

Abstract
The apolipoprotein E4 (APOE4) gene is a well-known risk factor for Alzheimer’s disease (AD) and other neurological disorders. Post-menopausal women with AD who express at least one APOE4 gene have more severe neuropathology and worsened cognitive scores than their non-expressing counterparts. Since 17β-estradiol down-regulates inflammation as part of its neuroprotective role, we examined the effect of 17β-estradiol on the response of microglia to immune activation as a function of APOE genotype. Our data show that the anti-inflammatory activity of 17β-estradiol is significantly reduced in APOE4 targeted replacement mice compared to APOE3 mice. A significant interaction between APOE genotype and the response to 17β-estradiol was observed for NO and cytokine production by immune activated microglia. The genotype specific effect was not restricted to brain macrophages since peritoneal macrophages from APOE4 ovariectomized mice also demonstrated a significant difference in 17β-estradiol responsiveness. ERβ protein levels in APOE4 microglia were higher than APOE3 microglia, suggesting a difference in post-translational protein regulation in the presence of the APOE4 gene. Overall, our data indicate that the APOE genotype may be a critical component in assessing the effectiveness of 17β-estradiol’s action and may impact the neuroprotective role of 17β-estradiol and of hormone replacement therapy on brain function when the APOE4 gene is expressed.
Keywords: Inflammation, estrogen, microglia, APOE4, ERα, ERβ, Nitric Oxide, peritoneal macrophage
1. Introduction
The presence of the APOE4 gene has been established as a risk factor for Alzheimer’s disease (AD) [16] and individuals who are heterozygous (3/4) or homozygous (4/4) for APOE4 demonstrate earlier cognitive changes and increased density of amyloid plaques and neurofibrillary tangles, characteristic neuropathological lesions in AD, than those individuals who do not carry an APOE4 gene [48;53]. The widespread association of APOE4 with enhanced risk and poorer outcomes in neurological diseases such as head injury, stroke and multiple sclerosis [9;24;29] suggests that the APOE4 gene may contribute to a common pathophysiological pathway in multiple types of CNS damage. Our previous studies demonstrate that apolipoprotein E protein regulates innate immunity in an isoform specific manner [7;12;14;62]. Expression of either one or two copies of the APOE4 gene is associated with increased in vitro activation of microglia, the CNS macrophage, and a pro-inflammatory state. Lynch et al. [32] have confirmed this finding in vivo by demonstrating increased mRNA and protein levels of inflammatory cytokines such as tumor necrosis factor-alpha (TNFα) and interleukin-6 (IL-6) in the brains of APOE4 targeted replacement mice injected with lipopolysaccharide (LPS). A similar enhancement of inflammation after LPS injection was also demonstrated in the hippocampi of transgenic APOE4 mice compared to transgenic APOE3 mice [43].
Our previous findings in targeted replacement APOE4 mice suggest that the inflammatory response may be different in male and female mice [7]. Although controversial, gender differences in AD have been observed [4;50]. Women have slightly, but significantly, more global neuropathology and worsened dementia ratings than men [4]. In large part, the gender differences can be correlated with APOE genotype. Women who express an APOE4 gene have lower hippocampal volumes, more senile plaque pathology and worsened cognitive scores than their counterparts who do not express APOE4 [4]. While the women included in the studies were post-menopausal, the roles of sex steroid status or hormone replacement therapy in the observed results were not examined.
Estrogenic compounds are particularly important since 17β-estradiol regulates the innate immune response of macrophages and serves as an anti-inflammatory agent [8;45;60;61]. For example, treatment of brain macrophages (microglia) and peripheral macrophages with physiological doses of 17β-estradiol reduces the release of pro-inflammatory factors such as nitric oxide (NO) and TNFα [8;13;61]. Furthermore, the loss of estrogen with ovariectomy or with aging results in a shift of macrophage function towards a pro-inflammatory state [2;45;46]. Part of the neuroprotective effect of estrogen in the brain that is lost during aging and in post-menopausal women with AD may, in fact, stem from loss of 17β-estradiol’s regulation of the innate immune response.
We have examined the effect of the APOE4 gene on 17β-estradiol’s anti-inflammatory action in brain macrophages (microglia) derived from targeted replacement (TR) mice expressing the human APOE4 gene at the mouse APOE locus compared to mice expressing the human APOE3 gene at the same locus [56]. Our data presented here demonstrate a significant difference in the responsiveness of microglia and peripheral macrophages to 17β-estradiol. These results demonstrate that the presence of an APOE4 gene reduces the effectiveness of 17β-estradiol as an anti-inflammatory agent.
2. Methods
2.1 Animals
Homozygous apolipoprotein E targeted replacement (APOE3 or APOE4) mice contain a targeted insertion of exons 2–4 of the human APOE3 or APOE4 genes that replace the corresponding genomic DNA at the mouse APOE locus [56]. All animals were bred from in-house stock and were housed under a standard light:dark cycle and received food and water ad libitum. Ages of the female mice used in this study ranged from 55–60 weeks of age and were within the age range for reproductive senescence in C57BL/6J mice (48–64 weeks for cessation of cycling and entrance into either persistent vaginal cornification or into persistent diestrus) [21].
2.2 Ovariectomy
Adult female mice were anesthetized using ip sodium pentobarbital (40–70 mg/kg) and a dorsal sterile surgical field prepared. A 2 cm dorsal midline incision was made with its cranial terminus 1.5 to 2.5 cm caudal to the 13th rib. After trimming fascia away, the muscle wall was pierced with forceps, 1.5 to 2 cm lateral to the spine. The ovary is located in a fat pad under the dorsal muscle mass. With blunt forceps, the ovary was drawn through the muscle and skin incisions, clamped and then ligated to the level of the fallopian tube. The ovary was removed, the uterine horn returned to the body cavity and the skin incision closed with 1–2 wound clips. The second ovary was removed similarly. Mice were returned to their normal environment for an additional 4 weeks before use in the experimental protocols. In some cases, a sterile, 60-day time release pellet containing 17β-estradiol (0.36 mg; Innovative Research of America, Sarasota FL) was subcutaneously implanted into ovariectomized females one day following ovariectomy.
2.3 17β-estradiol Radioimmunoassay
Prior to harvesting peritoneal macrophages, blood was collected via cardiac puncture in order to determine the serum 17β-estradiol concentrations and was measured using the 17β-estradiol 125I RIA Kit from ICN Diagnostics (Costa Mesa, CA) per the manufacturer’s instructions. Prior to measurement, each blood sample was extracted using diethyl ether (4ml diethyl ether; 0.300 ml saline/serum sample), nitrogen evaporated at 37°C and reconstituted into diluent buffer. Values of serum estradiol in animals receiving time-release pellets for the 4 week period were 92 ± 35 pg/ml and 109 ± 29 pg/ml for APOE3 and APOE4 females, respectively. Although there was a tendency for the average values to be greater in the ovariectomized females implanted with an estradiol pellet from intact females, these values were not significantly different. The measured values of estradiol represent the integrated values after 4 weeks of treatment with slow release pellets. It is possible, however, that serum estradiol levels may have fluctuated due to non uniform release from the implanted pellet. Serum estradiol levels were below the detectable range (less than 10 pg/ml) in ovariectomized females.
2.4 Peritoneal macrophage isolation and culture
Peritoneal macrophages were isolated as previously described [7]. Prior to harvesting, macrophages were elicited by intraperitoneal (ip) injection of each adult mouse with 5mM sodium periodate. This process is well known to promote the entry of circulating monocytic cells into the peritoneum. After 72 hours, the mice were killed and the peritoneal cavities were flushed 2 times with warm PBS to obtain macrophages. Lavage fluid containing cells was carefully extracted from the peritoneum and the fluid containing cells from 2 to 3 mice of the same genotype and gender was pooled. In this manner a sufficient number of macrophages were obtained for the experimental assays. The cells were pelleted by centrifugation at 1000 g and then re-suspended into phenol red–free, serum-free media (high glucose DMEM containing 2 mM glutamine and 50 μg/ml gentamycin). Cells were counted, plated directly into 96 well plates and cultured for 2 days in a humidified 5% CO2, 95% air atmosphere. During this time the macrophages attach to the plastic of the culture plate and spread, resembling typical tissue macrophages.
