

Abstract
We have shown previously that blockade of EGFR cooperates with a pan-selective inhibitor of PI3K in EGFR-driven glioma. In this communication, we tested EGFR-driven glioma differing in PTEN-status, treating with the EGFR inhibitor erlotinib and a novel dual inhibitor of PI3Kα and mTOR (PI-103). Erlotinib blocked proliferation only in PTENwt cells, without further impact by PI-103. Although erlotinib monotherapy showed little effect in PTENmt glioma, PI-103 greatly augmented the antiproliferative efficacy of erlotinib in this setting. To address the importance of PI3K-blockade, we showed in PTENmt glioma that combining PI-103 and erlotinib was superior to either monotherapy, or to therapy combining erlotinib with either rapamycin (an inhibitor of mTOR) or PIK-90 (an inhibitor of PI3Kα). These experiments demonstrate that a dual inhibitor of PI3Kα and mTOR augments the activity of EGFR-blockade, offering a mechanistic rationale for targeting EGFR, PI3Kα, and mTOR in the treatment of EGFR-driven, PTEN-mutant glioma.
Keywords: Glioma, EGFR, PTEN, mTOR, PI-103
Introduction
Both amplification of EGFR and activation of phosphatidylinositol-3 kinase (PI3K) feature prominently in glioma (Persson et al., 2007). Activation of PI3K may occur as a consequence of EGFR amplification, and in such cases should respond to inhibitors of EGFR. PI3K may also be activated independently of EGFR, through gain-of-function mutations in PI3K itself or from inactivation of the lipid phosphatase PTEN, a negative regulator of PI3K (Vogt et al., 2006; Zhao and Roberts, 2006). Because activation of PI3K is uncoupled from upstream amplification of EGFR in PTEN-mutant glioma, we previously tested inhibitors of PI3K and of EGFR in combination, demonstrating efficacy for this approach in EGFR-driven, PTEN-mutant glioma cell lines and xenografts (Fan et al., 2003). The clinical relevance of this work was recently corroborated by observations in patients treated with inhibitors of EGFR. In two retrospective analyses, tumors in which activation of PI3K was coupled to EGFR amplification responded to inhibition of EGFR (Haas-Kogan et al., 2005; Mellinghoff et al., 2005). In contrast, tumors in which PI3K was activated independently of EGFR responded poorly to EGFR inhibition. Collectively, these reports suggest that in tumors with EGFR amplification and PTEN inactivation (comprising half of EGFR-amplified glioma), combining inhibitors of EGFR and inhibitors of PI3K represents a promising therapy.
What are the hurdles preventing translation of this approach to patients? The eight mammalian PI3-kinases are grouped within three classes (Engelman et al., 2006). Although pan-selective inhibitors of PI3Ks have been critical to our current understanding, these compounds indiscriminately inhibit all known PI3Ks and are toxic in patients (Workman et al., 2006). To delineate the role of individual PI3Ks, we recently synthesized a series of isoform-selective inhibitors of PI3Ks, defined the structural basis for their specificity, and systematically enumerated their biochemical targets (Knight et al., 2006). These agents, in conjunction with similar compounds described by others (Camps et al., 2005; Hayakawa et al., 2007; Hayakawa et al., 2006; Jackson et al., 2005; Knight et al., 2004; Sadhu et al., 2003) collectively represent an important arsenal of tools for analysis of signaling through PI3K. Using this chemical array, we linked increased specificity to decreased toxicity for agents targeting within the PI3K family and identified PI3Kα as critical for proliferation in malignant glioma (Fan et al., 2006).
We noted particular efficacy in combining inhibitors of PI3Kα with inhibitors of mTOR, a serine-threonine kinase that is part of a protein complex critical for cell growth (Corradetti and Guan, 2006; Sabatini, 2006). This result was at first somewhat surprising, as mTOR is activated as a consequence of signaling through PI3K. PI3Ks phosphorylate phosphatidylinositol-4,5-bisphosphate (PIP2), generating phosphatidylinositol-4,5-trisphosphate (PIP3), which in turn activates Akt, a PH-domain containing serine-threonine kinase that signals through downstream effecters including mTOR, to suppress apoptosis, promote cell growth, and drive proliferation (Shaw and Cantley, 2006). Importantly, inhibitors of mTORC1 actually activate signaling through PI3K (Fan et al., 2006; Shi et al., 2005; Sun et al., 2005). Thus, the efficacy of mTOR blockade is achieved at the cost of driving other outputs of Akt signaling, contributing to the overall disappointing results observed using inhibitors of mTOR clinically. Combined blockade of PI3Kα and mTOR shuts down mTOR, and also abrogates the activation of PI3K observed using mTOR inhibitors as monotherapy. We recently validated this dual-inhibitor approach by combining rapamycin--an inhibitor of mTOR, with PIK-90--an inhibitor of PI3Kα, and also through testing a dual inhibitor of PI3Kα and of mTOR (PI-103) that was well tolerated and was highly effective against glioma xenografts (Fan et al., 2006).
