (s, C-4′), 68.13 (s, C-4″), 68.22 (s, C-4‴), 75.10 (s, C-5′), 72.20 (s, C-5″), 74.24 (s, C-5‴), 63.75 (s, C-6′), 61.91 (s, C-6″), 63.86 (s, C-6‴), 56.77 (s, CH2-4), 57.16 (s, CH2-5), 21.20, 20.95, 20.93, 20.91, 20.89, 20.87, 20.85, 20.75, 20.67, 20.62, 20.58, 20.54 (12s, 12×CH3CO), 19.76 (s, CH3-3) ppm. ESIMS: m/z 1178 [M+] (28%), 1179 [M+1] (12%). Anal. Calcd. for C50H64NO30F(%): C, 50.96, H, 5.48, N, 1.19; Found: C, 50.92, H, 5.44, N, 1.16.
3-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-galactopyranosyl)-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 11 (1.01 g, 78%), syrup, Rf 0.35 (1:3 cyclohexane-EtOAc), NMR (CDCl3), δH: 4.80 (1 H, d, J1′,2′ = 8.2 Hz, H-1′), 5.13 (1 H, dd, J2′,3′ = 9.8 Hz, H-2′), 5.36 (1 H, dd, J3′,4′ = 4.2 Hz, H-3′), 5.30 (1 H, dd, J3″,4″ = 3.6 Hz, H-4′), 4.01 (1 H, m, H-5′), 4.33 (1 H, dd, J5′,6a′ = 3.2 Hz, J6a′,6b′ = 10.0 Hz, H-6a′), 4.11 (1 H, dd, J5′,6b′ = 4.6 Hz, H-6b′), 4.71 (2 H, d, J1″,2″ = J1‴,2‴ = 2.4 Hz, H-1″, H-1‴), 4.74 (2 H, dd, J2″,3″ = J2‴,3‴ = 6.2 Hz, H-2″, H-2‴), 5.22 (2 H, dd, J3″,4″ = J3‴,4‴ = 3.8 Hz, H-3″, H-3‴), 3.95 (2 H, dd, J4″,5″ = J4‴,5‴ = 2.0 Hz, H-4″, H-4‴), 4.02 (2 H, m, H-5″, H-5‴), 4.11 (2 H, dd, J5″,6a″ = J5‴,6a‴ = 2.0 Hz, J6a″,6b″ = J6a″,6b″ = 7.4 Hz, H-6a″, H-6a‴), 4.07 (2 H, dd, J5″,6b″ = J5‴,6b‴ = 5.6 Hz, H-6b″, H-6b‴), 4.87 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 13.6 Hz, CH2-4a, CH2-5b), 4.67 (1 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 13.6 Hz, CH2-4b, CH2-5a), 2.33 (3 H, s, CH3-2), 2.07, 2.04, 2.03, 2.00, 1.99, 1.98, 1.97, 1.96, 1.95, 1.94, 1.93, 1.92 (36 H, 12s, 12×CH3CO); δC: 171.27, 171.23, 171.15, 171.06, 170.87, 170.83, 170.76, 170.63, 170.58, 170.44, 170.29, 170.25 (12×CH3CO), 153.06 (d, 3JF-C = 16.0 Hz, Py-C2), 149.41 (d, 4JF-C = 4.6 Hz, Py-C3), 145.38 (d, 3JF-C = 4.6 Hz, Py-C4), 117.21 (d, 2JF-C = 31.3 Hz, Py-C5), 159.55 (d, 1JF-C = 235.0 Hz, Py-C6), 103.62 (s, C-1′), 98.32 (s, C-1″), 98.61 (s, C-1‴), 70.75 (s, C-2′), 70.22 (s, C-2″), 70.26 (s, C-2‴), 71.83 (s, C-3′), 70.36 (s, C-3″), 70.39 (s, C-3‴), 69.38 (s, C-4′), 67.04 (s, C-4″), 68.60 (s, C-4‴), 72.54 (s, C-5′), 71.90 (s, C-5″), 72.45 (s, C-5‴), 61.47 (s, C-6′), 62.46 (s, C-6″), 63.53 (s, C-6‴), 56.44 (s, CH2-4), 56.46 (s, CH2-5), 21.29, 21.21, 21.19, 21.17, 21.13, 21.10, 21.08, 21.05, 21.00, 20.95, 20.90, 20.88 (12s, 12×CH3CO), 20.35 (s, CH3-3) ppm. ESIMS: m/z 1178 [M+] (20%), 1179 [M+1] (17%). Anal. Calcd. for C50H64NO30F(%): C, 50.96, H, 5.48, N, 1.19; Found: C, 50.94, H, 5.45, N, 1.16.