2.5 Microglial cultures
Enriched microglial cultures were prepared from postnatal day 0–2 APOE3 and APOE4 pups using previously published methods [17]. Briefly, whole brains were removed and placed into sterile 1X phosphate buffered saline (1X PBS) containing 100μg/ml penicillin/streptomycin and 0.5% fungizone (Invitrogen, Rockville, MD). Meninges were removed under a dissecting microscope and cortices were placed directly into microglial growth media (high glucose phenol-red free DMEM containing 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin streptomycin/0.5% fungizone, all from Invitrogen). The cortical tissue was dissociated using the Papain Dissociation System (Worthington Biochemicals Corp, Lakewood NJ) as described by the manufacturer and cells in suspension were plated into T-75 tissue culture flasks. Cells were grown at 37°C in a 95% air 5% CO2 humidified atmosphere for approximately 5 days at which time the culture media was replaced with fresh growth media containing 10% horse serum in place of fetal bovine serum (FBS). After an additional 3–5 days, loosely adherent cells were removed from the culture by shaking on a rotary shaker (240 rpm) for 2 hours. The cells were pelleted by centrifugation (1000g; 10 min) and resuspended into phenol red free, serum free medium. The cells were then plated into 6 or 96 well dishes and were cultured for an additional 48 hrs in serum free, phenol red free media. Once plated, serum withdrawal or extended culture in serum free medium does not adversely affect microglial survival (data not shown). Microglial or macrophage viability prior to the experimental protocols was assessed in sister cultures using the MTT assay as previously described. Viability was greater than 98% (data not shown). The percentage of microglia was periodically measured in sister cultures using lectin cytochemistry. In this case, cells were reacted with lectin, Griffonia simplicifolia (GS-1), coupled to peroxidase. Color was then developed using nickel-enhanced diaminobenzidine (DAB) according to the manufacturer’s instructions (SigmaFast DAB, Sigma) and cells were visualized using a Nikon inverted microscope (Nikon, Melville, NY). Cultures prepared as described above were greater than 95% microglia.
2.6 Immune activation in vitro
Microglial cultures were pre-treated with 17β-estradiol for 15 hrs (overnight) prior to immune activation. Stock solutions of 1 mM 17β-estradiol (Sigma) was prepared in sterile DMSO and diluted to a final concentration in the nanomolar range in serum free, phenol red free microglial media for the experimental protocols. DMSO concentrations in all final media were below 0.001% and considered to be negligible. The estrogen receptor antagonist ICI 182,780 (Tocris Cookson, Ellisville, MO) was prepared as a 1 mM stock in DMSO and used at a final concentration of 10nM. For detection of supernatant nitrite, 17β-estradiol - treated and untreated macrophages were immune activated for 20–24 hours using the combination of recombinant mouse interferon-γ (rmIFNγ) plus either lipopolysaccharide (LPS), a bacterial endotoxin prepared from E. coli serotype O11:B5 (100ng/ml) or polyinosinic-polycytidylic acid (PIC) (50 μg/ml), a double stranded RNA that serves as a viral mimetic (all from Sigma, St. Louis, MO), diluted into phenol red free, serum free microglial media. To maintain exposure of the pre-treated cells to 17β-estradiol, the same concentration of 17β-estradiol that was used in the pre-treatment protocol was also added to the media containing immune activators. For quantitative analysis of mRNA, 17β-estradiol - treated or untreated cells were immune activated for 3 hrs using the same activating agents as above.
2.7 Measurement of nitrite and protein
The supernatant levels of nitrite, the stable oxidation product of NO in biological solutions, was used as a measure of microglial NO production. Nitrite levels in microglial cultures were measured by chemiluminescence using a Sievers 280 Nitric Oxide Analyzer (Sievers, Boulder CO) and nitrite levels in peritoneal macrophages or were measured using the Griess reaction as described [7]. Standard curves were prepared from sodium nitrite diluted into the treatment media. Total protein (μg/well) was measured using the BCA method (Pierce, Rockford, IL) based on the manufacturer’s directions using BSA (μg/ml) as standard. Nitrite levels in each well were normalized to total μg protein and expressed as μmoles nitrite/μg protein.
2.8 Measurement of cytokine levels
Supernatant levels of TNFα or IL-6 were determined using an ELISA per the manufacturer’s instructions (BioSource International, Camarillo, CA). TMB-One (Promega, Madison, WI) was used for substrate color development and the reaction was stopped using 1M H2SO4. Absorbance was measured at 450 nm using a Molecular Devices microplate reader (Molecular Devices, Sunnydale, CA). Cytokine values were normalized to μg protein and presented as pg TNFα (or IL-6)/μg protein.
2.9 Western blot
Western blot from microglial or cortical lysates were performed in a standard manner. Antibodies for detection of ERα and ERβ were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA; ERα (MC-20) and used at a dilution of 1:1000 or from Affinity Bioreagents (Golden, CO; ERβ (PA1-311) and used at a dilution 1:500.
2.10 RNA extraction, preparation, quantitative RT-PCR
RNA was extracted from microglial cultures of each genotype using the RNA Easy Mini Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. RNA was quantified using a Beckman DV530 Life Science UV/Visible Spectrophotometer (Beckman-Coulter, Fullerton, CA). RNA was reverse transcribed to cDNA using High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) with MultiScribe Reverse Transcriptase and random primers. mRNA expression levels were determined by quantitative real-time PCR. Essentially, qPCR reactions were performed using 100ng cDNA per reaction with a ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems) and Taqman Assays-on-Demand Gene Expression primer/probe sets and the Taqman Universal PCR master mix (all from Applied Biosystems) for the following primer sets of mouse genes: nitric oxide synthase 2 (NOS2) (Genbank: NM_010927; Assay-on-Demand ID: Mm00440485_m1); estrogen receptor α (ERα; esr1) (Genbank: NM_007956; Assay-on-Demand ID: Mm00433149_m1); estrogen receptor β (ERβ; esr2) (Genbank: NM_010157; Assay-on-Demand ID: Mm00599819_m1); and eukaryotic 18S, a standard housekeeping gene, (Genbank: X03205; Assay-on-Demand ID: Hs99999901_s1). To confirm the primer sequences, PCR products from ERα and ERβ real-time PCR reactions were cloned into PCR2.1 –TOPO vector using the TOPO TA cloning kit (Invitrogen, Carlsbad, CA) and sequenced. Sequences of ERα and ERβ PCR products 100% matched corresponding Genbank sequences (NM_007956 for ERα and NM_010157 for ERβ) after a BLAST search (NCBI).
Relative mRNA quantitation was calculated using the 2−ΔΔCT method [36] by normalizing the value of the target gene for each sample to its endogenous 18S control value, and then normalizing these values to a baseline sample, designated as the calibrator. In these experiments, the APOE3 untreated microglia served as the calibrator for each gene analyzed.
2.11 Statistical analysis
For microglia and macrophage cultures, average values (± SEM) for each treatment condition were compared between similarly treated APOE3 or APOE4 cultures. Samples were assayed and averaged from at least 3 different litter groups (where a litter group is defined as those cells grown from different mouse litters; 3–5 mouse pups/litter), with a minimum of triplicate wells analyzed per experimental paradigm for each litter group. Data were analyzed using 2-way ANOVA with Bonferroni’s post hoc test where appropriate using the Prism 4.0 software package (Graphpad, San Diego, CA). Significance was set at P < 0.05 in all cases.
