

Abstract

To modulate and report the pharmacokinetics of peptide-based pharmaceuticals, a novel geminally perfluro-tert-butylated β-amino acid (βFa) and its Fmoc- and Boc- protected forms were designed and synthesized. βFa was incorporated into a model tripeptide via standard solid-phase chemistry. Both the amino acid (free and protected) and the tripeptide show a sharp singlet 19F NMR signal. Reversed-phase chromatography and 1-octanol/water partition measurements demonstrate that βFa is extremely hydrophobic.
We are interested in the design and synthesis of fluorinated amino acids as modulators and reporters of peptide pharmacokinetics. The potential benefits brought by fluorinated amino acids include prolonged in vivo half-life,1 enhanced membrane permeability2 and non-invasive detection via 19F magnetic resonance spectroscopy.3
A generic highly fluorinated amino acid that can dramatically increase the lipophilicity of a peptide and give a singlet sharp 19F NMR signal is highly desirable for pharmacokinetic modulation and reporting purposes. To this end, we designed a geminally perfluoro-tert-butylated β-amino acid (βFa, 1), as shown in Figure 1. We also need βFa in its Fmoc- and Boc-protected forms (2 and 3, respectively) for solid-phase peptide synthesis. The fluorine atoms are introduced into the amino acid through two symmetrically positioned perfluoro-tert-butyl groups. Such spherically symmetric arrangement of the 18 fluorine atoms in βFa ensures that they have identical chemical environment and avoids 19F-19F or 19F-1H coupling. As a result, we anticipate this amino acid to give a single 19F NMR signal. βFa is achiral and needs no side chain protection, significantly simplifying the synthesis of both the free amino acid and its protected forms. Considering that electron-withdrawing capacity of perfluoro-tert-butyl groups can potentially weaken the basicity of the amino group and hence its reactivity during solid-phase peptide synthesis, a β-amino acid is adopted to ensure sufficient separation between the amino group and the two perfluoro-tert-butyl groups. This also allows better steric accommodation of the bulky perfluoro-tert-butyl groups.
Figure 1.

Structures of target molecules.
To demonstrate the feasibility of βFa to be incorporated into peptides via solid-phase synthesis, we designed the following tripeptide, formyl-Gly-βFa-Gly-amide 4. This peptide serves dual purposes: first, to demonstrate that protected βFa can indeed be incorperated into peptides via solid-phase peptide synthesis; second, to demonstrate that βFa can indeed increase the hydrophobicity of a peptide. βFa is positioned in the middle of the tripeptide to demonstrate that βFa can be placed in any position of a peptide, not just the N-, C-termini. The N- and C-termini of the tripeptide are formylated and amidated, respectively. These modifications abolish terminal charges of the tripeptide so that they will not interfere with hydrophobicity and 1-octanol/water partition measurements. Note that the disentanglement of hydrophobic interactions from electrostatic interections in peptides/proteins is far from trivial.4 Two glycines, which have no side chains, flank the central βFa. The purpose is to abolish nearest neighbor interactions that can interfere with hydrophobicity measurements.5 Hence, this tripeptide provides a “clean” model system for hydrophobicity and 1-octanol/water partition measurements. A reference tripeptide 5 contains tryptophan (Trp) in place of βFa. Trp is the most hydrophobic one among the 20 natural amino acids5 and serves as an excellent reference point for hydorphobilicty measurements. Structures of the two tripeptides (4 and 5) are shown in Figure 2.
Figure 2.

Strucutres of two model tripeptides: formyl-Gly-βFa-Gly-amide (4) and formyl- Gly-L-Trp-Gly-amide (5).