These preclinical studies support the use of combination therapy directed against EGFR and PI3K in glioma, and in parallel demonstrate that inhibitors of PI3Kα cooperate with inhibitors of mTOR in this disease. The goal of our current work is to test inhibitors of EGFR in combination with a dual inhibitor of PI3Kα and mTOR in glioma. In this communication, we demonstrate that PI-103 cooperates with erlotinib in PTEN-mutant glioma, establishing a mechanistic rationale for blockade of EGFR, PI3Kα, and mTOR in the treatment of EGFR-driven, PTEN mutant glioma.
RESULTS
PTEN status and efficacy: erlotinib versus PI-103
To clarify the role of PTEN as a determinant of response to inhibitors of EGFR/PI3K/mTOR signaling, we transduced EGFR into the glioma cell lines LN229 and U87, and treated these with erlotinib, or with PI-103. In contrast to the PTENmt cell line U87:EGFR, LN229:EGFR cells (PTENwt) showed a prominent response to erlotinib (Fig 1A-B). Flow cytometric analysis demonstrated G0G1 arrest in LN229 cells (Fig 1C). In comparison, U87:EGFR cells showed a more modest response (Fig 1C). These data are consistent with results by others that PTEN status represents an important determinant of response to EGFR inhibitors (Haas-Kogan et al., 2005; Mellinghoff et al., 2005).
Fig 1.
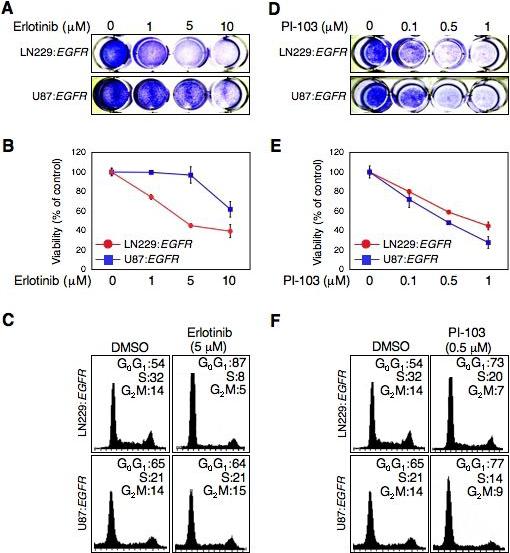
EGFR inhibitor erlotinib inhibits cell proliferation and induces G0G1 arrest dependent on PTEN status. In contrast, anti-proliferative effects of the dual PI3 kinase/mTOR inhibitor PI-103 were not dependent on PTEN status. LN229:EGFR (PTENwt) and U87:EGFR (PTENmt) cells were treated with erlotinib or PI-103. (A and D) Cell proliferation was visualized by crystal-violet staining of cells after treatment with erlotnib or PI-103 for 72h at dosages indicated. (B and E) Cell growth was determined using WST-1 assay after treatment with erlotinib or PI-103 for 72h at dosages indicated. Error between triplicate measurements for each value shown. (C and F) Cells were treated with erlotinib 5 μM or PI-103 0.5 μM for 24 hr. Cells were trypsinized and prepared for flow cytometric analysis of cell cycle distribution. Percentage of cells in G0G1, S, and G2M phases of the cell cycle is indicated.
Since mutation at PTEN should not interfere with pathways coupling PI3K to mTOR, we reasoned that PTEN status might be less important for the dual inhibitor PI-103. Consistent with this model, PI-103 was equipotent against PTENwt and PTENmt cells, leading to arrest at G0G1 (Fig 1 D-F). The response to this compound contrasted with the clear PTEN-dependence observed using erlotinib, and suggests that PTEN status is not a critical determinant of response to the dual PI3K/mTOR inhibitor PI-103.