3. Results
3.1 The anti-inflammatory effect of estrogen is reduced in APOE4 microglia
To examine the effect of 17β-estradiol on immune activation in APOE3 and APOE4 microglia, microglial cultures were treated overnight with physiological doses of 17β-estradiol (0.1–5 nM) or remained untreated. The cells were then immune activated by stimulation for an additional 24 hrs with lipopolysaccharide (LPS; 100 ng/ml) plus recombinant murine interferon-γ (IFNγ;100 U/ml) in the continuing presence of 17β-estradiol. The microglial inflammatory response was then detected by measuring the supernatant levels of nitrite (for NO production) and TNFα or IL-6 levels (for cytokine production) and the response of APOE4 microglia was compared to the response of APOE3 microglia. Figure 1A–1C shows nitrite and cytokine levels measured in the same cell supernatants collected from a single representative APOE3 or APOE4 litter group. This matched data set demonstrates that the APOE genotype systematically and significantly affects microglial nitrite and cytokine production. As shown in Fig 1, the levels of nitrite, TNFα or IL-6 are higher in APOE4 microglia compared to APOE3 microglia, a finding that is consistent with our previously published data [6;11]. However, these representative data also show a direct effect of APOE genotype on the anti-inflammatory action of 17β-estradiol. Microglia derived from APOE4 brain are less responsive to 17β–estradiol over a physiological range of concentrations. A significant interaction (p = 0.022) between the genotype and the response to 17β-estradiol was observed for nitrite production (Fig 1A) in the matched data but did not reach significance for either TNFα or IL-6 production (Fig 1B–1C). A strong interaction between genotype and the response to 17β-estradiol is more clearly observed in the grouped data shown in Figure 1D-1F. In this case, data are presented as the relative change from LPS + IFNγ alone (where 1.0 = 0 nM 17β-estradiol) for all litter groups. Relative values were used to reduce the variance observed in the absolute values of nitrite, TNFα or IL-6 produced by different litter groups in response to the same immune stimulation. Significant statistical interaction between the APOE genotype and the microglial anti-inflammatory response to 17β-estradiol was observed for nitrite, TNFα or IL-6 production (p = 0.0005 for IL-6; p <0.0001 for TNFα and p = 0.013 for nitrite). These data strongly indicate that APOE4 microglia are less responsive to 17β-estradiol compared to APOE3 microglia.
Figure 1. APOE genotype influences the effect of 17β-estradiol on immune activated microglia.

Cultured microglia from either APOE3 or APOE4 mice were pretreated with varying doses of 17β-estradiol (0.1 to 5 nM) followed by the addition of 100U/ml IFNγ and 100 ng/ml LPS. A–C: Supernatant nitrite and cytokine levels were measured in the same cell supernatants collected from a single representative APOE3 or APOE4 litter group. Significance was determined using 2-way ANOVA with the Bonferroni post-hoc test. (A) Supernatant nitrite levels demonstrated a significant interaction between APOE genotype and 17β-estradiol dose; p = 0.022; ** = p < 0.01 for 1 nM 17β-estradiol compared to LPS+IFNγ alone in APOE 3 microglia; * = p < 0.05 for 1 or 5 nM 17β-estradiol compared to LPS+IFNγ alone for APOE4 microglia. (B) Supernatant TNFα levels demonstrated a significant effect of genotype (p = 0.0001) and of estrogen dose (p = 0.037). (C) Supernatant IL-6 levels demonstrated a significant effect of genotype (p = 0.0001). D–F: Data represent the average relative change (±SEM) from LPS+IFNγ alone (=1.0) for 6–12 different litter groups, 3 wells cultured per litter group. (D) The change in supernatant nitrite levels demonstrated a significant interaction between APOE genotype and estrogen response (p= 0.0134);* = p < 0.05 for APOE3 vs APOE4; *** = p < 0.001 for APOE3 vs APOE4 (E) The change in supernatant TNFα levels demonstrated a significant interaction between genotype and estrogen dose (p < 0.0001); ** = p < 0.01 for APOE3 vs APOE4; *** = p < 0.001 for APOE3 vs APOE4. (F) The change in supernatant IL-6 levels demonstrated a significant interaction between genotype and estrogen dose (p < 0.0005); *** = p < 0.001 for APOE3 vs APOE4.
Since 17β-estradiol has been previously reported to reduce the expression of NOS2, the gene that codes for inducible nitric oxide synthase [3;60;61;68], we also measured changes in the level of mRNA for NOS2 in APOE3 and APOE4 microglia using quantitative RT-PCR. As predicted for immune induction [41], NOS2 mRNA expression dramatically increased over background levels on treatment of the microglia with LPS+IFNγ in both APOE3 and APOE4 microglia (Table 1). A significantly higher level of NOS2 mRNA was observed for APOE4 microglia compared to APOE3 microglia and is likely to underlie the increased production of NO in APOE4 microglia. However, pre-treatment with 1 nM 17β-estradiol had no effect on the stimulated NOS2 mRNA levels in either APOE3 or APOE4 microglia.
Table 1.
Effect of 17β-estradiol treatment on the fold increase in NOS2 mRNA in APOE3 and APOE4 microglia
| Untreated | LPS + IFNγ | LPS + IFNγ + 17β-estradiol | |
|---|---|---|---|
| APOE3 | 0.8 ± 0.15 (7) | 1391 ± 379 (3) | 2056 ± 547 (4) |
| APOE4 | 1.2 ± 0.3 (10) | 3479 ± 440* (10) | 3295 ± 394 (7) |
= p < 0.03 when comparing LPS + IFNγ treated APOE4 microglia compared to LPS + IFNγ treated APOE3 microglia. Data points represent the average (±SEM) fold increase in mRNA. (n)= number of litter groups assayed. LPS= 10 ng/ml; IFNγ = 10 U/ml; 17β-estradiol = 1 nM
3.2 Expression of ERα and ERβ mRNA and protein is APOE4 genotype dependent
To determine if the response to 17β–estradiol in APOE3 or APO4 microglia was mediated by the classical estrogen receptors ERα or ERβ, microglia were pre-treated with either 17β–estradiol or 17β–estradiol plus ICI 182,780, followed by immune activation in the continuing presence of 17β–estradiol with or without ICI. ICI 182,780 is a well-known global antagonist of ERα and ERβ [28], although it has recently been shown to act as an agonist for an estrogen-binding G-protein coupled receptor [71]. In APOE3 microglia, 17β–estradiol-mediated suppression of immune-activated NO production was blocked by ICI 182,780 (Fig. 2). However, nitrite levels in APOE3 microglia were not simply returned by ICI 182,780 treatment to the control values found in LPS + IFNγ-only treated cells, but instead significantly increased to levels greater than baseline. Under identical treatment conditions, immune activated APOE4 microglia were poorly responsive to 17β-estradiol and did not respond to ICI 182,780 at the concentrations studied. Using 2-way ANOVA, a significant interaction (p = 0.005) was observed between genotype and the microglial response to treatment suggesting that the APOE genotype strongly influences the effect of 17β-estradiol.
Figure 2. ICI 182,780 blocks 17β-estradiol in APOE3 microglia.
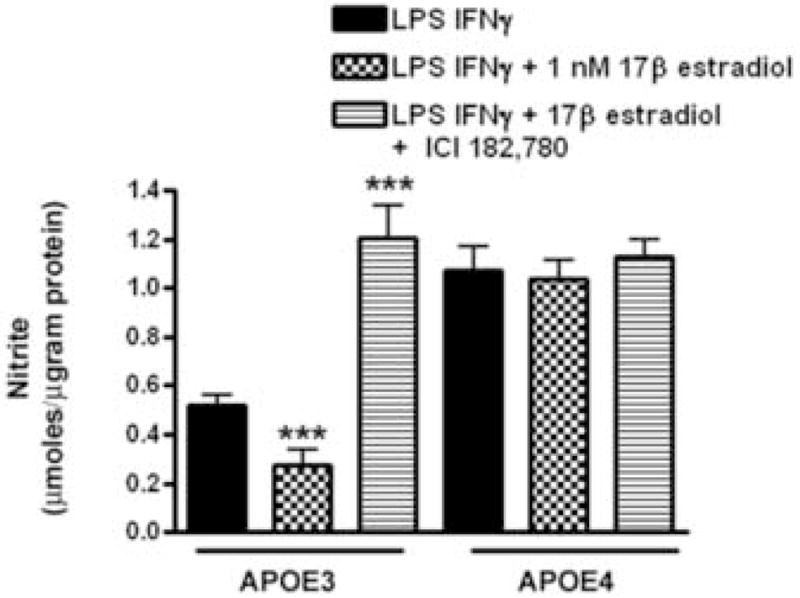
Microglia from APOE3 and APOE4 mice were pretreated with 17β-estradiol in the presence and absence of ICI 182,780 followed by the addition of LPS+ IFNγ. Average (±SEM) supernatant nitrite levels were determined and the effect of 17β-estradiol and ICI 182,780 treatment compared between APOE3 and APOE4 mice. Significance was determined using 2-way ANOVA with the Bonferroni post-hoc test. A significant interaction was observed between genotype and the response to treatment (p = 0.005); *** = p < 0.001 for treatments compared to LPS+ IFNγ alone.