The commercially available pentaerythritol (6) provides an ideal starting material for the synthesis of compound 1. Our synthesis commenced with the selective protection of pentaerythritol (Scheme 1). Protecting two of the four hydroxyl groups in pentaerythritol 6 with p-methoxylbenzaldehyde gave the diol 7 with good yield.6 As the acidity of the hydroxyl group in perfluoro-tert-butanol is enhanced by the three electron-withdrawing -CF3 groups, perfluoro-tert-butanol is a good substrate for the Mitsunobu reaction to form perfluoro-tert-butyl ethers.7 Thus, the Mitsunobu reaction was employed to introduce two perfluoro-tert-butyl groups into compound 7 in just one step to give fluorinated ether 8 with 98% yield after the reaction mixture was stirred at 45 °C for 30 h. Such a high yield was achieved by carrying out the reaction in a sealed vessel and in the presence of 4Å molecular sieve. Neither FC-72 (C6F14) nor HFE-7100 (a mixture of n-C4F9OCH3 and i-C4F9OCH3) could extract compound 8 from the acetonitrile/water (95%/5%) solution of the reaction mixture. Instead, standard flash chromatography was used to purify ether 8 with a 98% yield. The p-methoxybenzylidene acetal protecting group was cleaved off compound 8 by powdered aluminum chloride in the presence of anisole to give 1,3-diol 9 with quantitative yield. Treatment of 1,3-diol 9 with thionyl chloride gave a cyclic sulfite intermediate which was then oxidized by in situ generated ruthenium tetraoxide to give the cyclic sulfate 10 with an 84% yield in two steps. Ring opening of the cyclic sulfate 10 with sodium azide, followed by hydrolysis of the resulting sulfonic acid, gave the azido compound 11 with excellent yield which was then subjected to Jones oxidation to give the carboxylic acid 12 with an 85% yield. Hydrogenation of the azido group in compound 12 yielded the fluorinated amino acid 1 with excellent yield. This completes the synthesis of the free amino acid, as depicted in Scheme 1.
Scheme 1.

Synthesis of 1
To obtain the Fmoc-protected form of the amino acid, the amino group of compound 1 reacted with 9-fluorenylmethoxycarbonyl chloride (FmocCl) to give compound 2 with a 96% yield on a 13.3-gram scale, as depicted in Scheme 2.
Scheme 2.
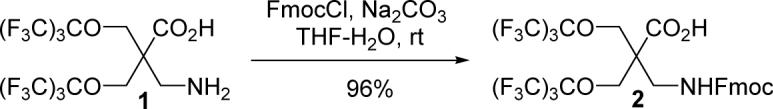
Synthesis of 2
To obtain the Boc-protected form of the amino acid, the azido group of compound 12 was reduced to the amino group which then reacted with di-tert-butyl dicarbonate (Boc2O) to give compound 3 with a 96% yield on a 1.3-gram scale, as depicted in Scheme 3.
Scheme 3.

Synthesis of 3
The two tripeptides (4 and 5) were synthesized using standard Fmoc chemistry on a solid support.8 Both the carboxyl group and the amino group of βFa showed good reactivity during solid-phase reaction. Hence, βFa can be incorporated into any position of a target peptide through its carboxyl group, amino group or both. Tripeptide 4 was purified by normal-phase HPLC while tripeptide 5 was purified by reversed-phase HPLC due to their different solubilities in water and methanol. The molecular mass and the purity of each tripeptide were verified by mass spectrometry and analytical HPLC, respectively.
The 19F NMR spectra of the free amino acid 1 and the tripeptide 4 are shown in Figure 3. As designed, all 18 fluorine atoms give a singlet sharp 19F signal (linewidth ∼0.01 ppm) in both the free amino acid 1 (left) and the tripeptide 4 (right). This is also the case for the two protected forms (compounds 2 and 3) of this amino acid (see Supporting Information).
Figure 3.
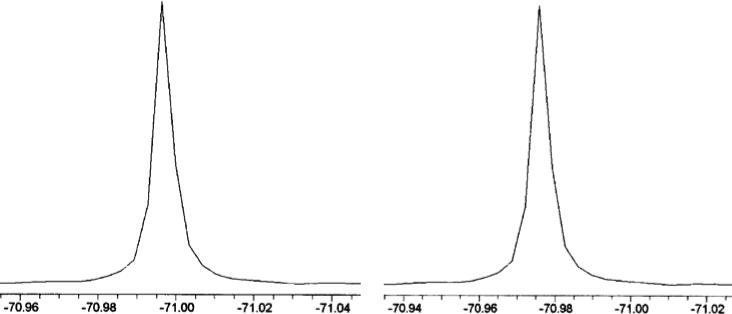
Chemical shift (ppm) of 19F NMR in free amino acid 1 (left) and tripeptide 4 (right) (376 MHz, CD3OD, C6F6 as internal standard).
The hydrophobicity of βFa was evaluated in the context of tripeptide 4, with tripeptide 5 serving as a reference point. To this end, we used the reversed-phase chromatography method developed by Hodges and coworkers, who determined the relative hydrophobicity order of 23 amino acids.5 Figure 4 shows the chromatogram of the co-injection of the two tripeptides. Clearly, tripeptide 4 is much more retentive than tripeptide 5 in reversed-phase chromatography, proving that βFa is much more hydrophobic than Trp, the most hydrophobic natural amino acid.