3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoro-pyridoxol 12 and 3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(α-D-mannopyranosyl)-6-fluoropyridoxol 13 Compounds 10, 11 (1.00 g, 0.85 mmol) were deacetylated as described above for 4, to yield 12 and 13 in quantitative yields.
3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(β-D-glucopyranosyl)-6-fluoropyridoxol 12 (0.57 g), foam solid, Rf 0.20 (1:4 MeOH-EtOAc), NMR (DMSO-d6), δH: 5.01 (1 H, d, J1′,2′ = 8.2 Hz, H-1′), 5.22 (1 H, dd, J2′,3′ = 9.0 Hz, H-2′), 4.92 (1 H, dd, J3′,4′ = 4.6 Hz, H-3′), 4.70 (1 H, dd, J4′,5′ = 2.6 Hz, H-4′), 3.91 (1 H, m, H-5′), 4.12 (1 H, dd, J5′,6a′ = 3.2 Hz, J6a′,6b′ = 10.2 Hz, H-6a′), 4.00 (1 H, dd, J5′,6b′ = 5.6 Hz, H-6b′), 5.14 (2 H, d, J1″,2″ = 10.0 Hz, H-1″, H-1‴), 4.82 (2 H, dd, J2″,3″ = J2‴,3‴ = 8.2 Hz, H-2″, H-2‴), 4.69 (2 H, dd, J3″,4″ = J3‴,4‴ = 3.4 Hz, H-3″, H-3‴), 4.93 (2 H, dd, J4″,5″ = J4‴,5‴ = 3.2 Hz, H-4″, H-4‴), 3.65 (2 H, m, H-5″, H-5‴), 3.55 (2 H, dd, J5″,6a″ = J5‴,6a‴ = 4.8 Hz, J6a″,6b″ = J6a‴,6b‴ = 12.0 Hz, H-6a″, H-6a‴), 3.31 (2 H, dd, J5″,6b″ = J5‴,6b‴ = 5.6 Hz, H-6b″, H-6b‴), 4.29 (1 H, d, JCH2-4a,CH2-4b = 7.6 Hz, CH2-4a), 4.36 (1 H, d, JCH2-4a,CH2-4b = 7.6 Hz, CH2-5a), 4.20 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 7.6 Hz, CH2-4b, CH2-5b), 4.18 ~ 3.65 (12 H, br, HO-2′, 3′, 4′, 6′, 2″, 3″, 4″, 6″, 2‴, 3‴, 4‴, 6‴, exchangeable with D2O), 2.42 (3 H, s, CH3-2); δC: 144.43 (d, 3JF-C = 15.0 Hz, Py-C2), 136.26 (d, 4JF-C = 3.8 Hz, Py-C3), 124.40 (d, 3JF-C = 3.8 Hz, Py-C4), 120.39 (d, 2JF-C = 32.8 Hz, Py-C5), 148.98 (d, 1JF-C = 259.7 Hz, Py-C6), 103.65 (s, C-1′), 101.76 (s, C-1″, C-1‴), 72.34 (s, C-2′), 71.28 (s, C-2″, C-2‴), 74.55 (s, C-3′), 73.88 (s, C-3″, C-3‴), 69.82 (s, C-4′), 68.89 (s, C-4″, C-4‴), 76.62 (s, C-5′), 77.29 (s, C-5″, C-5‴), 61.36 (s, C-6′), 60.95 (s, C-6″, C-6‴), 60.54 (s, CH2-4), 60.78 (s, CH2-5), 19.88 (s, CH3-3) ppm. ESIMS: m/z 673 [M+] (8%), 674 [M+1] (14%). Anal. Calcd. for C26H40NO18F(%): C, 46.34, H, 5.99, N, 2.08; Found: C, 46.32, H, 5.97, N, 2.07.
