

Abstract
The phosphatidylinositol (PI) 3-kinase is activated by the Type I and II interferon (IFN) receptors, but its precise role in the generation of IFN responses is not well understood. In the present study we used embryonic fibroblasts from mice with targeted disruption of the genes encoding for both the p85α and p85β regulatory subunits of PI3′ kinase (p85α-/-β-/-) to precisely define the role of the PI 3 kinase in the control of IFN-induced biological responses. Our data demonstrate that the PI 3 kinase plays dual regulatory roles in the induction of IFN-responses, by controlling both IFNα- and IFNγ- dependent transcriptional regulation of interferon sensitive genes (ISGs) and at the same time regulating the subsequent initiation of mRNA translation for such genes. This includes the Isg15, Cxcl10 and/or Irf7 genes, whose functions are important in the generation of the biological effects of IFNs. Consistent with this, the induction of IFN-antiviral responses is defective in double p85α/p85β knockout cells. Thus, integration of signals via the PI 3 kinase is a critical event during engagement of the IFN receptors that complements both the transcriptional activity of Jak-Stat pathways and controls initiation of mRNA translation.
Keywords: cytokines, signal transduction, interferon, mRNA translation
Introduction
Interferons (IFNs) are cytokines that play critical roles in the host defense against viral and parasitic infections, and in immunosurveilance against malignant cells (1-6). Because of these properties, IFNs have been used in the treatment of viral infections as well as various malignancies and neurological disorders (7-10). Over the years, there has been an increasing interest in understanding the signaling mechanisms downstream of the IFN receptors, since insights into the cellular events that mediate the antiviral and antitumor effects of IFNs may lead to the design of novel drugs that target specific signaling effectors. Among the signaling cascades and elements that transmit Type I and II IFN signals, the Jak-Stat pathway has been the most extensively studied. Different combinations of Jak kinases are associated with the different IFN receptors, and binding to these cell surface receptors results in activation of Jaks and downstream phosphorylation/ activation of the Stats. The activated Stats then translocate to the nucleus and trigger transcription of IFN sensitive genes (ISGs), via binding to specific elements in the promoters of the ISGs (1-6).
Distinct from the classic Jak-Stat pathways, multiple other signaling cascades are activated by IFNs, underscoring the complexity of the signaling mechanisms invoked by IFNs (11-19). For example, MAP kinase pathways, and in particular the p38 MAPK signaling pathway, are activated by Type I IFNs and are required for full transcriptional activation of ISGs and IFN-generated biological responses, separate from Stat-activation (11-15). Another major signaling pathway activated by IFNs is the PI3 kinase pathway. In mammals, IFNs and other cytokines engage class I PI3 kinases, which exist as inactive heterodimers of the regulatory (p85) and the catalytic (p110) subunits in the cytoplasm of cells (20). The p85 regulatory subunit is recruited to the cell membrane by activated receptor tyrosine kinases (RTK), and this induces a conformational change in the complex, resulting in reversal of the inhibitory effects on the p110 catalytic subunit and activation of its kinase domain (20). Adapters such as the insulin receptor substrate (IRS) proteins serve as docking proteins to regulate downstream engagement of PI3 kinase in response to IFNs (21-23).
In previous work, we demonstrated that activation of the PI3K by type I and II IFNs mediates activation of p70S6K and phosphorylation/de-activation of the translational repressor 4E-BP1 (24-26). These studies have raised the possibility that PI3 kinase participates in the regulation of mRNA translation in response to IFNs, but its precise role in mRNA translation for ISGs has not been directly established. In the present study we used mouse embryonic fibroblasts (MEFs) with targeted disruption of the genes for both the p85α and p85β regulatory subunits of PI3 kinase, to study its precise role in the generation of IFN responses. Altogether, our data establish that the PI3 kinase exhibits dual regulatory functions in IFN-signaling, controlling both transcription and mRNA translation of IFN-inducible genes, and plays a critical role in the generation of the antiviral effects of IFNs.
Materials and Methods
Cell lines and Reagents
Recombinant mouse IFNα4 was provided by Wellcome research laboratories (Kent, UK). Recombinant mouse IFNβ was provided by Dr. Darren Baker, Biogen Idec. Recombinant mouse IFNγ was from PBL Biomedical Laboratories (Piscataway, NJ). Immortalized MEFs from p85 knockout mice and the retroviral addback MEFs have been described previously (24, 25, 27). The MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum and antibiotics. A rabbit polyclonal antibody against mouse ISG15 was kindly provided by Dr. Dong-Er Zhang (Scripps Research Institute, La Jolla, CA). Another polyclonal antibody against ISG15 has been previously described (28). Antibodies against tubulin and IP10 were from Abcam (Cambridge, MA). Antibodies against of Akt, 4E-BP1 and their phosphorylated forms were from Cell Signaling Technologies (Beverly, MA).
Cell Lysis and Immunoblotting
For short-time treatments (up to 60 minutes), cells were starved overnight in serum free medium. Cells were treated with the indicated IFNs for the indicated times, and lysed in phosphorylation lysis buffer (PLB) supplemented with PMSF, aprotinin and orthovanadate, as previously described (25, 26, 28). Equal protein aliquots were resolved by SDS PAGE and immunoblotting using an enhanced chemiluminescence (ECL) method was performed as in our previous studies (24-26, 28).
Luciferase Reporter Assays
Luciferase reporter assays were performed as previously described (12, 26, 27). The ISRE-luciferase construct (29) was provided by Dr Richard Pine (Public Health Research Institute, New York, NY), while the 8X GAS construct (30) was provided by Dr Christofer Glass (University of California, San Deigo,CA). Cells were transfected with a β-galactosidase construct and either ISRE or 8X GAS-luciferase constructs using superfect (Qiagen). Forty eight hours post transfection cells were either left untreated or treated with interferon for six hours and luciferase activity was measured. β-galactosidase activity was used for normalization.