Figure 4.

Retention behavior of tripeptides 4 and 5 in reversed-phase HPLC.
The 1-octanol/water partition coefficients (Poct) of the two tripeptides were evaluated. Poct is a standard physicochemical parameter in assessing membrane permeability of peptides and other drugs.9 For 5 (formyl-Gly-Trp-Gly-amide), Poct = 1/9.5, as determined by analytical HPLC at 280 nm (Trp signal) of the 1-octanol and aqueous phases. Therefore, in spite of the hydrophobicity of Trp, 5 still prefers water to 1-octanol. As for 4 (formyl-Gly-βFa-Gly-amide), afrer equilibration, its existence in water can be detected by neither analytical HPLC nor by 19F NMR while its existence in 1-octanol can be readily detected by both analytical HPLC and 19F NMR (see Supporting Information). Based on this result, we conclude that, for 4, Poct >> 10. Hence, Poct(4) is over 100 times larger than Poct(5).
Results of the hydrophobicity measurement and the 1-octanol/water partition coffecient measurement are entirely consistent with each other, both showing that, in the context of a peptide, βFa is very hydrophobic and strongly prefers organic over aqueous phase. All these bode well for its potential use as a membrane permeability enhancer for peptide-based pharmaceuticals.
In conclusion, a novel highly fluorinated β-amino acid (βFa) has been designed and synthesized. The Fmoc- and Boc-protected forms of this amino acid have also been synthesized with high yield. The syntheses were highly efficient with an overall yield ca. 65% at multi-gram synthesis scale. βFa can be incorporated into peptides using standard solid-phase synthesis. As designed, all 18 fluorine atoms give a singlet sharp 19F NMR signal in both the free amino acid and a tripeptide context, auguring well for 19F-MRS monitoring. βFa is much more hydrophobic than Trp, the most hydrophobic natural amino acid, and results in much stronger preference of organic over aqueous phases.
Experimental Section
2-(4-Methoxy-phenyl)-5,5-bis-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxymethyl)-[1,3]dioxane 8
To a stirred mixture of compound 7 (24.4 g, 96.1 mmol), tripenylphosphine (75.5 g, 288.5 mmol) and 4Å molecular sieve (20.0 g) in tetrahydrofuran (500 mL) at 0 °C was added dropwise diethylazodicarboxylate (50.2 g, 288.2 mmol). After the addition, the reaction mixture was allowed to warm to room temperature and stirred for an additional 20 min. Then perfluoro-tert-butanol (68.0 g, 288.0 mmol) was added in one portion and the resulting mixture was stirred at 45 °C for 30 h in a sealed vessel. The mixture was evaporated to dryness and the residue was dissolved in ethyl ether (600 mL). After filtered through a pad of Cellite, the filtrate was washed with brine (300 mL), dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (n-hexane/ethyl acetate = 20/1) to give 8 as a solid (65.1 g, 98%). mp. 89−90 °C; 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 1H), 3.80 (s, 4H), 3.89 (s, 2H), 4.13 (s, 1H), 4.16 (s, 1H), 4.49 (s, 2H), 5.39 (s, 1H), 6.88−6.92 (m, 2H), 7.33−7.37 (m, 2H); 19F NMR (376 MHz, CDCl3) δ −73.42 (s); 13C NMR (100.7 MHz, CDCl3) δ 39.2, 55.3, 66.2, 67.7, 68.7, 102.4, 113.8, 120.1 (q, J = 292.6 Hz), 120.3 (q, J = 292.6 Hz), 127.3, 129.9, 160.3; MS (CI) m/z 691 (M++1, 100), 690 (M+, 17), 583 (22); HRMS (CI) Calcd for C21H17F18O5: 691.0787, Found: 691.0792.