Expression of ERα and/or ERβ has been previously reported for primary cultures of microglia and murine macrophage cell lines such as BV2 and N9 cells [3;19;59]. To determine if the genotype-specific differences in 17β-estradiol’s action could be explained by different profiles of ERs, we measured mRNA levels for ERα and ERβ in untreated and immune activated APOE3 and APOE4 microglia using quantitative RT-PCR. In all cases, untreated APOE3 microglia were used as the calibrator for the 2−ΔΔCT analysis. ERα mRNA levels were significantly higher (p < 0.01) in untreated APOE4 microglia compared to untreated APOE3 microglia while ERβ mRNA levels were the same for both genotypes (Fig. 3A, B). Stimulation with LPS+IFNγ for 3 hrs reduced ERβ mRNA in both APOE3 and APOE4 cells to the same level compared to no treatment (Fig 3B). Interestingly, immune stimulation altered the APOE genotype-specific difference in ERα mRNA levels. The high basal levels of ERα mRNA in APOE4 microglia were decreased by LPS+IFNγ treatment to the same level as observed in immune stimulated APOE3 microglia (Fig 3).
Figure 3. Expression of mRNA and protein for ERα and ERβ in microglia from APOE3 and APOE4 mice.

(A and B) Quantitative RT-PCR was used to measure the average fold change (±SEM) in ERα and ERβ mRNA in microglia using untreated APOE3 as the calibrator. (A) Basal (untreated) levels of ERα mRNA were significantly higher in APOE4 microglia. Significance was determined using 2-way ANOVA with the Bonferroni post-hoc test. A significant effect of genotype was observed (p = 0.0025) for n = 7–12 cultures from a minimum of 3 different litter groups per genotype; ** = p < 0.01 for APOE4 compared to APOE3. (B) No significant difference was observed between untreated or treated APOE3 and APOE4 ERβ mRNA levels. A significant effect of LPS+IFNγ treatment compared to untreated was observed for both APOE3 and APOE4 mRNA (*** = p< 0.001). (C) ERα (top panel) and ERβ (middle and bottom panels) protein levels in untreated microglial lysates were determined using Western blot (labeled- Mg) from APOE3 (labeled- 3) and APOE4 (labeled- 4) mice. For ERβ, blots are presented from 3 different untreated microglial litter groups. Permanently transfected neuronal cells derived from the N2a cell line expressing human APOE3 (labeled -3) or human APOE4 (labeled -4) [6] were used as controls to demonstrate ER antibody specificity. N2a cells only express ERα and do not express ERβ. GADPH served as a loading control.
Protein levels of estrogen receptors demonstrated a distinctly different profile. Untreated levels of ERα protein expression were the same in APOE3 and APOE4 microglia while untreated levels of ERβ protein were greater in APOE4 microglia compared to APOE3 microglia (Fig 3C). Protein levels were also measured in LPS +IFNγ-treated microglia after 20–24 hrs of treatment. Immune stimulated- ERα and ERβ protein expression levels were not different from untreated levels at this time point (data not shown) with ERβ levels remaining higher in APOE4 microglia compared to APOE3 microglia.
3.3 Peritoneal macrophages from ovariectomized mice also demonstrate an APOE genotype-specific difference in the response to 17β-estradiol
Since microglia were derived from neonatal, pre-pubertal animals, we examined the isoform specificity of 17β-estradiol on immune-stimulated macrophage function in aged, 50–66 week old ovariectomized (OVX) adult female mice. Baseline serum estrogen levels after ovariectomy were negligible (<10pg/ml) in both the APOE3 and APOE4 mice, thereby eliminating differences in circulating estrogen levels as a confounding factor. In addition, all female mice used in the study were within the age range for reproductive senescence in C57BL/6J mice (48–64 weeks for cessation of cycling and entrance into either persistent vaginal cornification or into persistent diestrus) [20]. Similarly to the microglial experiments, we treated peritoneal macrophages isolated from APOE3 and APOE4 ovariectomized female mice with immune activators and measured supernatant nitrite or cytokine levels in the presence and absence of exogenously added 17β-estradiol (Fig 4). As shown in Figure 4 NO production by immune stimulated macrophages derived from APOE3 OVX mice was significantly higher than NO production by macrophages derived from APOE4 OVX mice. Since the level of NO generated by macrophages from OVX mice reflects the effect of the chronic lack of exposure to circulating estrogens, the higher levels of NO production in APOE3 compared to APOE4 macrophages is consistent with the removal of a larger “suppressive” factor controlling NO production in APOE3 macrophages. Increased pro-inflammatory cytokine production by macrophages derived from ovariectomized mice compared to intact mice has been previously demonstrated [45]. Treatment with 1nM 17β-estradiol significantly reduced nitrite levels by approximately 50% in LPS + IFNγ –activated APOE3 macrophages but did not significantly reduce nitrite levels in macrophages from APOE4 mice. In addition to using LPS + IFNγ as the immune stimulation, we also stimulated with a viral mediator of the immune response, polycytidylic-polyinosinic acid (PIC) coupled with IFNγ. PIC is a well-known induction agent for TLR3-type responses in contrast to TLR4-type responses that are typical of a response to LPS [51]. 17β-estradiol also significantly reduced PIC + IFNγ-stimulated nitrite production in APOE3 macrophages but did not affect nitrite levels produced in APOE4 macrophages at the concentration studied (Fig 4B).
Figure 4. Peritoneal macrophages from ovariectomized APOE4 mice also demonstrate decreased responsiveness to exogenous treatment with 17β-estradiol.

Cultured peritoneal macrophages from ovariectomized APOE3 or APOE4 mice were pretreated with 1nM 17β-estradiol followed by the addition of 100 ng/ml LPS plus 100 U/ml IFNγ (4A: LPS + IFNγ) or 50μg/ml PIC + 100 U/ml IFNγ (4B; PIC + IFNγ) in the continuing presence of 1 nM 17β-estradiol. Data are presented as the average supernatant nitrite levels (± SEM) and a 2-way ANOVA with the Bonferroni post-hoc test was used to determine significant differences. A significant interaction was observed between APOE genotype and the 17β-estradiol dose for either LPS + IFNγ or PIC + IFNγ-stimulated conditions (p < 0.0001). *** = p < 0.001 compared to LPS + IFNγ alone; # = p < 0.05 for APOE3 compared to APOE4; ## = p < 0.01 for APOE3 compared to APOE4.