3-O-(β-D-galactopyranosyl)-α4, α5-di-O-(α-D-mannopyranosyl)-6-fluoropyridoxol 13 (0.57 g), foam solid, Rf 0.26 (1:4 MeOH-EtOAc), NMR (DMSO-d6), δH: 5.00 (1 H, d, J1′,2′ = 8.0 Hz, H-1′), 5.23 (1 H, dd, J2′,3′ = 10.0 Hz, H-2′), 5.16 (1 H, dd, J3′,4′ = 3.8 Hz, H-3′), 5.08 (1 H, dd, J3″,4″ = 3.2 Hz, H-4′), 4.21 (1 H, m, H-5′), 4.51 (1 H, dd, J5′,6a′ = 3.6 Hz, J6a′,6b′ = 10.2 Hz, H-6a′), 4.31 (1 H, dd, J5′,6b′ = 4.8 Hz, H-6b′), 4.84 (2 H, d, J1″,2″ = J1‴,2‴ = 2.6 Hz, H-1″, H-1‴), 4.68 (2 H, dd, J2″,3″ = J2‴,3‴ = 6.0 Hz, H-2″, H-2‴), 5.02 (2 H, dd, J3″,4″ = J3‴,4‴ = 3.6 Hz, H-3″, H-3‴), 4.05 (2 H, dd, J4″,5″ = J4‴,5‴ = 2.2 Hz, H-4″, H-4‴), 3.94 (2 H, m, H-5″, H-5‴), 4.21 (2 H, dd, J5″,6a″ = J5‴,6a‴ = 2.4 Hz, J6a″,6b″ = J6a″,6b″ = 8.4 Hz, H-6a″, H-6a‴), 4.17 (2 H, dd, J5″,6b″ = J5‴,6b‴ = 6.5 Hz, H-6b″, H-6b‴), 4.77 (2 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 11.6 Hz, CH2-4a, CH2-5b), 4.57 (1 H, d, JCH2-4a,CH2-4b = JCH2-5a,CH2-5b = 11.6 Hz, CH2-4b, CH2-5a), 2.45 (3 H, s, CH3-2), 4.30 ~ 3.70 (12 H, br, HO-2′, 3′, 4′, 6′, 2″, 3″, 4″, 6″, 2‴, 3‴, 4‴, 6‴, exchangeable with D2O); δC: 151.07 (d, 3JF-C = 15.3 Hz, Py-C2), 148.61 (d, 4JF-C = 4.8 Hz, Py-C3), 144.27 (d, 3JF-C = 3.6 Hz, Py-C4), 116.29 (d, 2JF-C = 32.0 Hz, Py-C5), 157.25 (d, 1JF-C = 233.5 Hz, Py-C6), 103.68 (s, C-1′), 98.56 (s, C-1″), 98.68 (s, C-1‴), 71.76 (s, C-2′), 70.66 (s, C-2″), 70.86 (s, C-2‴), 72.38 (s, C-3′), 71.46 (s, C-3″), 71.32 (s, C-3‴), 70.48 (s, C-4′), 66.87 (s, C-4″), 67.90 (s, C-4‴), 73.64 (s, C-5′), 72.20 (s, C-5″), 72.65 (s, C-5‴), 62.77 (s, C-6′), 63.56 (s, C-6″), 64.83 (s, C-6‴), 57.54 (s, CH2-4), 58.41 (s, CH2-5), 20.12 (s, CH3-3) ppm. ESIMS: m/z 673 [M+] (5%), 674 [M+1] (9%). Anal. Calcd. for C26H40NO18F(%): C, 46.34, H, 5.99, N, 2.08; Found: C, 46.31, H, 5.97, N, 2.05.