Quantitative RT-PCR (TaqMan)
Cells were treated with 5000 IU/ml of IFNα or 2500 IU/ml of IFNγ for 6 hours and RNA was isolated using RNeasy kit (Qiagen). 1μg RNA was reverse transcribed using Omniscript reverse transcriptase and oligo dT primer (Qiagen). Real-time RT-PCR for the Isg15 and Ip10 genes was carried out on ABI7900 sequence detection system (Applied Biosystem) using commercially available FAM labeled probes and primers (Applied Biosystem). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization. ΔCt values (target gene Ct minus GAPDH Ct) for each triplicate sample were averaged, and ΔΔCt was calculated. mRNA amplification was determined by formula 2-ΔΔCt as previously described (28). Relative quantitation of mRNA levels was plotted as fold increase over untreated samples.
Isolation of polysomal RNA and Quantitative RT-PCR
p85α+/+ β+/+ and p85α-/- β-/- MEFs were treated with mIFNα or mIFNγ for 40 hours, and isolation of polysomal RNA and quantitative RT-PCR on the polysomal fractions were performed as previously described (28). Briefly, cell pellets were lysed in hypotonic lysis buffer (5mM Tris pH7.5, 2.5mM MgCl2, 1.5mM KCl) supplemented with protease inhibitors (Calbiochem), RNAse-In (Ambion), 1mM DTT, and 100 μg/ml of cycloheximide. TritonX 100 and sodium deoxycholate were added to the lysates to a final concentration of 0.5% each. The lysates were clarified by centrifugation and supernatants were layered over 10-50% continuous sucrose gradient. After ultracentrifugation, fractions were collected monitoring the absorbance at 254nm as a function of gradient depth. The polysomal fractions were pooled and total RNA from pooled fractions was isolated using RNAqueous-micro kit from Ambion (Austin, TX). Total polysomal RNA for each experimental condition was quantitated and equal amounts of RNA were reverse transcribed into cDNA using the Omniscript RT kit and Oligo (dT) primers (Qiagen). Real-time PCR for the Isg15, Irf7 or Cxcl10 gene was carried out using commercial available FAM labeled probes and primers (Applied Biosystem) and GAPDH was used for normalization. To further confirm the results, real-time PCR for the Isg15 and Cxcl10 genes was also repeated using tubulin for normalization. mRNA amplification was determined by formula 2-ΔΔCt as described above and relative quantitation of mRNA levels was plotted as fold increase as compared with untreated samples.
Antiviral Assays
The antiviral effects of mouse IFNα were determined in assays using encephalomyocarditis virus (EMCV) as the challenge virus, as in our previous studies (12, 26, 28).
Results
The PI 3 kinase pathway is required for activation of Akt by IFNs
In initial studies, we sought to determine whether engagement of the PI3 kinase is required for downstream phosphorylation of Akt, whose function is required for activation of pathways that regulate initiation of mRNA translation by IFNs (28). For this purpose, we used mouse embryonic fibroblasts (MEFs) derived from mice with targeted disruption of both the p85α and p85β subunits of the PI3 kinase (p85α-/-β-/-). p85α+/+β+/+ and p85α-/-β-/- MEFs were treated with either mouse IFNα (Fig 1A) or mouse IFNγ (Fig 1B), and cell lysates were processed for immunoblotting with an antibody that recognizes the phosphorylated form of Akt on Ser 473. Treatment of the p85α+/+β+/+ MEFs with either IFNα or IFNγ resulted in strong phosphorylation of Akt on serine 473, but such phosphorylation was defective in p85α-/-β-/- MEFs (Fig. 1A and B). Similar results were also seen when the IFNα-inducible phosphorylation of Akt on threonine 308, the PDK1 phosphorylation site (31), was compared in p85α+/+β+/+ and p85α-/-β-/- MEFs treated with mouse IFNα. Thus, engagement of PI3 kinase is essential for downstream phosphorylation and activation of Akt by both Type I and II IFNs.
Figure1. IFN-dependent phosphorylation/activation of Akt is PI3 kinase dependent.
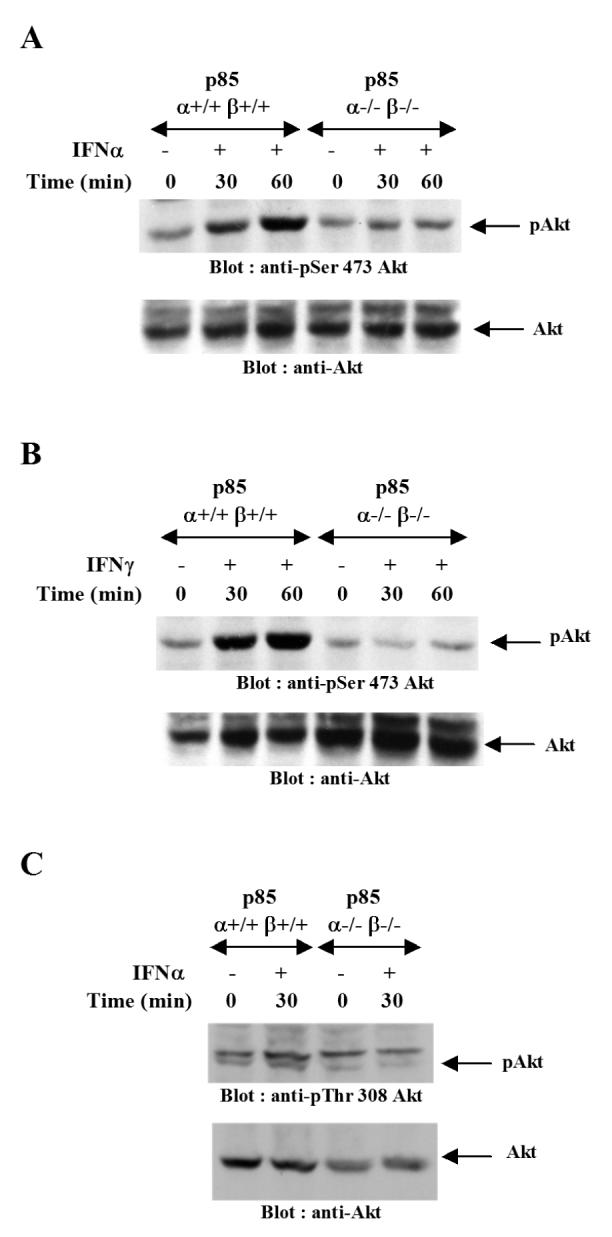
A. p85 α+/+β+/+ and p85 α-/-β-/- MEFs were treated with mouse IFNα for the indicated times. The cells were lysed and equal amounts of protein were resolved by SDS-PAGE and immunoblotted with an antibody against phosphorylated form of Akt on Ser 473 (top panel). The same blot was stripped and re-probed with an anti-Akt antibody (lower panel). B. p85 α+/+β+/+ and p85 α-/-β-/- MEFs were treated with mouse IFNγ for the indicated times. Equal amounts of protein were resolved by SDS-PAGE and immunoblotted with an antibody against phosphorylated form of Akt on Serine 473 (top panel). The same blot was stripped and re-probed with an anti- Akt antibody (lower panel). C. p85 α+/+β+/+ or p85 α-/-β-/- mouse embryonic fibroblasts (MEFs) were treated with mouse IFNα for the indicated times. Equal amounts of protein were resolved by SDS-PAGE and immunoblotted with an antibody against the phosphorylated form of Akt on Thr 308 (top panel). The same blot was stripped and re-probed with an anti-Akt antibody (lower panel).