We also determined if circulating (endogenous) levels of 17β-estradiol would affect the function of peritoneal macrophages in APOE3 and APOE4 mice in a genotype specific manner. For these experiments we isolated and cultured peritoneal macrophages from aged, female mice that had been exposed to continuous replacement of 17β-estradiol using a time-release pellet implanted after ovariectomy (OVX + Est. Pellet). Macrophage responses to immune stimulation from these mice were then compared to macrophages derived from untreated ovariectomized mice (OVX). As shown in Figure 5A and 5B, immune stimulated supernatant nitrite levels were significantly lower in cultured macrophages from OVX mice treated with time release 17β-estradiol pellets compared to untreated OVX mice from both genotypes. However, macrophages from APOE4 female mice did not respond to the endogenous 17β-estradiol treatment in the same manner as macrophages from APOE3 mice. Treatment with 17β-estradiol pellets reduced the average nitrite levels in APOE3 macrophages by approximately 79% and 54% for LPS + IFNγ and PIC + IFNγ, respectively while only a 40% and 26% drop was observed in macrophages from APOE4 mice. These data suggest that 17β-estradiol replacement is less effective in APOE4 macrophages. Statistical analysis revealed a significant interaction between APOE genotype and the response to treatment (p= 0.014 for LPS + IFNγ treatment and p < 0.0001 for PIC + IFNγ treatment), indicating that the observed changes were dependent on APOE genotype.
Figure 5. Nitrite production in peritoneal macrophages derived from ovariectomized APOE3 and APOE4 mice after replacement of endogenous estrogen.

Peritoneal macrophages from ovariectomized (OVX) mice and OVX mice treated with a slow release estrogen pellet (OVX + Est Pellet) were immune activated with LPS+IFNγ (A) or PIC+IFNγ (B). Average (±SEM) supernatant nitrite concentrations were determined and significant differences were calculated using 2-way ANOVA with the Bonferroni post-hoc test. A significant interaction was observed between APOE genotype and the treatment condition for both types of immune induction (p < 0.0001). # = p < 0.05 for APOE3 compared to APOE4; ## = p < 0.01 for APOE3 compared to APOE4; * = p < 0.05 for OVX + Est pellet compared to OVX alone; *** = p < 0.001 for OVX + Est pellet compared to OVX alone.
A similar effect was observed for TNFα production in immune stimulated macrophages derived from OVX and OVX+Est. Pellet mice (Fig 6A; B). TNFα levels were decreased by 17β-estradiol replacement in both APOE3 and APOE4 macrophages compared to OVX alone. In this case, TNFα production was reduced by 71% and 81% for LPS + IFNγ and PIC + IFNγ immune induction, respectively. Similar treatment in OVX + Est. Pellet APOE4 mice, however, produced only a 59% drop in TNFα for LPS + IFNγ activation and a 54% drop in TNFα for PIC + IFNγ activation. The genotype specific differences for both induction paradigms was judged as significant using 2-way ANOVA followed by Bonferroni post hoc analysis. In addition, we found a significant interaction (p<0.0001) between APOE genotype and the treatment in PIC+IFNγ activated macrophages. These data indicate that the effectiveness of endogenous 17β-estradiol replacement was reduced in APOE4 ovariectomized mice.
Figure 6. TNFα levels in peritoneal macrophages derived from ovariectomized APOE3 and APOE4 mice after replacement of endogenous estrogen.

Average supernatant levels of TNFα (±SEM) were measured in PIC+IFNγ or LPS+IFNγ-treated macrophages cultured from OVX or OVX + Est. Pellet mice. For treatment with LPS+IFNγ (A), a significant effect of genotype (p = 0.024) or of treatment was observed (p < 0.0001). No significant interaction was found. For treatment with PIC+IFNγ (B), a significant interaction between genotype and treatment was observed (p < 0.0001). # = p < 0.05 for APOE3 compared to APOE4; ## = p < 0.01 for APOE3 compared to APOE4; *** = p < 0.001 for OVX + pellet compared to OVX alone.
4. Discussion
17β-estradiol is well known to protect the brain from various types of insults or injury, including ischemic stroke [27;39;65], beta-amyloid-mediated neurotoxicity [35;66] and oxidative damage [5;58]. Part of 17β-estradiol’s neuroprotective mechanism stems from its ability to enhance neurite extension and synaptic remodeling, increase growth and survival factors, and reduce mitochondrial damage and oxidative stress [25;33;63;70]. Since neuroinflammation is an important component of injury or disease in the CNS [20;55], the anti-inflammatory role of 17β-estradiol is also an important factor in CNS pathophysiology. 17β-estradiol -mediated suppression of immune activation in microglia, the CNS macrophage, has now been well established and includes decreased production of cytokines such as TNFα, of proteases and of NO [3;8;13;45;60;61]. Our data confirm this previously published work and show that both microglia and peritoneal macrophages from mice expressing the human APOE3 gene are down-regulated by 17β-estradiol. For peritoneal macrophages, direct treatment with 17β-estradiol or endogenous replacement of 17β-estradiol via a slow release pellet reduced immune activation in normal (APOE3) macrophages. As further shown in our study, the reduction of microglial NO and cytokines occurred within a range of 17β-estradiol concentrations (0.5 to 5 nM) typically associated with normal physiological levels of estradiol activity and was blocked by ICI 182,780, a pan-estrogen receptor blocker [28]. The effect of ICI 182,780, however, appeared to be more complex than a pure ER antagonist. In microglia, co-treatment with 17β-estradiol plus ICI 182,780 not only blocked 17β-estradiol -mediated suppression of NO production but actually increased the level of NO to values above the baseline. The presence of a robust immune response in 17β-estradiol plus ICI 182,780-treated microglia indicate that ICI 182,780 did not directly damage microglial function. Rather, these data imply that ICI 182,780 acts on additional pathways to influence immune-activated NO production. Zhao et al [71] have shown that ICI 182,780 can act as an ER agonist in neurons where it activates ERK and Akt signaling pathways. Since 17β-estradiol has also been shown to have pro-inflammatory effects that include increasing the production of IFNγ and increasing NOS2 expression, [26;30], part of the observed effect of ICI 182,780 in our study may involve activation of pro-inflammatory signaling pathways.
In contrast to other published data [3;60], we did not observe an 17β-estradiol - mediated reduction in NOS2 mRNA despite a 40% reduction in NO production initiated by 17β-estradiol treatment. Part of the difference may be due to the type of immune activation used in our study. We used co-treatment of LPS and IFNγ as our standard immune activation paradigm whereas other studies used LPS only [3;60]. IFNγ works synergistically with immune activators such as LPS or PIC to generate a maximal immune response [1]. Thus, it is possible that detection of statistically significant changes in NOS2 mRNA mediated by 17β-estradiol under conditions of maximal induction are technically more difficult. Although we used quantitative real time PCR to detect NOS2 mRNA changes, we cannot completely rule out assay sensitivity as a factor in our results. However, there is another compelling alternate explanation. Using gene screen technology, Fertuck et al. [22] have recently shown that 17β-estradiol induces arginase I (AGI), an enzyme that utilizes arginine to produce ornithine, the first step in polyamine production. Since polyamines are well known to have critical roles in cell physiology, including enhancement of transcription factor binding and coactivator function, modulation of polyamines is a likely outcome of estrogen’s action on cells. Importantly, the arginase enzyme competes directly with NOS for arginine, their common substrate, and has been clearly shown to reduce NO production as a consequence of this competition [15;31]. Thus, it is tempting to speculate that 17β-estradiol may actually upregulate arginase 1 rather than down-regulating NOS2 in APOE3 microglia as part of its enhanced anti-inflammatory activity compared to APOE4 microglia. We cannot rule out, however, that other post-translational modifications that regulate iNOS function may also occur.
Our data on APOE3 microglia and peritoneal macrophages confirm and extend the role of 17β-estradiol in immune regulation. The uniqueness of our study, however, centers on the comparison of 17β-estradiol -mediated immune regulation in mice expressing only the human APOE3 gene to mice expressing only the human APOE4 gene. Our data clearly demonstrate that the anti-inflammatory activity of 17β-estradiol is reduced in APOE4 mice. The strong, statistically significant interaction between APOE genotype and the response to 17β-estradiol was observed for nitrite and cytokine production by immune activated microglia. The genotype specific effect was not restricted to brain macrophages since APOE4 peritoneal macrophages also demonstrated a significant reduction in 17β-estradiol responsiveness. However, the change in the 17β-estradiol response in peritoneal macrophages from adult ovariectomized APOE4 mice was not as robust as observed for microglia from APOE4 mice. The exact reasons for this difference are unclear but may be related to the age of the animal or to prior exposure to circulating steroid hormones, including 17β-estradiol. Finally, the genotype specific effect was observed with immune activation involving either TLR4 (LPS) or TLR3 (PIC) receptors suggesting that multiple pathways may be altered in the presence of the APOE4 gene.