3-O-Benzyl-6-fluoropyridoxol 14
To a well stirred CH2Cl2 (10 mL)-H2O (10 mL) biphasic mixture (pH 10 ~ 11) of 1 (0.50 g, 2.67 mmol) and TBAB (0.10 g, 0.31 mmol), a solution of benzyl bromide (0.51 g, 2.94 mmol, 1.1 equiv.) in CH2Cl2 (10 mL) was added dropwise over a period of 4 ~ 5h, while the reaction temperature was maintained at 50 °C, and the stirring continued for an additional hour. Products were extracted (CH2Cl2, 4×20 mL), washed free of alkali, dried (Na2SO4), and concentrated, the residue was purified by column chromatography on silica gel with 1:2 cyclohexane-EtOAc to afford major product 14 (0.56 g, 76%), white crystalline, Rf 0.38 (1:2 cyclohexane-EtOAc), NMR (CDCl3), δH: 7.39 (5 H, m, Ar-H), 4.90 (2 H, s, PhCH2), 4.75 (2 H, d, JH-5,HO-5 = 5.4 Hz, CH2-5), 4.72 (2 H, d, JH-4,HO-4 = 6.0 Hz, CH2-4), 3.57 (1 H, t, JH-5,HO-5 = 5.4 Hz, α5-OH, exchangeable with D2O), 3.49 (1 H, t, JH-4,HO-4 = 6.0 Hz, α4-OH, exchangeable with D2O), 2.44 (3 H, s, CH3-2); δC: 151.34 (d, 3JF-C = 9.6 Hz, Py-C2), 146.97 (d, 4JF-C = 2.9 Hz, Py-C3), 149.55 (d, 3JF-C = 3.1 Hz, Py-C4), 119.09 (d, 2JF-C = 20.8 Hz, Py-C5), 156.30 (d, 1JF-C = 216.2 Hz, Py-C6), 136.33, 128.96, 128.88, 128.57 (Ph-C), 55.99 (s, PhCH2, CH2-4), 56.76 (s, CH2-5), 19.31 (s, CH3-2) ppm. Anal. Calcd. for C15H16NO3F(%): C, 64.96, H, 5.82, N, 5.05; Found: C, 64.95, H, 5.79, N, 5.04.
3-O-Benzyl-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoro-pyridoxol 15 and 3-O-Benzyl-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-manno-pyranosyl)-6-fluoropyridoxol 16
Glycosylation of 14 (0.46 g, 2.0 mmol) with 8 or 9 (1.83 g, 4.45 mmol, 1.1 equiv.) was carried out as for 3, 10 and 11 to give 15 and 16, respectively.
3-O-Benzyl-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 15 (0.32 g, 95%), syrup, Rf 0.35 (3:2 cyclohexane-EtOAc), NMR (CDCl3), δH: 7.41 (5 H, m, Ar-H), 5.36 (1 H, d, J1′,2′ = 8.2 Hz, H-1′), 5.41 (1 H, d, J1″,2″ = 8.2 Hz, H-1″), 5.14 (2 H, dd, J2′,3′ = J2″,3″ = 7.4 Hz, H-2′, H-2″), 4.45 (2 H, dd, J3′,4′ = J3″,4″ = 3.3 Hz, H-3′, H-3″), 4.84 (2 H, dd, J4′,5′ = J4″,5″ = 3.8 Hz, H-4′, H-4″), 3.96 (2 H, m, H-5′, H-5″), 4.80 (2 H, dd, J5′,6a′ = J5″,6a″ = 2.6 Hz, J6a′,6b′ = J6a″,6b″ = 10.1 Hz, H-6a′, H-6a″), 4.10 (2 H, dd, J5′,6b′ = J5″,6b″ = 3.0 Hz, H-6b′, H-6b″), 4.94 (2 H, s, PhCH2), 4.55 (1 H, d, JCH2-4a,CH2-4b = 10.4 Hz, CH2-4a), 4.48 (1 