Erlotinib blocks mTOR in PTENwt cells
To explore downstream targets mediating the response of glioma cells to EGFR blockade, we performed immunoblotting of PTENwt and PTENmt cells in response to erlotinib using phospho-specific antibodies. Treatment of cells with EGF led to equivalent responses in MAP-kinase signaling irrespective of PTEN status, as indicated by levels of p-Erk (Fig 2). Inhibition of EGFR impacted levels of p-Erk similarly in both cell lines, consistent with pathways linking EGFR to MAP kinase signaling that were not impacted by PTEN status (Fig 2A). In contrast, although treatment with EGF led to activation of p-Akt in PTENwt cells (which showed increased levels of p-Akt), EGF showed little impact on p-Akt in PTENmt cells. PTENmt cells had high levels of p-Akt at baseline, consistent with activation of PI3K through loss of PTEN, a negative regulator of PI3K signaling. These high levels of p-Akt were only modestly affected by treatment of PTENmt cells with erlotinib, and stood in contrast to comparable experiments treating PTENwt cells with this compound (Fig 2A), again supporting a model in which loss of PTEN effectively uncouples activation of PI3K/Akt from upstream signaling through EGFR.
Fig 2.
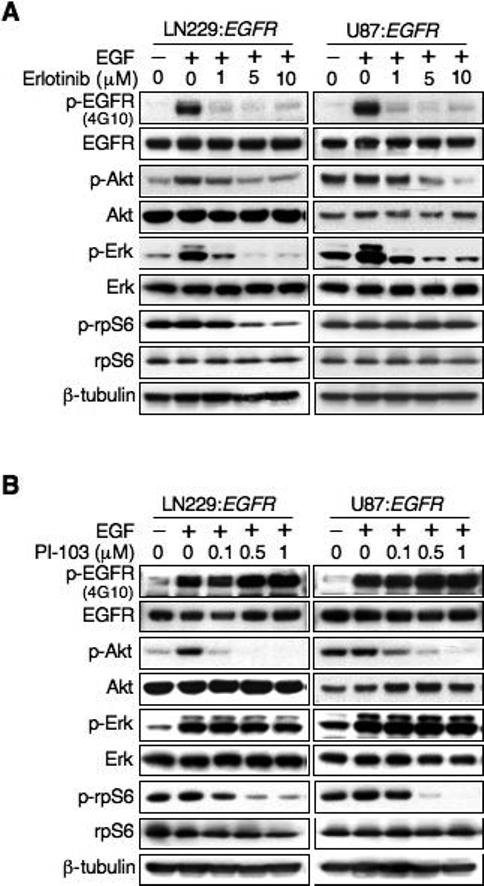
Impact of PTEN status on biochemical response to Erlotinib or PI-103 treatment. (A) The EGFR inhibitor erlotinib inhibits signaling through Akt and mTOR in a PTEN dependent manner. LN229:EGFR (PTENwt) and U87:EGFR (PTENmt) cells were treated with erlotinib or PI-103 at dosages shown for 24 hr. EGF was added 15 min prior to harvest, and lysates immunoblotted as indicated. Erlotinib therapy led to decreased Erk signaling in both cell lines, but impacted levels of p-Akt and the mTOR target p-rpS6 kinase only in cells wild-type for PTEN. (B) The dual PI3Kα/mTOR inhibitor PI-103 blocks both Akt and mTOR irrespective of PTEN status. Experimental conditions were identical to (A). Although U87:EGFR cells had higher base-line signaling through p-Akt, treatment with PI-103 led to dose dependent blockade of both p-Akt and p-rpS6, without appreciably impacting levels of p-Erk.
To address the response of mTOR signaling, we analyzed the mTOR target ribosomal protein S6 kinase (rpS6). At baseline, levels of p-rpS6 were prominent in both cell lines, apparently unaffected by PTEN status, and similarly (in these cells, grown in 10% FBS) unaffected by treatment with EGF. In contrast, PTEN status was important in determining whether blockade of EGFR affected levels of p-rpS6. In PTENwt cells, treatment with erlotinib led to decreased levels of p-rpS6, which changed in parallel with p-Akt (Fig 2A). Importantly, treatment of PTENmt cells with erlotinib did not impact levels of p-rpS6, even at doses sufficiently high to block signaling through p-Akt. These data demonstrate that PTEN links EGFR to mTOR, that inhibition of EGFR can block signaling through mTOR in PTENwt but not in PTENmt cells, and suggest that blockade of mTOR correlates with efficacy of EGFR inhibitors.