In previous work, we had demonstrated that engagement of PI3 kinase is required for Type I and II IFN-induced phosphorylation of p70 S6 kinase (24, 25). Also, studies with the pharmacological inhibitors had suggested that phosphorylation and de-activation of translation repressor 4E-BP1 may occur downstream of PI3 kinase and mTOR (24-26). To definitively address this issue, we sought to determine whether IFN-induced phosphorylation of 4E-BP1 is defective in p85α-/-β-/- MEFs. As shown in Fig. 2A and 2B, both Type I and II IFN-treatment induced strong phosphorylation of 4E-BP1 in p85α+/+β+/+ MEFs on Thr 70 (Fig. 2A and B) or Thr 37/46 (Fig. 2C and D), but such phosphorylation was reduced but not entirely eliminated in p85α-/-β-/- MEFs (Fig. 2A-D).
Figure 2. Requirement of p85α and β regulatory subunits of the PI3 kinase for Type I and II IFN-dependent phosphorylation of 4E-BP1.

A. p85 α+/+β+/+ and p85 α-/-β-/- MEFs were treated with mouse IFNα for the indicated times. Equal protein aliquots were resolved by SDS-PAGE and immunoblotted with antibody against the phosphorylated form of 4EBP1 on Thr 70 (top panel). The same blot was subsequently stripped and re-probed with an anti- 4E-BP1 antibody (middle panel). The signals for phospho-4E-BP1 and 4EBP1 or GAPDH controls from 3 independent experiments, including the experiment shown above, were quantitated by densitometry, and the intensity of phospho 4E-BP1 relative to 4E-BP1 or GAPDH controls was calculated. Data are expressed as the means of ratios of phospho-4E-BP1 to 4E-BP1 or GAPDH levels ± S.E. for each experimental condition (Bottom panel). B. p85 α+/+β+/+ and p85 α-/-β-/- MEFs were treated with mouse IFNγ for the indicated times. Equal protein aliquots were resolved by SDS-PAGE and immunoblotted with antibodies against the phosphorylated form of 4EBP1 on Thr 70 (top panel). Equal protein aliquots from the same experiment were resolved on a separate gel and probed with anti- 4E-BP1 antibody (middle panel). The signals for phospho-4E-BP1 and 4EBP1 or GAPDH controls from 3 independent experiments, including the experiment shown above, were quantitated by densitometry, and the intensity of phospho-4E-BP1 relative to 4E-BP1 or GAPDH controls was calculated. Data are expressed as the means of ratios of phospho-4E-BP1 to 4E-BP1 or GAPDH levels ± S.E. for each experimental condition (Bottom panel). C-D p85 α+/+β+/+ and p85 α-/-β-/- MEFs were treated with mouse IFNα (C) or mouse IFNγ (D) for the indicated times. Equal protein aliquots were resolved by SDS-PAGE and immunoblotted with antibodies against the phosphorylated form of 4E-BP1 on Thr37/46, as indicated. The same blots were subsequently stripped and re-probed with an anti-4E-BP1 antibody as indicated. The signals for phospho-4E-BP1 and 4E-BP1 or GAPDH controls from 4 (C) or 3 (D) independent experiments, including the ones shown above, were quantitated by densitometry, and the intensity of phospho-4E-BP1 relative to 4EBP1 or GAPDH controls was calculated. Data are expressed as the means of ratios of phospho-4E-BP1 to 4E-BP1 or GAPDH levels ± S.E. for each experimental condition.
Regulation of IFN-inducible expression of IFN-stimulated gene products by the PI 3 kinase
We next sought to determine the role PI3 kinase in expression of protein products that are important for generation of the biological effects of IFNs. We examined the expression of ISG15, a type I IFN inducible protein that plays an important role in ISGylation of proteins and generation of antiviral responses (32, 33). In addition, we studied the role of p85 in expression of CXCL10, a Type II IFN induced protein that plays a role in IFNγ-induced apoptosis and inhibition of viral replication (34). p85α+/+β+/+ and p85α-/-β-/- MEFs were treated with mouse IFNα or mouse IFNβ, and cell lysates were resolved by SDS-PAGE and immunoblotted with an anti ISG15 antibody. There was strong induction of ISG15 in p85α+/+β+/+ cells, but such induction was defective in p85α-/-β-/- cells (Fig. 3A and B), establishing a requirement for the PI 3 kinase in the expression of ISG15 protein. Similarly, the IFNγ-inducible expression of CXCL10 was defective in p85α-/-β-/- cells (Fig. 3C). To definitively establish the requirement of PI3 kinase in IFN-inducible expression of ISG15, experiments were performed in which ISG15 induction was compared in p85α-/-β-/- MEFs and p85α-/-β-/- MEFs, in which p85α or p85β were ectopically re-expressed. As shown in Fig. 4A, ectopic expression of p85α or p85β in p85α-/-β-/- cells, partially restored the ability of IFNα to induce ISG15 expression (Fig. 4A). Similarly, the ability of mouse IFNβ to induce ISG15 was also partially restored in cells complemented with p85α or p85β (Fig. 4B).
Figure 3. Type I or II IFN-induced expression of ISG15 and CXCL10 is defective in the absence of the p85α and p85β subunits of the PI3 kinase.