H, d, JCH2-4a,CH2-4b = 10.4 Hz, CH2-4b), 4.60 (1 H, d, JCH2-5a,CH2-5b = 11.0 Hz, CH2-5a), 4.52 (1 H, d, JCH2-5a,CH2-5b = 11.0 Hz, CH2-5b), 2.37 (3 H, s, CH3-2), 2.00, 1.99, 1.98, 1.97, 1.96, 1.95, 1.94, 1.93 (24 H, 8s, 8×CH3CO); δC: 170.84, 170.76, 170.31, 170.29, 170.26, 169.95, 169.92, 169.84 (8×CH3CO), 152.18 (d, 3JF-C = 14.5 Hz, Py-C2), 142.64 (d, 4JF-C = 4.6 Hz, Py-C3), 150.12 (d, 3JF-C = 3.8 Hz, Py-C4), 116.20 (d, 2JF-C = 32.0 Hz, Py-C5), 157.40 (d, 1JF-C = 234.3 Hz, Py-C6), 136.37, 129.00, 128.94, 128.87, 128.16, 127.77 (Ph-C), 100.23 (s, C-1′), 100.41 (s, C-1″), 71.41 (s, C-2′, C-2″), 72.08 (s, C-3′), 72.19 (s, C-3″), 68.34 (s, C-4′), 68.51 (s, C-4″), 72.86 (s, C-5′), 72.93 (s, C-5″), 61.86 (s, C-6′), 61.98 (s, C-6″), 60.98 (s, CH2-4), 61.28 (s, CH2-5), 20.88, 20.85, 20.82, 20.75, 20.73, 20.60, 20.59, 20.58 (8s, 8×CH3CO), 19.43 (s, CH3-3) ppm. ESIMS: m/z 937 [M+] (35%), 938 [M+1] (25%). Anal. Calcd. for C43H52NO21F(%): C, 55.05, H, 5.59, N, 1.49; Found: C, 55.03, H, 5.57, N, 1.48.
3-O-Benzyl-α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 16 (0.30 g, 90%), syrup, Rf 0.40 (3:2 cyclohexane-EtOAc), NMR (CDCl3), δH: 7.38 (5 H, m, Ar-H), 5.38 (1 H, d, J1′,2′ = 2.6 Hz, H-1′), 5.41 (1 H, d, J1″,2″ = 2.6 Hz, H-1″), 5.36 ~ 3.95 (18 H, m, H-2′, 3′, 4′, 5′, 6′, 2″, 3″, 4″, 5″, 6″, PhCH2, CH2-4, CH2-5), 2.38 (3 H, s, CH3-2), 2.02, 2.00, 1.99, 1.98, 1.97, 1.96, 1.95, 1.94 (24 H, 8s, 8×CH3CO); δC: 171.25, 171.18, 170.89, 170.85, 170.78, 170.66, 170.60, 170.48 (8×CH3CO), 153.28 (d, 3JF-C = 15.8 Hz, Py-C2), 145.48 (d, 4JF-C = 4.8 Hz, Py-C3), 150.16 (d, 3JF-C = 3.8 Hz, Py-C4), 116.30 (d, 2JF-C = 31.0 Hz, Py-C5), 157.77 (d, 1JF-C = 206.8 Hz, Py-C6), 98.42 (s, C-1′), 100.03 (s, C-1″), 72.60 ~ 56.54 (13C, C-2′, 3′, 4′, 5′, 6′, 2″, 3″, 4″, 5″, 6″, PhCH2, CH2-4, CH2-5), 21.23, 20.94, 20.92, 20.90, 20.88, 20.86, 20.84, 20.80, 20.78 (8s, 8×CH3CO), 18.37 (s, CH3-3) ppm. ESIMS: m/z 937 [M+] (32%), 938 [M+1] (20%). Anal. Calcd. for C43H52NO21F(%): C, 55.05, H, 5.59, N, 1.49; Found: C, 55.01, H, 5.55, N, 1.45.
α4, α5-Di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 17 and α4, α5-di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 18 A mixture of 15 or 16 (0.29 g, 0.30 mmol) and Pd-C (5%, 50 mg) in MeOH (40 mL) was stirred for 24h at r.t. under H2 (25 psi). Evaporated filtrate gave 17, 18 in quantitative yields.