2-Azidomethyl-3-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxy)-2-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxymethyl)-propan-1-ol 11
Sodium azide (4.4 g, 66.9 mmol) was added to a stirred solution of compound 10 (21.2 g, 33.4 mmol) in dimethylformaldehyde (120 mL). The reaction mixture was stirred at 60 °C for 4 h. The solvent was removed under vacuo and the residue was dissolved in tetrahydrofuran (120 mL). Sulfuric acid (0.87 mL) and water (0.32 mL) was added to the stirred tetrahydrofuran solution and the resulting mixture was stirred at room temperature for an additional 1 h. After removing the solvent, the residue was redissolved in dichloromethane (200 mL) and extracted with perfluorohexane (100 mL 4 times). The combined extraction was washed with dichloromethane (10 mL) and concentrated under vacuo to give the pure azide 11 as a clear oil (19.3 g, 97%). 1H NMR (400 MHz, CDCl3) δ 3.47 (s, 2H), 3.63 (s, 2H), 4.02 (s, 4H); 19F NMR (376 MHz, CDCl3) δ −73.21 (s); 13C NMR (100.7 MHz, CDCl3) δ 45.8, 49.8, 60.2, 66.8, 79.6 (m), 120.2(q, J = 293.3 Hz); MS (CI) m/z 598 (M++1, 72), 570 (100); HRMS (CI) Calcd for C13H10F18N3O3: 598.0435, Found: 598.0418.
2-Aminomethyl-3-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxy)-2-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxymethyl)-propionic acid 1
A mixture of Palladium on carbon (2.5 g) in methanol (200 mL) was degassed for 2 min and stirred under a hydrogen atmosphere for 30 min. A solution of acid 12 (10.7 g, 17.5 mmol) in methanol (10 mL) was then added and the resulting mixture was stirred at room temperature under a hydrogen atmosphere for additional 30 h. After solvent removal, the resulting residue was purified by flash column chromatography on silica gel (methanol/dichloromethane = 10/1) to give the amino acid 1 as a solid (10.1 g, 98%). mp. 182−184 °C; 1H NMR (400 MHz, CD3OD) δ 2.99 (s, 2H), 4.22 (d, J = 9.2 Hz, 2H), 4.49 (d, J = 8.4 Hz, 2H); 19F NMR (376 MHz, CD3OD) δ −71.00 (s); 13C NMR (100.7 MHz, CD3OD) δ 41.7, 52.1, 69.9, 80.9 (m), 121.7 (q, J = 292.6 Hz), 175.0; MS (CI) m/z 586 (M++1, 100); HRMS (CI) Calcd for C13H10F18NO4: 586.0322, Found: 586.0285.
2-[(9H-Fluoren-9-ylmethoxycarbonylamino)-methyl]-3-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxy)-2-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxymethyl)-propionic acid 2
To a stirred solution of amino acid 1 (10.1 g, 17.2 mmol) in tetrahydrofuran (100 mL) and water (100 mL) was added sodium carbonate (4.6 g, 42.9 mmol). After all the sodium carbonate was dissolved, the resulting mixture was cooled to 0 °C and 9-fluorenylmethyl chloroformate (6.7 g, 25.9 mol) was added in three portions. The resulted reaction mixture was stirred at room temperature overnight. The solvent was removed under vacuo and the residue was purified by flash column chromatography on silica gel (n-hexane/ethyl acetate = 5/1) to give the acid 2 as a white solid (13.3 g, 96%). mp. 104−105 °C; 1H NMR (400 MHz, CD3OD) δ 3.45 (s, 2H), 4.16 (m), 4.27 (m, 4H), 4.46 (d, J = 8.8 Hz, 2H) 7.26 (t, J = 7.2 Hz, 2H), 7.35 (t, J = 7.2 Hz, 2H), 7.60 (d, J = 7.6 Hz, 2H), 7.74 (d, J = 7.2 Hz, 2H); 19F NMR (376 MHz, CD3OD) δ −71.00 (s); 13C NMR (100.7 MHz, CD3OD) δ 36.9, 42.8, 53.7, 68.2, 69.0, 80.9 (m), 120.9, 121.7 (q, J = 292.6 Hz), 126.2, 128.1, 128.8, 142.6, 145.2, 158.8, 174.4; MS (CI) m/z 808 (M++1, 100); HRMS (CI) Calcd for C28H20F18NO6: 808.1003, Found: 808.1010.