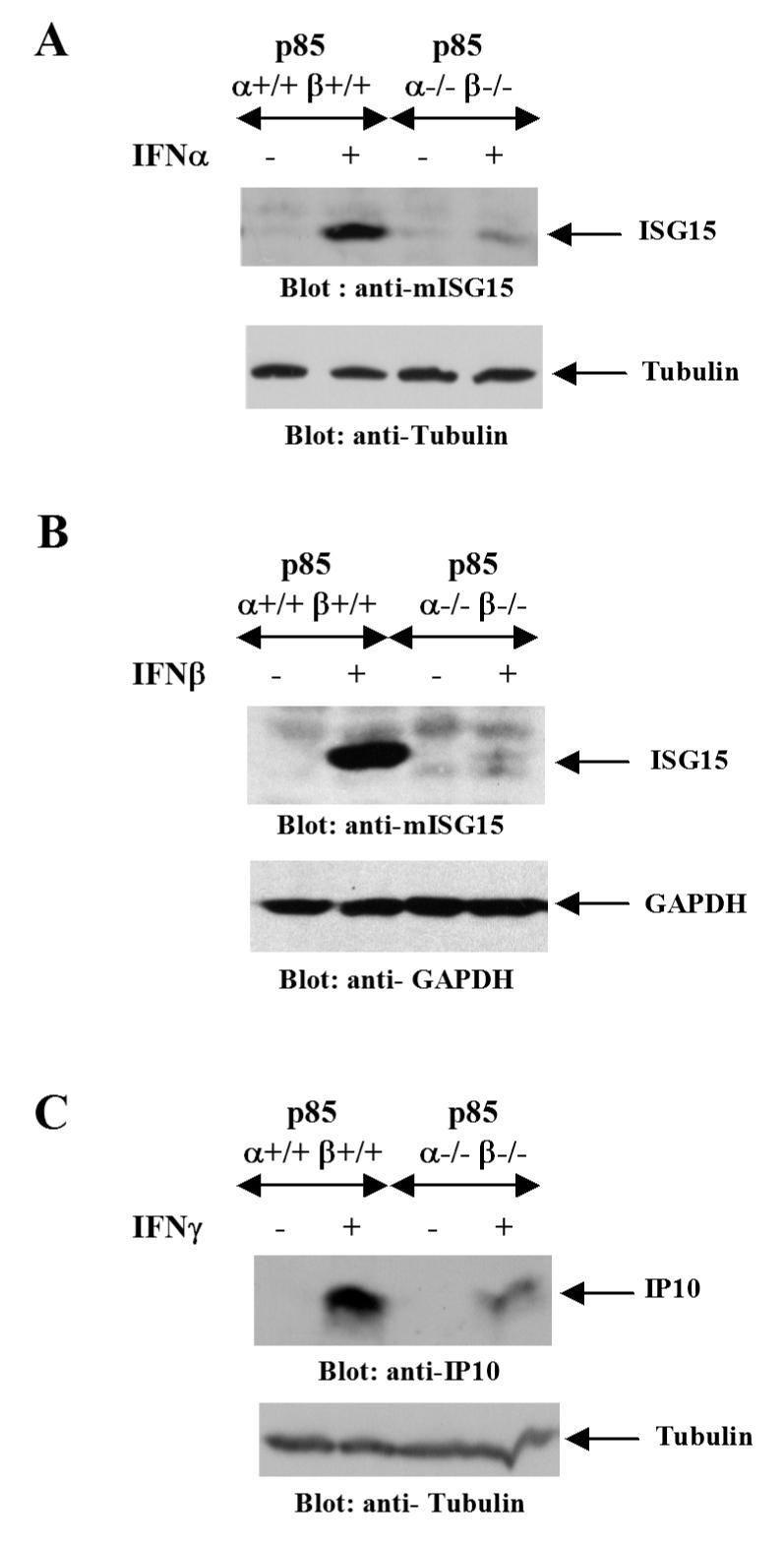
A. p85 α+/+β+/+ or p85 α-/-β-/- MEFs were treated with mouse IFNα for 48 hours and, after cell lysis, equal protein aliquots were resolved by SDS-PAGE and immunoblotted with an anti-ISG15 antibody (Top panel). The same blot was reprobed with anti-tubulin antibody to control for protein loading (Lower panel). B. p85 α+/+β+/+ or p85 α-/-β-/- MEFs were treated with mouse IFNβ for 24 hours and, after cell lysis, equal protein aliquots were resolved by SDS-PAGE and immunoblotted with an anti-ISG15 antibody (Top panel). The same blot was probed with an anti-GAPDH antibody as a control for loading (Lower panel). C. p85 α+/+β+/+ or p85 α-/-β-/- MEFs were treated with mouse IFNγ for 48 hours and, after cell lysis, equal protein aliquots were resolved by SDS-PAGE and immunoblotted with an anti-CXCL10 (IP10) antibody (Top panel). The same blot was reprobed with anti-tubulin antibody as a control for loading (Lower panel).
Figure 4. Restoration of ISG15 expression in p85α/p85β double knock-out MEFs by ectopic re-expression of the p85α or p85β.