α4, α5-Di-O-(2, 3, 4, 6-tetra-O-acetyl-β-D-glucopyranosyl)-6-fluoropyridoxol 17 (0.26 g), syrup, Rf 0.28 (1:3 cyclohexane-EtOAc), NMR (CDCl3), δH: 7.33 (1 H, s, HO-3, exchangeable with D2O), 5.30 (1 H, d, J1′,2′ = 8.4 Hz, H-1′), 5.35 (1 H, d, J1″,2″ = 8.4 Hz, H-1″), 5.09 (2 H, dd, J2′,3′ = J2″,3″ = 7.6 Hz, H-2′, H-2″), 4.35 (2 H, dd, J3′,4′ = J3″,4″ = 3.4 Hz, H-3′, H-3″), 4.80 (2 H, dd, J4′,5′ = J4″,5″ = 3.6 Hz, H-4′, H-4″), 3.89 (2 H, m, H-5′, H-5″), 4.77 (2 H, dd, J5′,6a′ = J5″,6a″ = 2.4 Hz, J6a′,6b′ = J6a″,6b″ = 10.6 Hz, H-6a′, H-6a″), 4.05 (2 H, dd, J5′,6b′ = J5″,6b″ = 3.2 Hz, H-6b′, H-6b″), 4.51 (1 H, d, JCH2-4a,CH2-4b = 10.3 Hz, CH2-4a), 4.45 (1 H, d, JCH2-4a,CH2-4b = 10.3 Hz, CH2-4b), 4.57 (1 H, d, JCH2-5a,CH2-5b = 11.1 Hz, CH2-5a), 4.49 (1 H, d, JCH2-5a,CH2-5b = 11.1 Hz, CH2-5b), 2.35 (3 H, s, CH3-2), 1.99, 1.98, 1.97, 1.96, 1.95, 1.94, 1.93, 1.91 (24 H, 8s, 8×CH3CO); δC: 170.82, 170.78, 170.65, 170.58, 170.46, 169.85, 169.82, 169.80 (8×CH3CO), 152.28 (d, 3JF-C = 14.2 Hz, Py-C2), 148.28 (d, 4JF-C = 3.2 Hz, Py-C3), 142.69 (d, 3JF-C = 4.8 Hz, Py-C4), 116.26 (d, 2JF-C = 32.2 Hz, Py-C5), 157.56 (d, 1JF-C = 231.4 Hz, Py-C6), 100.35 (s, C-1′), 100.54 (s, C-1″), 71.37 (s, C-2′, C-2″), 72.18 (s, C-3′), 72.29 (s, C-3″), 68.38 (s, C-4′), 68.56 (s, C-4″), 72.83 (s, C-5′), 72.88 (s, C-5″), 61.82 (s, C-6′), 61.89 (s, C-6″), 60.90 (s, CH2-4), 61.19 (s, CH2-5), 20.85, 20.83, 20.82, 20.80, 20.78, 20.76, 20.73, 20.65 (8s, 8×CH3CO), 19.32 (s, CH3-3) ppm. ESIMS: m/z 847 [M+] (30%), 848 [M+1] (21%). Anal. Calcd. for C36H46NO21F(%): C, 50.99, H, 5.47, N, 1.65; Found: C, 50.96, H, 5.45, N, 1.62.
α4, α5-Di-O-(2, 3, 4, 6-tetra-O-acetyl-α-D-mannopyranosyl)-6-fluoropyridoxol 18 (0.26 g), syrup, Rf 0.27 (1:3 cyclohexane-EtOAc), NMR (CDCl3), δH: 7.33 (1 H, s, HO-3, exchangeable with D2O), 5.33 (1 H, d, J1′,2′ = 2.7 Hz, H-1′), 5.37 (1 H, d, J1″,2″ = 2.7 Hz, H-1″), 5.45 ~ 4.07 (16 H, m, H-2′, 3′, 4′, 5′, 6′, 2″, 3″, 4″, 5″, 6″, CH2-4, CH2-5), 2.35 (3 H, s, CH3-2), 2.01, 2.00, 1.99, 1.98, 1.97, 1.96, 1.95, 1.94 (24 H, 8s, 8×CH3CO); δC: 171.33, 171.21, 170.85, 170.83, 170.76, 170.61, 170.56, 170.53 (8×CH3CO), 153.67 (d, 3JF-C = 15.8 Hz, Py-C2), 149.08 (d, 4JF-C = 3.0 Hz, Py-C3), 145.68 (d, 3JF-C = 4.6 Hz, Py-C4), 118.23 (d, 2JF-C = 31.2 Hz, Py-C5), 157.59 (d, 1JF-C = 223.1 Hz, Py-C6), 98.67 (s, C-1′), 100.33 (s, C-1″), 72.8 ~ 56.56 (12C, C-2′, 3′, 4′, 5′, 6′, 2″, 3″, 4″, 5″, 6″, CH2-4, CH2-5), 20.99, 20.97, 20.93, 20.90, 20.88, 20.86, 20.84, 20.80 (8s, 8×CH3CO), 18.45 (s, CH3-3) ppm. ESIMS: m/z 847 [M+] (25%), 848 [M+1] (18%). Anal. Calcd. for C36H46NO21F(%): C, 50.99, H, 5.47, N, 1.65; Found: C, 50.97, H, 5.44, N, 1.63.