The effect of 17β-estradiol on macrophage function is dependent on the interaction of 17β-estradiol with ERα and ERβ, members of the nuclear receptor superfamily [40;69]. To detect any differences in ERα or ERβ that may contribute to the observed APOE genotype specific effect, we examined the basal and immune stimulated levels of mRNA and protein expression for ERα and ERβ in APOE3 and APOE4 microglia. Two major APOE genotype specific differences were observed. Basal (untreated) ERα mRNA levels were significantly higher in APOE4 microglia compared to APOE3 microglia. Despite the approximate 80% increase in mRNA, however, basal ERα protein expression was similar in APOE3 and APOE4 microglia. Messenger RNA levels for ERβ were the same in APOE3 and APOE4 microglia but ERβ protein levels were higher in APOE4 microglia. Overall, these data show that protein expression levels do not follow mRNA levels. The failure of ERα protein levels to change in response to increased ERα mRNA levels was also observed in microglia by Vegeto et al [59], where immune stimulation produced similar diverse results between mRNA and protein levels. In our study, despite equivalent expression of ERβ mRNA in LPS+IFNγ-treated APOE3 and APOE4 microglia, the ERβ protein levels remained higher in treated APOE4 cells. Overall, our data suggest that ERβ protein levels in microglia are altered by the presence of an APOE4 gene. The recent discovery of multiple ERβ isoforms [34] may eventually provide insight into the apparent dichotomy between ER mRNA and protein expression levels in microglia.
ERβ has been implicated in the anti-inflammatory responses mediated by 17β-estradiol and is critical to aspects of neuronal survival [3;64]. If this is the case, then the high ERβ protein levels observed in the APOE4 microglia are inconsistent with the depressed responsiveness to estrogen-immune regulation observed in APOE4 macrophages. However, high levels of ERβ may disrupt normal estrogen-mediated signaling. Studies of ERs show that in cells that co-express ERβ and ERα, ERβ acts as dominant regulator of estrogen signaling by inhibiting ERα transcriptional activation [36;38]. The increased level of ERβ in APOE4 microglia, thus, may block ERα signaling and its associated neuroprotection [57;65]. High levels of ERβ may also disrupt estrogen-mediated regulation of apolipoprotein E, itself. Stone [54] and others [49;64] have shown that 17β-estradiol treatment increases the production and release of apoliporotein E from astrocytes and microglia. More recently, Wang et al [64] have shown that ERα activation is responsible for up-regulation of apolipoprotein E in HTT cells and in primary neuronal cultures while ERβ down-regulates apolipoprotein E production. Thus, the high levels of ERβ in APOE4 microglia compared to APOE3 microglia may account, at least in part, for the lower level of apolipoprotein E protein that has been observed in APOE4 mice brain [47]. However, there are many potential points within the immune activation signaling pathway that may be defective in APOE4 microglia compared to APOE3 microglia. The fact that immune activation using either TLR4 or TLR3 are similarly affected by the APOE4 genotype suggests that common downstream pathways may be regulated in a genotype-specific manner.
Regardless of the exact mechanism, the functional changes observed in the response to 17β-estradiol in APOE4 microglia and macrophages are likely to impact inflammation in the brain as well as in other regions of the body. If our data in mice can be translated to the human population, then we would suggest that women with an APOE4 gene may also demonstrate altered 17β-estradiol responsiveness. Studies have now shown a significantly greater risk for cognitive loss and dementia in women compared to men expressing an APOE4 gene [10;23;42]. This isoform specific difference is associated with a significant reduction of hippocampal volume in women with mild cognitive impairment or AD [23]. Decreased hippocampal volume is also observed in healthy 60 year old women expressing an APOE4 gene compared to women who only express the APOE3 gene [11]. Importantly, Yaffe et al. [67] have reported that estrogen therapy in postmenopausal women was associated with less cognitive decline only if they did not express an APOE4 gene. The inability of estrogen replacement therapy to compensate for the lost responsiveness in the presence of the APOE4 gene implies that simply adding estrogens back may not accomplish the goals of reactivating the protective, anti-inflammatory effects of estrogens. Thus, the APOE genotype may be a critical component in assessing the effectiveness of different estrogen replacement therapies in human populations.
Acknowledgments
This work was supported by a grant from NIH to CAC (AG023802-01), MPV (AG019780), and CMB (Minority Research Supplement to P50 AG05128) and an NSF Predoctoral Fellowship (CMB).
Footnotes
Disclosure Statement: MP Vitek is a principal in Cognosci Inc, Research Triangle Park, NC. The work in this manuscript is independent of this interest and no conflict exists. The remaining authors have no disclosures or conflicts of interest
Animal Guidelines: All animal work was carried out under approved animal welfare guidelines mandated by the Duke University Medical Center IACUC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adams D. Regulation of macrophage function by interferon-γ. In: Baron S, Coppenhaver D, Dianzani F, Fleischmann W, Hughes T, Klimpel G, Niesel D, Stanton G, Tyring S, editors. Interferon. Galveston, TX: University of Texas Medical Branch; pp. 341–351. [Google Scholar]
- 2.Attansio R, Gust D, Wilson M, Meeker T, Gordon T. Immunomodulatory effects of estrogen and progesterone replacement in a nonhuman primate model. J Clin Immunol. 2002;22:263–269. doi: 10.1023/a:1019997821064. [DOI] [PubMed] [Google Scholar]
- 3.Baker A, Brautigam V, Watters JJ. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor beta. Endocrinology. 2004;145:5021–5032. doi: 10.1210/en.2004-0619. [DOI] [PubMed] [Google Scholar]
- 4.Barnes L, Wilson R, Bienias J, Schneider J, Evans D, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psych. 2006;62:685–691. doi: 10.1001/archpsyc.62.6.685. [DOI] [PubMed] [Google Scholar]
- 5.Behl C, Widmann M, Trapp T, Holsboer F. 17β-estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Comm. 2006;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- 6.Bellosta S, Nathan BP, Orth M, Dong L-M, Mahley RW, Pitas RE. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J Biol Chem. 1995;270:27063–27071. doi: 10.1074/jbc.270.45.27063. [DOI] [PubMed] [Google Scholar]
- 7.Brown CM, Wright E, Colton C, Sullivan P, Laskowitz D, Vitek M. Apolipoprotein E isoform mediated regulation of nitric oxide release. Free Radic Biol Med. 2002;32:1071–1075. doi: 10.1016/s0891-5849(02)00803-1. [DOI] [PubMed] [Google Scholar]
- 8.Bruce-Keller A, Keeling J, Keller J, Huang F, Calmondola S, Mattson M. Antiinflammatory effects of estrogen on microglia activation. Endocrinology. 2000;141:3646–3656. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- 9.Chapman J, Vinokurov S, Achiron A, Karussis D, Mitosek-Sewczyk K, Birnbaum M, Michaelson D, Korczyn A. APOE genotype is a major predictor of long term progression of disability in MS. Neurology. 2001;56:312–316. doi: 10.1212/wnl.56.3.312. [DOI] [PubMed] [Google Scholar]