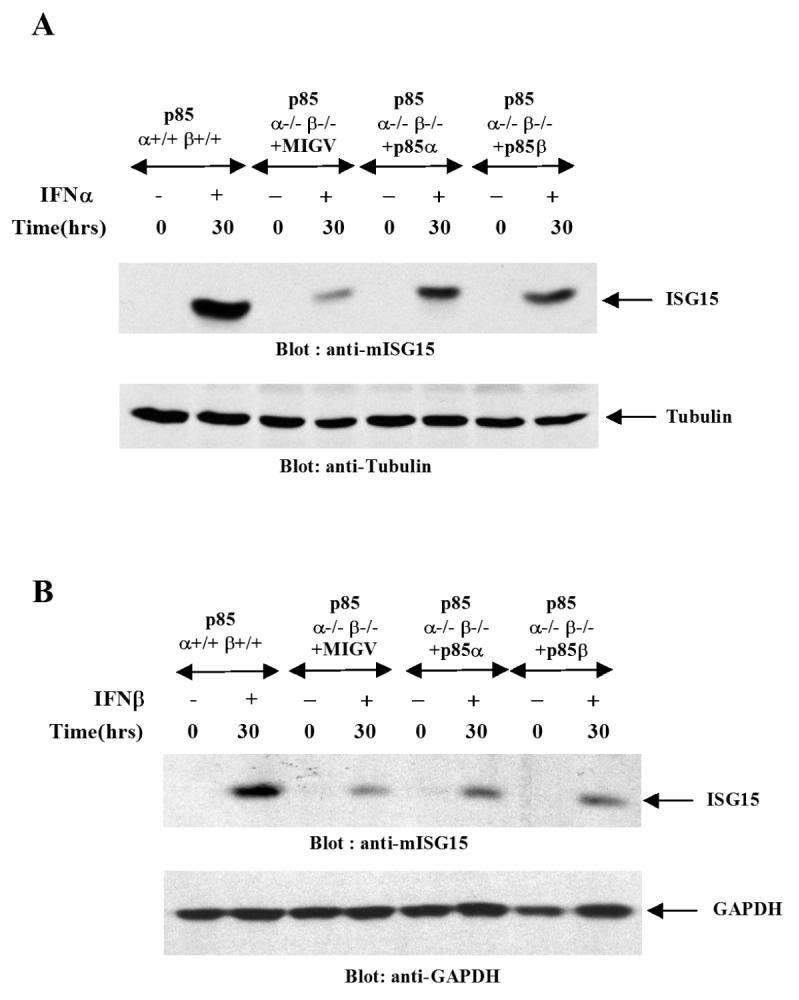
A. p85 α+/+β+/+ or p85 α-/-β-/- MEFs transduced with retroviral constructs encoding either p85α isoform (pMIG-p85α) or p85β isoform (pMIG-p85β) or with control vector alone (pMIG) were treated with mouse IFNα as indicated. Equal protein aliquots were resolved by SDS PAGE and immunoblotting was done with an anti- ISG15 antibody (Top panel). The blot was reprobed with anti-tubulin antibody to control for protein loading (Lower panel). B. p85 α+/+β+/+ or p85α-/-β-/- MEFs transduced with retroviral constructs encoding either the p85α isoform (pMIG-p85α) or p85β isoform (pMIG-p85β) or control vector alone (pMIG) were treated with mouse IFNβ as indicated. Equal protein aliquots were resolved by SDS PAGE and immunoblotted with an anti-ISG15 antibody (Top panel). The same blot was probed with anti- GAPDH antibody as a control for protein loading (Lower panel).
Altogether, our data established that PI3 kinase activity is essential for induction of Type I and II IFN-regulated gene products that play key roles in the generation of the biological effects of IFNs. To better understand the mechanisms by which such effects occur, we proceeded to determine whether such regulation occurs at the transcriptional or post-transcriptional levels, or both. We first examined whether there are differences between wild-type MEFs and double p85α/p85β knockouts in terms of IFN-inducible gene transcription via ISRE or GAS elements, in luciferase promoter assays. p85α+/+β+/+ or p85α-/-β-/- MEFs were transfected with an ISRE or 8x GAS luciferase constructs and after treatment with IFNα or IFNγ, luciferase activity was measured. As shown in Fig. 5, there was induction of luciferase activity in p85α+/+β+/+ MEFs in response to IFNα and IFNγ, but very little activity was seen in p85α-/-β-/- MEFs (Fig. 5A and B), suggesting that defective gene transcription may account, in part, for the decreased expression of the ISG15 or CXCL10 proteins. Consistent with this, when gene transcription of the Isg15 and/or Cxcl10 genes was directly assessed in p85α+/+β+/+ and p85α-/-β-/- MEFs, using real-time RT-PCR, there was a significant defect in such transcriptional activation in the knockout cells (Fig. 5C and D).
Figure 5. The function of the PI3 kinase is essential for Type I and II IFN-dependent gene transcription.
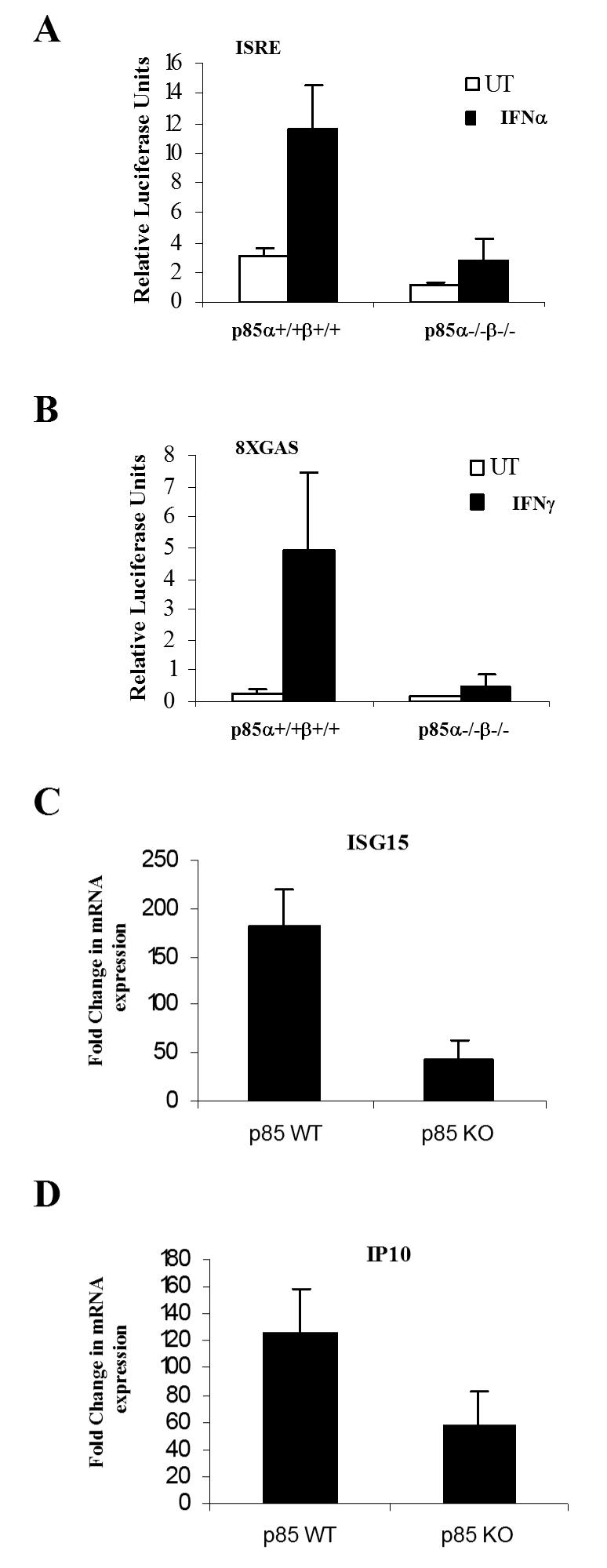
A-B. p85α+/+β+/+ and p85α-/-β-/- MEFs were transfected with a β-galactosidase expression vector and either an ISRE (A) or 8xGAS (B) luciferase plasmids. 48 hours after transfection, triplicate cultures were either left untreated or treated with IFNα (A) or IFNγ (B) for 6 hours, and luciferase reporter assays were carried out. The data are expressed as relative luciferase units for each condition, normalized for β-galactosidase activity. Data represent means ± S.E. values of two independent experiments for panels A and B. C. p85 α+/+β+/+ and p85 α-/-/β-/- MEFs were incubated for 6 hours at 37°C in the absence or presence of mouse IFNα. Expression of mRNA for the Isg15 gene was evaluated by quantitative RT-PCR (TaqMan). GAPDH was used for normalization. Data are expressed as -fold increase over untreated samples and represent means ± S.E. of 4 experiments. D. p85 α+/+β+/+ and p85 α-/-/β-/- MEFs were incubated for 6 hours at 37 °C in the absence or presence of mouse IFNγ. Expression of mRNA for the Cxcl10 gene was evaluated by quantitative RT-PCR (TaqMan). GAPDH was used for normalization. Data are expressed as -fold increase over IFNγ-untreated samples and represent means ± S.E. of 4 experiments.
Requirement of the PI 3 kinase for IFN-inducible mRNA translation and antiviral responses