Acknowledgments
Supported by grants from the DOD Breast Cancer Initiative IDEA award DAMD17-03-1-0343, and the Cancer Imaging Program, NIH P20 CA 86354 (pre-ICMIC). NMR experiments were conducted at the Mary Nell and Ralph B. Rogers NMR Center, an NIH BTRP facility #P41-RR02584. We thank Dr. Li Liu for preparing the breast cancer cells.
References
- 1.Pocsi I, Taylor SA, Richardson AC, Smith BV, Price RG. Comparison of several new chromogenic galactosides as substrates for various beta-D-galactosidases. Biochim Biophys Acta. 1993;1163:54–60. doi: 10.1016/0167-4838(93)90278-y. [DOI] [PubMed] [Google Scholar]
- 2.Heuermann K, Cosgrove J. S-Gal: an autoclavable dye for color selection of cloned DNA inserts. Biotechniques. 2001;30:1142–1147. doi: 10.2144/01305pf01. [DOI] [PubMed] [Google Scholar]
- 3.Buller CJ, Zang XP, Howard EW, Pento JT. Measurement of beta-galactosidase tissue levels in a tumor cell xenograft model. Methods & Findings in Exp Clin Pharmacol. 2003;25:713–716. doi: 10.1358/mf.2003.25.9.793338. [DOI] [PubMed] [Google Scholar]
- 4.Tung CH, Zeng Q, Shah K, Kim DE, Schellingerhout D, Weissleder R. In vivo imaging of beta-galactosidase activity using far red fluorescent switch. Cancer Res. 2004;64:1579–1583. doi: 10.1158/0008-5472.can-03-3226. [DOI] [PubMed] [Google Scholar]
- 5.Lee KH, Byun SS, Choi JH, Paik JY, Choe YS, Kim BT. Targeting of lacZ reporter gene expression with radioiodine-labeled phenylethyl-beta- d-thiogalactopyranoside. Eur J Nucl Med Mol Imaging. 2004;31:433–438. doi: 10.1007/s00259-003-1395-7. [DOI] [PubMed] [Google Scholar]
- 6.Louie AY, Huber MM, Ahrens ET, Rothbacher U, Moats R, Jacobs RE, Fraser SE, Meade TJ. In vivo visualization of gene expression using magnetic resonance imaging. Nature Biotechnol. 2000;18:321–325. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- 7.Cui W, Otten P, Li Y, Koeneman K, Yu J, Mason RP. A novel NMR approach to assessing gene transfection: 4-fluoro-2-nitrophenyl-β-D-galactopyranoside as a prototype reporter molecule for β-galactosidase. Magn Reson Med. 2004;51:616–620. doi: 10.1002/mrm.10719. [DOI] [PubMed] [Google Scholar]
- 8.Yu JX, Ma Z, Li Y, Koeneman KS, Liu L, Mason RP. Synthesis and Evaluation of a Novel Gene Reporter Molecule: Detection of β-galactosidase activity Using 19F NMR of a Fluorinated Vitamin B6 conjugate. Med Chem. 2005;1:255–262. doi: 10.2174/1573406053765495. [DOI] [PubMed] [Google Scholar]
- 9.Yu JX, Otten P, Ma Z, Cui W, Liu L, Mason RP. A Novel NMR Platform for Detecting Gene Transfection: Synthesis and Evaluation of Fluorinated Phenyl β-D-Galactosides with Potential Application for Assessing LacZ Gene Expression. Bioconj Chem. 2004;15:1334–1341. doi: 10.1021/bc049936d. [DOI] [PubMed] [Google Scholar]
- 10.Yu JX, Kodibagkar V, Cui W, Mason RP. 19F: a versatile reporter for non-invasive physiology and pharmacology using magnetic resonance. Curr Med Chem. 2005;12:818–848. doi: 10.2174/0929867053507342. [DOI] [PubMed] [Google Scholar]