- 10.Chen T, Wang L, Farrar W. Interleukin 6 activates androgen receptor mediated gene expression through a signal transducer and activator of transcription 3 dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000;60:2132–2135. [PubMed] [Google Scholar]
- 11.Cohen R, Small C, Lalonde F, Friz B, Sunderland T. Effect of apolipoprotein E genotype on hippocampal volume loss in aging healthy women. Neurol. 2001;57:2223–2228. doi: 10.1212/wnl.57.12.2223. [DOI] [PubMed] [Google Scholar]
- 12.Colton C, Brown C, Czapiga M, Vitek M. Apolipoprotein-E allele specific regulation of nitric oxide production. New York Acad Sci. 2002;962:212–225. doi: 10.1111/j.1749-6632.2002.tb04070.x. [DOI] [PubMed] [Google Scholar]
- 13.Colton C, Brown C, Vitek M. Sex steroids, APOE genotype and the innate immune system. Neurobiol Aging. 2005;26:363–372. doi: 10.1016/j.neurobiolaging.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Colton C, Needham L, Brown C, Cook D, Rasheed K, Burke J, Strittmatter W, Schmeckel D, Vitek MP. APOE genotype specific differences in human and mouse macrophage nitric oxide production. J Neuroimmunol. 2004;147:62–67. doi: 10.1016/j.jneuroim.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 15.Colton CA, Xu Q, Burke J, Bae S, Wakefield J, Nair A, Strittmatter W, Vitek MP. Disrupted spermine homeostasis: a novel mechanism in polyglutamine-mediated aggregation and cell death. J Neurosci. 2004;24:7118–7127. doi: 10.1523/JNEUROSCI.1233-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corder E, Robertson K, Lannfelt L, Bogdanovic N, Eggertsen G, Wilkins J, Hall C. HIV-infected subjects with the E4 allele for APOE have excess dementia and peripheral neuropathy. Nat Med. 1998;4:1182–1184. doi: 10.1038/2677. [DOI] [PubMed] [Google Scholar]
- 17.Czapiga M, Colton CA. Microglial function in human APOE3 and APOE4 transgenic mice. Altered arginine transport. J Neuroimmunol. 2002;4673:1–8. doi: 10.1016/s0165-5728(02)00394-6. [DOI] [PubMed] [Google Scholar]
- 18.DAgostino P, Milano S, Barbera C, DiBella G, LaRosa M, Ferlazzo V, Farruggio R, Miceli D, Miele M, Castagnetta L, Cillari E. Sex hormones modulate inflammatory mediators produced by macrophages. Ann N Y Acad Sci. 1999;8876:426–429. doi: 10.1111/j.1749-6632.1999.tb07667.x. [DOI] [PubMed] [Google Scholar]
- 19.Dimayauga F, Reed J, Carnero G, Wang C, Dimayuga E, Dimayuga V, Perger A, Wilson M, Keller J, Bruce- Keller A. Estrogen and brain inflammation: Effects on microglial expression of MHC, costimulatory molecules and cytokines. J Neuroimmunol. 2005;161:123–136. doi: 10.1016/j.jneuroim.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 20.Eikelenboom P, Rosemuller JM, Van Muiswinkel F. Inflammation and Alzheimer’s disease: relationships between pathogenic mechanisms and clinical expression. Exp Neurol. 1998;154:89–98. doi: 10.1006/exnr.1998.6920. [DOI] [PubMed] [Google Scholar]
- 21.Felicio L, Nelson J, Finch C. Longitudinal studies of estrous cyclicity in aging C57Bl/6J mice: II. Cessation of cyclicity and the duration of persistent vaginal cornification. Biol Reprod. 1984;31:446–453. doi: 10.1095/biolreprod31.3.446. [DOI] [PubMed] [Google Scholar]
- 22.Fertuck K, Eckel J, Gennings C, Zacharewski T. Identification of temporal patterns of gene expression in the uteri of immature, ovariectomized mice following exposure to ethynylestradiol. Physiol Genomics. 2003;15:127–141. doi: 10.1152/physiolgenomics.00058.2003. [DOI] [PubMed] [Google Scholar]
- 23.Fleisher A, Grundman M, Jack C, Petersen R, Taylor C, Kim H, Schiller D, Bagwell V, Sencakova D, Weiner M, DeCarli C, DeKosky S, vanDyck C, Thal L. Sex, Apolipoprotein E e4 status and hippocampal volume in mild cognitive impairment. Arch Neurol. 2005;62:953–957. doi: 10.1001/archneur.62.6.953. [DOI] [PubMed] [Google Scholar]
- 24.Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, Babaey S, Yehuda B, Groswasser Z. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurol. 1999;52:248. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 25.Gandy S. Estrogen and neurodegeneration. Neurochem Res. 2004;28:1003–1008. doi: 10.1023/a:1023246921127. [DOI] [PubMed] [Google Scholar]
- 26.Han G, Magee T, Khorram O. Regulation of nitric oxide synthase isoforms by estrogen in the human endometrium. Fertil Steril. 2005;84(Suppl):1220–1227. doi: 10.1016/j.fertnstert.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 27.Horsburgh K, McCulloch J, Nilsen M, Roses A, Nicoll J. Increased neuronal damage and apoE immunoreactivity in human apolipoprotein E - E4 isoform-specific transgenic mice after global ischemia. Eur J Neurosci. 2000;12:4309–4317. [PubMed] [Google Scholar]
- 28.Howell A, Osborne C, Morris C, Wakeling A. ICI 182,780 (Faslodex): development of a novel, pure antiestrogen. Cancer. 2000;89:817–825. doi: 10.1002/1097-0142(20000815)89:4<817::aid-cncr14>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Jordan B, Relkin N, Ravdin L, Jacobs A, Bennett A, Gandy S. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. J Amer Med Soc. 1997;278:2143–2145. [PubMed] [Google Scholar]
- 30.Karpuzoglu E, Ahmed SA. Estrogen regulation of nitric oxide and inducible nitric oxide synthase (iNOS) in immune cells: implications for immunity, autoimmune diseases, and apoptosis. Nitric Oxide. 2006;15:177–186. doi: 10.1016/j.niox.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Kepka-Lenhart D, Mistry S, Wu G, Morris S. Arginase I: a limiting factor for nitric oxide and polyamine synthesis by activated macrophages. Am J Physiol Reg Inte Comp Physiol. 2000;279:R2237–R2242. doi: 10.1152/ajpregu.2000.279.6.R2237. [DOI] [PubMed] [Google Scholar]
- 32.Knoferl M, Jarrar D, Angele M, Ayala A, Schwacha M, Bland K, Chaudry I. 17 beta-Estradiol normalizes immune responses in ovariectomized females after trauma-hemorrhage. Am J Physiol Cell Physiol. 2001;281:C1131–C1138. doi: 10.1152/ajpcell.2001.281.4.C1131. [DOI] [PubMed] [Google Scholar]
- 33.Lee S, McEwen B. Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications. Annu Rev Pharmacol Toxicol. 2001;41:569–591. doi: 10.1146/annurev.pharmtox.41.1.569. [DOI] [PubMed] [Google Scholar]
- 34.Leung Y, Mak P, Hassan S, Mo SM. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proc Natl Acad Sci U S A. 2006;103:13162–13167. doi: 10.1073/pnas.0605676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Shen Y, Yang L-B, Lue L, Finch C, Rogers J. Estrogen enhances uptake of amyloid b protein by microglia derived from the human cortex. J Neurochem. 2000;75:1447–1454. doi: 10.1046/j.1471-4159.2000.0751447.x. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Huang J, Yi P, Bambara R, Hilf R, Muyan M. Single chain estrogen receptors (ERs) reveal that the ERα/β heterodimer emulates functions of the ERα dimer in genomic estrogen signaling pathways. Mol Cell Biol. 2004;24:7681–7694. doi: 10.1128/MCB.24.17.7681-7694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak K, Schmittgen T. Analysis of relative gene expression data using real time quantitative PCR and the 2(−ΔΔC (T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Matthews J, Gustafsson J. Estrogen signaling: A subtle balance between ERa and ERb. Molecular Inventions. 2003;3:281–292. doi: 10.1124/mi.3.5.281. [DOI] [PubMed] [Google Scholar]
- 39.McCullough L, Hurn PD. Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrinol Metab. 2003;14:228–235. doi: 10.1016/s1043-2760(03)00076-6. [DOI] [PubMed] [Google Scholar]