In subsequent experiments, we sought to directly determine whether mRNA translation for IFN-sensitive genes is defective in cells with targeted disruption of the p85α and p85β genes. For this purpose, polysomes from mouse IFNα-treated p85α+/+β+/+ and p85α-/-β-/- MEFs were isolated. The polysomal profiles from wild-type or p85α/β knockout cells, prior to and after IFNα-treatment, are shown in Fig. 6A. There was an abundance of Isg15 mRNA in polysomes isolated from IFNα- treated p85α+/+β+/+ cells, but not p85α-/-β-/- cells (Fig. 6 B, C), establishing that mRNA translation for the Isg15 gene is defective in the absence of PI3 kinase activity. Similarly, the polysomal distribution of Irf7 mRNA in p85α+/+β+/+ versus p85α-/-β-/- MEF cells was also examined. This assessment was particularly relevant, as IRF7 plays an important role in antiviral and immune responses and is a regulator of IFN production in innate immune responses (35-37), while recent studies have shown that the 5′ UTR of Irf7 is highly structured and its translation is de-repressed in 4E-BP1 and 4E-BP2 knockout mice (37). There was strong induction of Irf7 mRNA in polysomes isolated from IFNα-treated p85α+/+ β+/+ cells but such an increase was not seen in p85α-/-β-/- cells (Fig. 6D). Similar results were also seen when p85α+/+β+/+ and p85α-/-β-/- MEFs were treated with mIFNγ and the abundance of Cxcl10 mRNA in polysomal fractions was evaluated. There was increased expression of Cxcl10 polysomal mRNA in p85α+/+β+/+ MEFs, but such an increase was not seen in p85α-/-β-/- MEFs (Fig. 7A-C). Altogether, these data established that mRNA translation of IFN-responsive genes is defective in cells with targeted disruption of both the p85α and p85β subunits of PI3 kinase, underscoring the relevance of the PI3 kinase pathway in the process.
Figure 6. Activation of the PI3 kinase is essential for Type I IFN-regulated mRNA translation of the Isg15 and Irf7 genes.

A. p85α+/+β+/+ and p85α-/-/β-/- MEFs were either left untreated or treated with mouse IFNα for 40 hours. Cell lysates were separated on 10-50% sucrose gradient and optical density (OD) at 254nm was recorded. The OD254 nm is shown as a function of gradient depth for each treatment. B-D. Polysomal fractions were collected as indicated in A and RNA was isolated. Subsequently, quantitative real time RT-PCR assays to determine Isg15 (B-C) or Irf7 (D) mRNA expression in polysomal fractions was carried out, using GAPDH (B and D) or Tubulin (C) for normalization. Data are expressed as fold increase over IFNα-untreated samples and represent means ± S.E. for 3 independent experiments.
Figure 7. Engagement of PI3 kinase is essential for Type II IFN-dependent mRNA translation of the Cxcl10 gene.

A. p85α+/+β+/+ and p85α-/-β-/- MEFs were either left untreated or treated with mIFNγ for 40 hours. Cell lysates were separated on 10-50% sucrose gradient and optical density (OD) at 254nm was recorded. The OD254 nm is shown as a function of gradient depth for each treatment. B-C. Polysomal fractions were collected as indicated in A and RNA was isolated. Subsequently, quantitative real time RT-PCR assays to determine Cxcl10 mRNA expression in polysomal fractions was carried out, using GAPDH (B) or Tubulin (C) for normalization. Data are expressed as fold increase over IFNγ-untreated samples and represent means ± S.E. for 2 independent experiments.
To directly determine whether such defective gene transcription and mRNA translation seen in p85α-/-β-/- MEFs has consequences in the generation of the antiviral effects of IFNα, the ability of IFNα to inhibit replication of encephalomyocarditis virus (EMCV) was assessed. As shown in Fig. 8, p85α+/+β+/+ MEFs were much more sensitive to the antiviral effects of IFNs, as compared to p85α-/-β-/- MEFs, establishing an important regulatory role for the PI3 kinase in control of IFN-generated antiviral responses.
Figure 8. Defective IFNα-dependent antiviral responses in cells with targeted disruption of the genes for the p85α and 85β regulatory subunits of the PI3 kinase.