We next asked whether PTEN status was a determinant of the biochemical response to the dual inhibitor PI-103. Both PTENwt and PTENmt cells showed significant blockade of p-Akt in response to PI-103 (Fig 2B), whereas levels of p-Erk were only minimally affected. While base-line levels of p-rpS6 showed minor differences comparing PTENwt and PTENmt cells, the response to PI-103 was qualitatively similar. These experiments are consistent with results in Fig 1, demonstrating that PI-103 was equipotent in blocking proliferation in both PTENwt and PTENmt cells, suggesting that PTEN status did not correlate with response to this agent.
Erlotinib cooperates with PI-103 to arrest cells
Collectively, data in Figs 1-2 argues that mutation at PTEN uncouples EGFR from downstream signaling through PI3K and mTOR, suggesting that blockade of PI3K and/or mTOR could enhance the efficacy of EGFR inhibition in PTENmt glioma. To address the efficacy of this combination, we treated cells with erlotinib in combination with PI-103 (Fig 3). As expected, erlotinib led to G0G1 arrest in PTENwt cells, and was minimally augmented by PI-103. In contrast, erlotinib had little impact as monotherapy in PTENmt cells. Erlotinib did show efficacy when combined with low-dose (100 nM) PI-103 in this setting however, with combination therapy effectively promoting arrest at G0G1 (Fig 3A).
Fig 3.
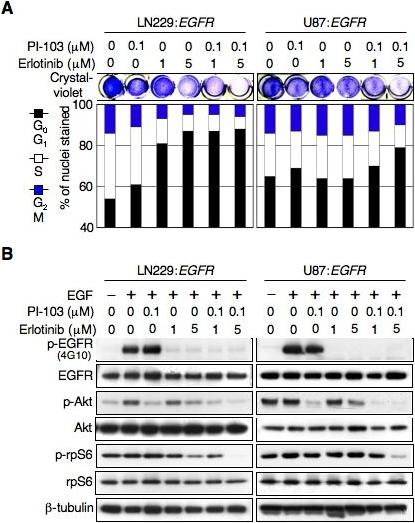
Inhibitors of EGFR cooperate with the dual PI3Kα/mTOR inhibitor PI-103 to arrest growth of PTEN-mutant human glioma cells. LN229:EGFR (PTENwt) and U87:EGFR (PTENmt) cells were treated with erlotinib, PI-103, or erlotinib plus PI-103 at indicated dosages. (A) Proliferation was visualized by crystal-violet staining of cells after treatment for 72 hr (top panel). Flow cytometry analysis (bar graph-bottom panel) shows effects on cell cycle after treatment for 24 hr. Percentage of cells in G0G1, S, and G2M phases of the cell cycle is indicated. (B) Cells were treated same as in (A). EGF was added 15 min prior to harvest. Lysates were subjected to immunoblot analysis with antisera indicated.
Immunoblotting experiments further reinforced results in Fig 2A, demonstrating that PTEN status correlated with the ability of erlotinib monotherapy to impact signaling through mTOR (Fig 3B). As monotherapy, erlotinib could block signaling through mTOR most effectively in PTENwt cells. Addition of PI-103 to erlotinib in these cells only minimally altered the efficiency through which erlotinib monotherapy blocked signaling through mTOR. In contrast, mutation at PTEN was an important and negative determinant of erlotinib's ability to impact activation of mTOR. Whereas treatment of PTENmt cells with erlotinib minimally affected levels of the mTOR target p-rpS6, addition of PI-103 to erlotinib in these cells led to effective blockade of p-rpS6 (Fig 3B). These observations again support a model in which PTEN status correlates with the ability of EGFR inhibitors to impact signaling through mTOR, and supports combining PI-103 with erlotinib in EGFR-driven PTENmt tumors.