- 11.Yu JX, Liu L, Kodibagkar VD, Cui W, Mason RP. Synthesis and Evaluation of Novel Enhanced Gene Reporter Molecules: Detection of β-Galactosidase Activity Using 19F NMR of Trifluoromethylated Aryl β-D-Galactopyranosides. Bioorg Med Chem. 2006;14:326–333. doi: 10.1016/j.bmc.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Kodibagkar V, Yu J, Liu L, Brown B, Hetherington HP, Gerard RD, Mason RP. ISMRM 13th Scientific Meeting; Miami Beach, Florida, USA. 2005. 19F CSI of gene-reporter molecule OFPNPG. [Google Scholar]
- 13.He S, Mason RP, Hunjan S, Mehta VD, Arora V, Katipally R, Kulkarni PV, Antich PP. Development of Novel 19F NMR pH Indicators: Synthesis and Evaluation of a Series of Fluorinated Vitamin B6 Analogs. Bioorg Med Chem. 1998;6:1631–1639. doi: 10.1016/s0968-0896(98)00104-7. [DOI] [PubMed] [Google Scholar]
- 14.Rye CS, Withers SG. Curr Opinion Chem Biology. 2000;4:573. doi: 10.1016/s1367-5931(00)00135-6. [DOI] [PubMed] [Google Scholar]
- 15.Sinnott ML. Catalytic mechanism of enzymic glycosyl transfer. Chem Rev. 1990;90:1171–1202. [Google Scholar]
- 16.Pauling L. The nature of the chemical bond. Cornell University; Ithaca, NY: 1980. [Google Scholar]
- 17.McCarter JD, Adam MJ, Withers SG. Binding energy and catalysis. Fluorinated and deoxygenated glycosides as mechanistic probes of Escherichia coli (lacZ) β-galactosidase. Biochem J. 1992;286:721–727. doi: 10.1042/bj2860721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Street IP, Armstrong CR, Withers SG. Hydrogen bonding and specificity. Fluorodeoxy sugars as probes of hydrogen bonding in the glycogen phosphorylase-glucose complex. Biochemistry. 1986;25:6021. doi: 10.1021/bi00368a028. [DOI] [PubMed] [Google Scholar]
- 19.Withers SG. 1998 Hoffmann la Roche Award Lecture - Understanding and exploiting glycosidases. Can J Chem. 1999;77:1. [Google Scholar]
- 20.Thoden JB, Hegeman AD, Wesenberg G, Chapeau MC, Frey PA, Holden HM. Structural analysis of UDP-sugar binding to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry. 1997;36:6294. doi: 10.1021/bi970025j. [DOI] [PubMed] [Google Scholar]
- 21.White A, Tull D, Johns K, Withers SG, Rose DR. Crystallographic observation of a covalent catalytic intermediate in a beta-glycosidase. Nature Struct Biol. 1996;3:149. doi: 10.1038/nsb0296-149. [DOI] [PubMed] [Google Scholar]
- 22.Mason RP. Transmembrane pH gradients in vivo: measurements using fluorinated vitamin B6 derivatives. Curr Med Chem. 1999;6:481–499. [PubMed] [Google Scholar]
- 23.Lemieux RU. In: Methods in Carbohydrates Chemistry. Whistler ML, Wolfrom RL, editors. II. Academic Press Inc; London: 1963. pp. 223–230. [Google Scholar]
- 24.Mehta VD, Kulkarni PV, Mason RP, Constantinescu A, Aravind S, Goomer N, Antich PP. 6-Fluoropyridoxol: a novel probe of cellular pH using 19F NMR spectroscopy . FEBS Letters. 1994;349:234–238. doi: 10.1016/0014-5793(94)00675-x. [DOI] [PubMed] [Google Scholar]