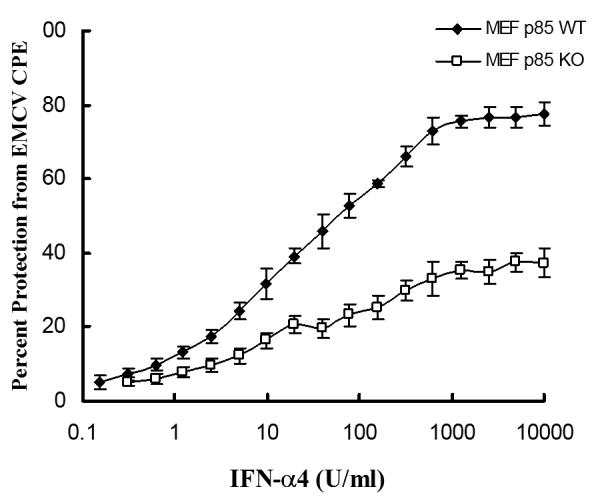
p85α+/+β+/+ and p85α-/-/β-/- MEFs were incubated in triplicate with the indicated doses of mouse IFNα. The cells were subsequently challenged with EMCV, and cytopathic effects (CPE) were quantified. Data are expressed as percent protection from CPE of EMCV.
Discussion
Beyond the classical Jak-Stat pathways, IFNs activate several other signaling cascades in normal and malignant cells. Such pathways either complement the function of Jak-Stat pathways or mediate IFN-dependent biological responses independently of them (4, 7, 38, 39). Among the alternate pathways activated by IFNs, the PI3 kinase pathway appears to play a prominent role in the generation of biological responses (16). After the original description of activation of the PI3 kinase pathway by IFNα (21), extensive work over the years has established important functions for PI3 kinase and its effectors in the generation of IFN-responses in various types of normal and malignant cells. These include regulatory effects on induction of apoptosis in neutrophils (40, 41), B lymphocytes (42) and astrocytes (43); and promotion of IFN-dependent monocyte adhesion (44). In addition, there is recent evidence that the PI3 kinase can mediate pro-apoptotic signals in multiple myeloma cells (45). Other recent work has shown that PI 3K activity is involved in IFNγ-dependent regulation of enteric epithelial permeability (46), as well as Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells (47). Interestingly, recent studies have also demonstrated that the function of the PI 3 kinase is important for nuclear translocation of IRF7 and Type I IFN production in response to TLR activation of pre-dendritic cells (48).
Despite the emerging evidence supporting a key role for the PI3K in IFN-signaling, the precise mechanisms by which this kinase regulates IFN-responses remain to be defined. Previous work in the Type II IFN system using pharmacological inhibitors has suggested that activation of the PI3K pathway may be required for IFNγ-dependent gene transcription, possibly by modifying serine phosphorylation of Stat1 (49). Other earlier studies have also shown that that the activation of this pathway by the Type I IFN receptor occurs independently of activation of Stat-pathways (50), and it is mediated by the engagement of insulin receptor substrate (IRS) proteins, IRS1 and IRS2 (21-23). Recent evidence has shown that this pathway regulates phosphorylation/ activation of the p70 S6 kinase and downstream phosphorylation of the S6 ribosomal protein, in response to both Type I and II IFNs (24, 25), raising the possibility that it regulates pathways that control initiation of mRNA translation. However, any potential regulatory effects of the PI3K on IFN-dependent mRNA translation had not been directly established, while the potential sequence of signaling events leading to such responses have not been clarified.
In the present study we provide direct evidence that activation of the PI3 kinase by the Type I and II IFN receptors plays critical and essential roles in the regulation of mRNA translation by IFNs. In studies using knockout cells for both the α and β isoforms of the p85 regulatory subunit of the PI3K, we found that the IFNα- or IFNγ- inducible phosphorylation/ activation of Akt requires upstream activation of the PI3 kinase. Similar to results from recent studies using Akt1/Akt2 double knockout cells, in which we demonstrated that Akt activation is essential for the IFN-activation of mTOR and its effectors (28), the current findings support a mechanism by which PI3K may control mTOR activation and mRNA translation in response to IFNs. We found that expression of key IFN-inducible proteins, that regulate the biological effects of IFNs, such as ISG15 and CXCL10, are defective in the absence of the regulatory subunits of the PI3 kinase. Importantly, we were able to directly demonstrate that mRNA translation, as assessed in polysomal mRNA fractions, for the Isg15, Irf7 and Cxcl10 genes is defective in cells lacking p85α and p85β. Thus, there is an essential requirement for PI3 kinase activity in the initiation of mRNA translation by both Type I and II IFNs.
Our data also demonstrated that there is defective transcription of IFN-stimulated genes in PI3 kinase knockout cells, as shown both by luciferase reporter assays and in real-time RT-PCR experiments directly examining transcription of the Isg15 and CxCl10 genes. Interestingly, although the effects of PI3K on IFN-dependent mRNA translation are mediated by Akt activation and downstream engagement of mTOR, the effects on transcription apparently involve an Akt-independent mechanism. This is shown by our previous studies that have demonstrated that IFNα- or IFNγ-dependent gene transcription is intact in Akt1/2 double knockout cells (28). Although the precise downstream signals that mediate such responses remain to be identified, it is possible that they are mediated by activation of different PKC-isoforms that may regulate gene transcription by distinct mechanisms (51, 52, 53), or via engagement of other Akt-independent signals. Beyond members of the PKC-family proteins, another group of kinases known to be regulated in an Akt-independent manner downstream of PI3K includes members of the Tec family of proteins (Btk, Itk and Rlk) (4). Interestingly, a member of this family of proteins, Bmx, has been previously shown to induce activation of the Stat-pathway (54), while recent studies in prostate cells suggest that it is also induced in a PI3K-dependent manner (55). Future studies to determine a potential involvement of Tec kinases and/or other elements in the control of IFN-dependent transcriptional activation will be of interest and may provide additional insights on the regulatory effects of PI3K in IFN-signaling.