Blockade of EGFR, PI3Kα, and mTOR in glioma
Having demonstrated that that PI-103 can augment the response to erlotinib in PTENmt cells, we next asked whether all three targets of these agents (EGFR, PI3Kα, and mTOR) were critical to achieve maximal proliferative blockade. We treated PTENmt cells with erlotinib in combination with the pure PI3Kα inhibitor PIK-90, the mTOR inhibitor rapamycin, combination therapy using both PIK-90 and rapamycin, or the dual PI3Kα/mTOR inhibitor PI-103. Measures of overall viability and of proliferation were consistent in showing that blockade of mTOR cooperated with inhibition of EGFR, and that further blockade of PI3Kα led to maximal proliferation block (Fig 4A, compare columns 7 and 9, p<0.0001-Student's t-test). TUNEL and subG1 fractions demonstrated no significant difference in apoptosis among these therapies (not shown). Immunoblotting experiments (Fig 4B) were aligned with results in Fig 4A. While cooperative inhibition of EGFR and of mTOR led to decreased levels of both p-EGFR and p-rpS6, treatment with rapamycin actually increased levels of p-Akt. Further inhibition of PI3Kα was required to efficiently block p-Akt in the setting of efficient blockade of both p-EGFR and p-rps6. These results demonstrate that blockade of EGFR and of mTOR cooperate in the treatment of EGFR-driven, PTENmt glioma, and that further efficacy can be achieved through concomitant blockade of PI3Kα.
Fig 4.
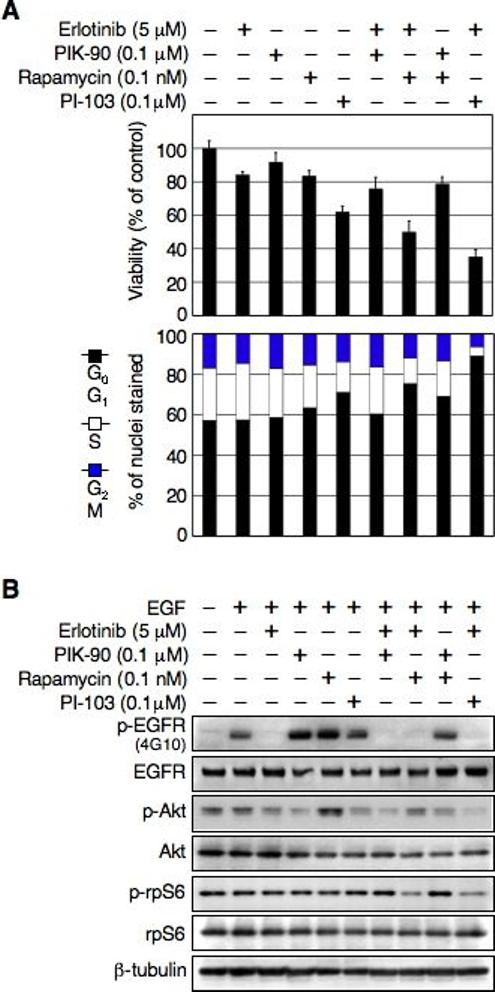
Cooperative inhibition of EGFR and mTOR are critical to arrest growth of PTENmt human glioma cells. (A) U87:EGFR (PTENmt) cells were treated with erlotinib, PIK-90, rapamycin, PI-103, erlotinib plus PIK-90, erlotinib plus rapamycin, PIK-90 plus rapamycin, or erlotinib plus PI-103 at dosages indicated. Viable cells were measure by WST-1 assay after treatment for 72 hr. Error was calculated among triplicate measurements (top panel). Flow cytometry analysis (bar graph-bottom panel) shows effects on cell cycle after treatment for 24 hr. Percentage of cells in G0G1, S, and G2M phases of the cell cycle is indicated. (B) Cells were treated as in (A). EGF was added 15 min prior to harvest. Lysates were subjected to immunoblot analysis with antisera indicated.
DISCUSSION
The malignant gliomas show intrinsic resistance to most medical therapies, contributing to the poor prognosis associated with these tumors (Persson et al., 2007). The association of EGFR amplification with high-grade glioblastoma multiforme tumors therefore led to early optimism that EGFR inhibition would be beneficial in glioma. This initial optimism was mitigated however, by the realization that only a subset of patients with EGFR-amplified glioma actually responded to blockade of EGFR (Nicholas et al., 2006). The failure of this approach in the majority of patients with EGFR-amplified glioma could stem from inefficient blockade of the receptor or from inability to reverse signaling abnormalities associated with EGFR amplification, even in the setting of adequate blockade of p-EGFR. Loss of PTEN is a likely contributor to this failure, as loss of PTEN effectively blocks the ability of EGFR inhibitors to impact downstream signaling through PI3K and ultimately through mTOR.