3-tert-Butoxycarbonylamino-2,2-bis-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethoxymethyl)-propionic acid 3
A mixture of Palladium on carbon (200 mg) in methanol (20 mL) was degassed for 2 min and stirred under hydrogen atmosphere for 30 min. A solution of acid 12 (1.2 g, 2.0 mmol) and di-tert-butyl dicarbonate (872 mg, 4.0 mmol) in methanol (5 mL) was then added and the resulting mixture was stirred at room temperature under an atmosphere of hydrogen gas for 30 h. After solvent removal, the resulting residue was purified by flash column chromatography on silica gel (n-hexane/ethyl acetate = 5/1) to give the amino acid 3 as a solid (1.32 g, 96%). mp. 128−130 °C; 1H NMR (400 MHz, CD3OD) δ 1.36 (s, 9H), 3.31 (s, 2H), 4.17 (d, J = 8.0 Hz, 2H), 4.39 (d, J = 8.0 Hz, 2H); 19F NMR (376 MHz, CD3OD) δ −71.01 (s); 13C NMR (100.7 MHz, CD3OD) δ 28.8, 42.8, 54.1, 69.6, 80.7, 81.1 (m), 121.9 (q, J = 293.3 Hz), 158.1, 177.2; MS (CI) m/z 686 (M++1, 10), 644 (100); HRMS (CI) Calcd for C18H18F18NO6: 686.0847, Found: 686.0815.
Supplementary Material
Acknowledgement
This work was supported by grants from the NIH (EB002880 and EB004416) and the Sidney Kimmel Foundation for Cancer Research. Y. B.Yu is a Kimmel scholar.
Footnotes
Supporting Information Available: Experimental procedures and product characterization for compounds 7, 9, 10, 12, synthesis and partition procedures for 4, 5, copies of 1H, 19F and 13C NMR spectra for compounds 8, 9, 10, 11, 12, 1, 2, 3, copies of HRMS spectra for compounds 1, 2, 3, copies of 1H NMR and HPLC spectra for compounds 4, 5, copy of 19F NMR spectra for compound 4, and copies of HPLC spectra of partition test for compounds 4, 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hsieh K–H, Needleman P, Marshall GR. J. Med. Chem. 1987;30:1097–1100. doi: 10.1021/jm00389a021. [DOI] [PubMed] [Google Scholar]
- 2.a Ortial S, Durand G, Poeggeler B, Polidori A, Pappolla MA, Böker J, Hardeland R, Pucci B. J. Med. Chem. 2006;49:2812–2820. doi: 10.1021/jm060027e. [DOI] [PubMed] [Google Scholar]; b Perino S, Contino-Pépin C, Jasseron S, Rapp M, Maurizis J-C, Pucci B. Bioorg. Med. Chem. Lett. 2006;16:1111–1114. doi: 10.1016/j.bmcl.2005.11.107. [DOI] [PubMed] [Google Scholar]; c Park BK, Kitteringham NR, O'Neill PM. Ann. Rev. Pharmacol. Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]; d Gerebtzoff G, Li-Blatter X, Fischer H, Frentzel A, Seelig A. ChemBioChem. 2004;5:676–684. doi: 10.1002/cbic.200400017. [DOI] [PubMed] [Google Scholar]
- 3.Wolf W, Presant CA, Waluch V. Adv. Drug Del. Rev. 2000;41:55–74. doi: 10.1016/s0169-409x(99)00056-3. [DOI] [PubMed] [Google Scholar]
- 4.a Yu Y, Monera OD, Hodges RS, Privalov PL. J. Mol. Biol. 1996;255:367–372. doi: 10.1006/jmbi.1996.0030. [DOI] [PubMed] [Google Scholar]; b Yu Y, Monera OD, Hodges RS, Privalov PL. Biophys. Chem. 1996;59:299–314. doi: 10.1016/0301-4622(95)00131-x. [DOI] [PubMed] [Google Scholar]
- 5.Kovacs JM, Mant CT, Hodges RS. Biopolymers. 2006;84:283–297. doi: 10.1002/bip.20417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguilera B, Romero-Ramirez L, Abad-Rodriguez J, Corrales G, Nieto-Sampedro M, Fernandez-Mayoralas A. J. Med. Chem. 1998;41:4599–4606. doi: 10.1021/jm980365i. [DOI] [PubMed] [Google Scholar]
- 7.Sebesta DP, O'Rourke SS, Pieken WA. J. Org. Chem. 1996;61:361–362. [Google Scholar]
- 8.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; New York: 2000. pp. 1–75. [Google Scholar]
- 9.a Abbruscato T, Williams S, Misicka A, Lipkowski AW, Hryby VJ, Davis TP. J. Pharmacol. Exp. Therapeut. 1996;276:1049–1057. [PubMed] [Google Scholar]; b Gentry CL, Egleton RD, Gillespie T, Abbruscato TJ, Bechowski HB, Hruby VJ, Davis TP. Peptides. 1999;20:1229–1238. doi: 10.1016/s0196-9781(99)00127-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.