- 40.McDonnell D. The molecular determinants of estrogen receptor pharmacology. Maturitas. 2004;48(sup 1):S7–S12. doi: 10.1016/j.maturitas.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Moncada S, Higgs E. Endogenous nitric oxide: Physiology, pathology and clinical relevance. Eur J Clin Invest. 1991;21:361–374. doi: 10.1111/j.1365-2362.1991.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 42.Mortensen E, Hogh P. A gender difference in the association between APOE genotype and age related cognitive decline. Neurol. 2001;57:89–95. doi: 10.1212/wnl.57.1.89. [DOI] [PubMed] [Google Scholar]
- 43.Nathan B, Barsukova A, Shen F, McAsey M, Struble R. Estrogen facilitates neurite extension via apolipoprotein E in cultured adult mouse cortical neurons. Endocrinology. 2004;145:3065–3073. doi: 10.1210/en.2003-1707. [DOI] [PubMed] [Google Scholar]
- 44.Ophir G, Amariglio N, Jacob-Hirsch J, Elkon R, Rechavi G, Michaelson DM. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-kappaB signaling cascade. Neurobiol Dis. 2005;20(3):709–18. doi: 10.1016/j.nbd.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 45.Pfeilschifter J, Koditz R, Pfohl M, Schatz H. Changes in proinflammatory cytokine activity after menopause. Endo Rev. 2002;23:90–119. doi: 10.1210/edrv.23.1.0456. [DOI] [PubMed] [Google Scholar]
- 46.Porter V, Greendale G, Schocken M, Zhu X, Effros R. Immune effects of hormone replacement therapy in post-menopausal women. Exp Gerontol. 2001;36:311–326. doi: 10.1016/s0531-5565(00)00195-9. [DOI] [PubMed] [Google Scholar]
- 47.Ramaswamy G, Xu Q, Huang Y, Weisgraber K. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25:10658–10663. doi: 10.1523/JNEUROSCI.1922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roses AD. Apolipoprotein E affects the rate of Alzheimer Disease expression. β-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease. J Neuropath Exp Neurol. 1994;53:429–437. doi: 10.1097/00005072-199409000-00002. [DOI] [PubMed] [Google Scholar]
- 49.Rozovsky I, Hoving S, Anderson C, O’Callaghan J, Finch C. Equine estrogens induce apolipoprotein E and glial fibrillary acidic protein in mixed glial cultures. Neurosci Lett. 2002;323:191–194. doi: 10.1016/s0304-3940(02)00146-5. [DOI] [PubMed] [Google Scholar]
- 50.Ruitenberg A, Ott A, vanSweiten J, Hofman A, Breteler M. Incidence of dementia: does gender make a difference? Neurobiol Aging. 2001;22:575–580. doi: 10.1016/s0197-4580(01)00231-7. [DOI] [PubMed] [Google Scholar]
- 51.Sabroe I, Read R, Whyte M, Dockrell D, Vogel S, Dower S. Toll like receptors in health and disease: complex questions remain. J Immunol. 2003;171:1630–1635. doi: 10.4049/jimmunol.171.4.1630. [DOI] [PubMed] [Google Scholar]
- 52.Savita R. Sex steroid hormones modulate the activation of murine peritoneal macrophages: receptor mediated modulation. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 1998;119:199–204. doi: 10.1016/s0742-8413(97)00207-7. [DOI] [PubMed] [Google Scholar]
- 53.Schmechel D, Saunders A, Strittmatter W, Crain B, Hulette C, Joo S, Pericak-Vance M, Goldgarber D, Roses AD. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1996;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stone D, Rozovsky I, Morgan T, Anderson C, Hajian H, Finch C. Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp Neurol. 1997;143:313–318. doi: 10.1006/exnr.1996.6360. [DOI] [PubMed] [Google Scholar]
- 55.Streit W. Microglia as neuroprotective immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- 56.Sullivan P, Mezour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous athersclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe2. J Clin Invest. 1998;102:130–135. doi: 10.1172/JCI2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki S, Brown CM, Wise PM. Mechanisms of neuroprotection by estrogen. Endocrine. 2006;29:209–215. doi: 10.1385/ENDO:29:2:209. [DOI] [PubMed] [Google Scholar]
- 58.Tang M, Subbiah MTR. Estrogens protect against hydrogen peroxide and arachidonic acid induced DNA damage. Biochim Biophys Acta. 1996;1299:155–159. doi: 10.1016/0005-2760(95)00227-8. [DOI] [PubMed] [Google Scholar]
- 59.Vegeto E, Belcredito S, Etterei S, Ghisletti S, Brusadelli A, Meda C, Krust A, Dupont S, Ciana P, Chambion P, Maggi A. Estrogen receptor-α mediates the brain antiinflammatory activity of estradiol. Proc Nat Acad Sci USA. 2003;100:9614–9619. doi: 10.1073/pnas.1531957100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciani P, Maggi A. Estrogen prevents the lipopolysaccharide induced inflammatory response in microglia. J Neurosci. 2001;21:1809–1818. doi: 10.1523/JNEUROSCI.21-06-01809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vegeto E, Ghisletti S, Meda C, Etteri S, Belcredito S, Maggi A. Regulation of the lipopolysaccharide signal transduction pathway by 17β-estradiol in macrophage cells. J Steroid Biochem Mol Biol. 2004;91:59–66. doi: 10.1016/j.jsbmb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 62.Vitek M, Snell J, Chernyshev O, Colton C. Modulation of nitric oxide production in human macrophages by apolipoprotein E and amyloid beta peptide. Biophys Biochem Res Comm. 1997;240:391–394. doi: 10.1006/bbrc.1997.7408. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Green P, Simpkins J. Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitropropionic acid in SK-N-SH human neuroblastoma cells. J Neurochem. 2001;77:804–811. doi: 10.1046/j.1471-4159.2001.00271.x. [DOI] [PubMed] [Google Scholar]
- 64.Wang J, Irwin R, Brinton RD. Activation of estrogen receptor alpha increases and estrogen receptor beta decreases apolipoprotein E expression in hippocampus in vitro and in vivo. Proc Natl Acad Sci U S A. 2006;103:16983–88. doi: 10.1073/pnas.0608128103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wise PM, Dubal D, Rau S, Brown C, Suzuki S. Are estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the women’s health initiative. Endo Rev. 2005;26:308–312. doi: 10.1210/er.2004-0014. [DOI] [PubMed] [Google Scholar]
- 66.Xu H, Gouras G, Greenfield J, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic J, Seeger M, Relkin N, Liao F, Checler F, Buxbaum J, Chait B, Thinakaran G, Sisodia S, Wang R, Greengard P, Gandy S. Estrogen reduces neuronal generation of Alzheimer beta amyloid peptides. Nature Med. 1998;4:447–451. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]
- 67.Yaffe K, Haan M, Byers A, Tangen C, Kuller L. Estrogen use, APOE and cognitive decline: evidence of gene-environment interaction. 2000;54:1949–1954. doi: 10.1212/wnl.54.10.1949. [DOI] [PubMed] [Google Scholar]
- 68.You H, Kim J, Jeong HG. 17β-estradiol increases inducible nitric oxide synthase expression in macrophages. Biochem Biophys Res Commun. 2003;303:1129–1134. doi: 10.1016/s0006-291x(03)00477-7. [DOI] [PubMed] [Google Scholar]
- 69.Zhang D, Trudeau V. Integration of membrane and nuclear estrogen receptor signaling. Comp Biochem Physiol A Mol Integr Physiol. 2006;144:306–315. doi: 10.1016/j.cbpa.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Tounekti O, Akerman B, Goodyer C, LeBlanc A. 17β-estradiol induces an inhibitor of active caspases. J Neurosci. 2001;21:176. doi: 10.1523/JNEUROSCI.21-20-j0007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao L, O’Neill K, Brinton R. Estrogenic agonist activity of ICI 182,780 (Faslodex) in hippocampal neurons: implications for basic science understanding of estrogen signaling and development of estrogen modulators with a dual therapeutic profile. J Pharmacol Exp Ther. 2006;319:1124–1132. doi: 10.1124/jpet.106.109504. [DOI] [PubMed] [Google Scholar]