In this communication, we present a preclinical approach aimed at reversing signaling abnormalities associated with EGFR amplification, offering a mechanistic rationale to combine inhibitors of EGFR and of mTOR to effect proliferation blockade in patients with EGFR-amplified, PTENmt glioma. We demonstrated efficacy for inhibitors of EGFR as monotherapy in glioma cells wild-type for PTEN, and that the antiproliferative effect of EGFR inhibitors correlated with the ability of these agents to impact levels of mTOR.
In contrast to PTENwt cells, erlotinib treatment of PTENmt cells did not appreciably impact proliferation and specifically did not impact mTOR, even when inhibitors of EGFR were used at doses sufficiently high to block p-Akt (Fig 2). Although erlotinib had little measurable activity as monotherapy in PTENmt cells, erlotinib clearly augmented the efficacy of PI-103 as measured both by blockade of mTOR and of proliferation (Fig. 4). Intriguingly, the ability of PI-103 and erlotinib to impact mTOR again was observed in a setting where combination therapy did not appreciably alter levels of p-Akt in comparison with PI-103 alone (Fig 4). The dissociation of Akt from mTOR in PTENmt glioma has also been observed by others (Wang et al., 2006), and suggests the presence of Akt-independent regulators of mTOR.
The failure of inhibitors of EGFR to impact mTOR signaling in PTENmt glioma also provides a rationale to combine inhibitors of EGFR and mTOR. While targeting both kinases simultaneously led to decreased proliferation in comparison with targeting either EGFR or mTOR alone (Fig 4), blockade of mTOR by rapamycin actually led to increased levels of p-Akt (Fig 4B, lane 5). The activation of p-Akt by rapamycin and its analogues has been described previously in primary human tumors (O'Reilly et al., 2006; Shi et al., 2005; Sun et al., 2005). Addition of an mTOR inhibitor effectively blocks mTOR, but at the cost of activating other targets of PI3K and Akt.
In response to the failure of EGFR inhibitors to block PI3K, Akt, or mTOR in PTENmt glioma, and because mTOR inhibitors actually activate the PI3K/Akt axis, we tested inhibitors of EGFR and of mTOR in combination with inhibitors of PI3Kα. The combinatorial inhibition of three targets effectively blocked signaling (Fig 3), and was more effective than any two targeted therapies in combination as measured both biochemically and through flow cytometric analyses (Fig 4). It is intriguing in this regard that even using approaches that blocked EGFR, PI3Kα, and mTOR in combination, and in the setting of efficient inhibition of Akt—an important mediator of anti-apoptotic signaling (Luo et al., 2003)—we failed to observe any appreciable apoptosis in any glioma cell lines tested (Fig 4 and data not shown). Thus, while the ability to translate our findings to patients awaits the development of isoform-specific inhibitors of PI3K that are well tolerated clinically, the ability to develop targeted agents that induce cytotoxic rather than cytostatic responses in the malignant gliomas represents a more formidable challenge, and one that may be critical to the long-term efficacy of these approaches in patients.
We are grateful to Russ Pieper and Cynthia Cowdry for cell lines, and Lou Chesler, Chris Hackett, and Matt Grimmer for critical review. This work, dedicated to the late Jeffrey P. Weiss, was supported by grants from the Burroughs Wellcome Fund, The Brain Tumor Society, The National Brain Tumor and Samuel G. Waxman Foundations, The Sandler Family, and the NCI SPORE Program.
METHODS
Growth inhibition, cell staining, and flow cytometry
LN229 and U87 cells transduced with EGFR as described (Fan and Weiss, 2005), were grown in media containing 10% FBS. PTENwt and PTENmt cells transduced with ΔEGFR were also examined in experiments similar to those in Figs 1-4, with comparable results (not shown). Erlotinib tablets (Genentech) were ground to powder, dissolved in aqueous HCl and aqueous phase extracted with ethyl acetate. The combined organic extracts were dried over sodium sulphate and concentrated to yield pure erlotinib. PIK-90 and PI-103 were synthesized as described (Knight et al., 2006). For viabilty, 105 cells were seeded in 12-well plates in the presence of erlotinib, PI-103, PIK-90, rapamycin (Cell Signaling), erlotinib plus PIK-90, erlotinib plus rapamycin, PIK-90 plus rapamycin or erlotinib plus PI-103 for 3 d. Cell viability was determined using a WST-1 assay (Roche). For crystal-violet staining, 105 cells were seeded in 12 well plates +/− PI-103, erlotinib, or PI-103 plus erlotinib. After 3d, cells were washed with water and stained with crystal-violet for 5 min. Flow cytometry was as previously described (Fan et al., 2006).
Immunoblotting
Membranes were blotted with antisera to p-Akt (Ser473), Akt, p-Erk (Ser202/204), p-S6 ribosomal protein (Ser235/236), and S6 ribsomal protein (Cell Signaling), 4G10, β-tubulin (Upstate Biotechnology), Erk2, or EGFR (Santa Cruz). Immunoblotting and detection were as previously described (Fan et al., 2002).
REFERENCES
- Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, Martin T, Gretener D, Perrin D, Leroy D, Vitte PA, Hirsch E, Wymann MP, Cirillo R, Schwarz MK, Rommel C. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;9:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. 2006;25:6347–6360. doi: 10.1038/sj.onc.1209885. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QW, Specht KM, Zhang C, Goldenberg DD, Shokat KM, Weiss WA. Combinatorial efficacy achieved through two-point blockade within a signaling pathway-a chemical genetic approach. Cancer Res. 2003;63:8930–8938. [PubMed] [Google Scholar]
- Fan QW, Weiss WA. RNA interference against a glioma-derived allele of EGFR induces blockade at G2M. Oncogene. 2005;24:829–837. doi: 10.1038/sj.onc.1208227. [DOI] [PubMed] [Google Scholar]
- Fan QW, Zhang C, Shokat KM, Weiss WA. Chemical genetic blockade of transformation reveals dependence on aberrant oncogenic signaling. Curr Biol. 2002;12:1386–1394. doi: 10.1016/s0960-9822(02)01070-9. [DOI] [PubMed] [Google Scholar]
- Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, Baumber R, Lamborn KR, Kapadia A, Malec M, Berger MS, Stokoe D. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- Hayakawa M, Kaizawa H, Kawaguchi K, Ishikawa N, Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S, Raynaud FI, Waterfield MD, Parker P, Workman P. Synthesis and biological evaluation of imidazo[1,2-a]pyridine derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem. 2007;15:403–412. doi: 10.1016/j.bmc.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Okada M, Ohta M, Tsukamoto SI, Parker P, Workman P, Waterfield M. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem. 2006 doi: 10.1016/j.bmc.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, Sturgeon SA, Prabaharan H, Thompson PE, Smith GD, Shepherd PR, Daniele N, Kulkarni S, Abbott B, Saylik D, Jones C, Lu L, Giuliano S, Hughan SC, Angus JA, Robertson AD, Salem HH. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005;11:507–514. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Chiang GG, Alaimo PJ, Kenski DM, Ho CB, Coan K, Abraham RT, Shokat KM. Isoform-specific phosphoinositide 3-kinase inhibitors from an arylmorpholine scaffold. Bioorg Med Chem. 2004;12:4749–4759. doi: 10.1016/j.bmc.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth T, Weiss WA, Williams R, Shokat KM. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- Nicholas MK, Lukas RV, Jafri NF, Faoro L, Salgia R. Epidermal growth factor receptor-mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res. 2006;12:7261–7270. doi: 10.1158/1078-0432.CCR-06-0874. [DOI] [PubMed] [Google Scholar]
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A, Fan Q-W, Phillips J, Weiss WA. Glioma. Elsevier; San Diego: 2007. [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sadhu C, Dick K, Tino WT, Staunton DE. Selective role of PI3K delta in neutrophil inflammatory responses. Biochem Biophys Res Commun. 2003;308:764–769. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005;4:1533–1540. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- Vogt PK, Bader AG, Kang S. PI 3-kinases: hidden potentials revealed. Cell Cycle. 2006;5:946–949. doi: 10.4161/cc.5.9.2725. [DOI] [PubMed] [Google Scholar]
- Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, Mischel PS. Mammalian Target of Rapamycin Inhibition Promotes Response to Epidermal Growth Factor Receptor Kinase Inhibitors in PTEN-Deficient and PTEN-Intact Glioblastoma Cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- Workman P, Clarke PA, Guillard S, Raynaud FI. Drugging the PI3 kinome. Nat Biotechnol. 2006;24:794–796. doi: 10.1038/nbt0706-794. [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Roberts TM. PI3 kinases in cancer: from oncogene artifact to leading cancer target. Sci STKE. 2006;2006:pe52. doi: 10.1126/stke.3652006pe52. [DOI] [PubMed] [Google Scholar]