Our studies also provide direct and definitive evidence establishing that the PI3K plays an essential role in the generation of the antiviral effects of IFNα, as shown by the defective induction of antiviral responses against EMCV in p85α/β double knockout cells. Interestingly, another study in which pharmacological inhibitors were used in multiple myeloma cells, suggested that PI3K regulates transcription of a subset of IFNα-stimulated genes that may be involved in the induction of apoptosis (56), although this needs to be confirmed in studies involving genetic approaches. Altogether, our work establishes that PI3K plays dual regulatory roles in IFN-signaling, controlling both transcription and mRNA translation of ISGs, and that such function is essential for the induction of the antiviral effects of IFNs in vitro and, possibly, in vivo. Interestingly, it is well established that in response to growth factors and other mitogenic signals, the PI 3′ kinase pathway mediates cell proliferative and/or anti-apoptotic responses (20, 57). The precise mechanisms that determine the specificity of signals during engagement of this pathway by IFNs remains to be established. It is possible that IFNs compete with growth factors for the use of the same signaling elements required for initiation of cap-dependent translation (i.e. inactivation of 4E-BP1 and subsequent activation of the eIF4E complex) downstream of activation of the PI 3K/Akt pathway. It is also likely that initiation of IFN-dependent gene transcription for specific ISGs and the resulting influx of messages for mRNA translation, accompanied by the timely IFN-dependent activation of the mTOR pathway, results in mRNA translation of specific gene products whose transcription is regulated by IFNs. Our studies provide some evidence for the existence of such a mechanism, but additional work will be required to definitively establish the validity of such a hypothesis in the future.
Footnotes
This work was supported by NIH grants CA77816, CA100579, CA121192, AG029138 and HL082946, a Merit Review Grant from the Department of Veterans Affairs, and Canadian Institutes of Health Research grant MOP 15094.
References
- 1.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu. Rev. Biochem. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 2.Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1420. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 3.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 4.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 5.Darnell JE. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 6.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 7.van Boxel-Dezaire AH, Rani MR, Stark GR. A collaborative study on larval excretory/secretory antigens of Toxocara canis for the immunodiagnosis of human toxocariasis with ELISA. Immunity. 2006;25:361–372. [Google Scholar]
- 8.Parmar S, Platanias LC. Interferons: mechanisms of action and clinical applications. Curr. Opin. Oncol. 2003;15:431–439. doi: 10.1097/00001622-200311000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Javed A, Reder AT. Therapeutic role of beta-interferons in multiple sclerosis. Pharmacol. Ther. 2006;110:35–56. doi: 10.1016/j.pharmthera.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Loutfy MR, Blatt LM, Siminovitch KA, Ward S, Wolff B, Lho H, Pham DH, Deif H, LaMere EA, Chang M, Kain KC, Farcas GA, Ferguson P, Latchford M, Levy G, Dennis JW, Lai EK, Fish EN. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA. 2003;290:3222–3228. doi: 10.1001/jama.290.24.3222. [DOI] [PubMed] [Google Scholar]
- 11.Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, Young PR, Fish EN, Platanias LC. Activation of the p38 mitogenactivated protein kinase by type I interferons. J. Biol. Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Sassano A, Majchrzak B, Deb DK, Levy DE, Gaestel M, Nebreda AR, Fish EN, Platanias LC. Role of p38alpha Map kinase in Type I interferon signaling. J. Biol. Chem. 2004;279:970–979. doi: 10.1074/jbc.M309927200. [DOI] [PubMed] [Google Scholar]
- 13.Mayer IA, Verma A, Grumbach IM, Uddin S, Lekmine F, Ravandi F, Majchrzak B, Fujita S, Fish EN, Platanias LC. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J. Biol. Chem. 2001;276:28570–28577. doi: 10.1074/jbc.M011685200. [DOI] [PubMed] [Google Scholar]
- 14.Verma A, Deb DK, Sassano A, Kambhampati S, Wickrema A, Uddin S, Mohindru M, Van Besien K, Platanias LC. Cutting edge: activation of the p38 mitogen-activated protein kinase signaling pathway mediates cytokine-induced hemopoietic suppression in aplastic anemia. J. Immunol. 2002;168:5984–5988. doi: 10.4049/jimmunol.168.12.5984. [DOI] [PubMed] [Google Scholar]
- 15.Katsoulidis E, Li Y, Yoon P, Sassano A, Altman J, Kannan-Thulasiraman P, Balasubramanian L, Parmar S, Varga J, Tallman MS, Verma A, Platanias LC. Role of the p38 mitogen-activated protein kinase pathway in cytokine-mediated hematopoietic suppression in myelodysplastic syndromes. Cancer Res. 2005;65:9029–9037. doi: 10.1158/0008-5472.CAN-04-4555. [DOI] [PubMed] [Google Scholar]
- 16.Kaur S, Uddin S, Platanias LC. The PI3′ kinase pathway in interferon signaling. J. Interferon Cytokine Res. 2005;12:780–787. doi: 10.1089/jir.2005.25.780. [DOI] [PubMed] [Google Scholar]
- 17.Kaur S, Parmar S, Smith J, Katsoulidis E, Li Y, Sassano A, Majchrzak B, Uddin S, Tallman MS, Fish EN, Platanias LC. Role of protein kinase C-delta (PKC-delta) in the generation of the effects of IFN-alpha in chronic myelogenous leukemia cells. Exp. Hematol. 2005;33:550–557. doi: 10.1016/j.exphem.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 18.Uddin S, Majchrzak B, Wang PC, Modi S, Khan MK, Fish EN, Platanias LC. Interferon-dependent activation of the serine kinase PI 3′-kinase requires engagement of the IRS pathway but not the Stat pathway. Biochem. Biophys. Res. Commun. 2000;270:158–162. doi: 10.1006/bbrc.2000.2402. [DOI] [PubMed] [Google Scholar]
- 19.Uddin S, Fish EN, Sher DA, Gardziola C, White MF, Platanias LC. Activation of the phosphatidylinositol 3-kinase serine kinase by IFN-alpha. J. Immunol. 1997;158:2390–2397. [PubMed] [Google Scholar]
- 20.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 21.Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3′-kinase. J. Biol. Chem. 1995;270:15938–15941. doi: 10.1074/jbc.270.27.15938. [DOI] [PubMed] [Google Scholar]
- 22.Platanias LC, Uddin S, Yetter A, Sun XJ, White MF. The type I interferon receptor mediates tyrosine phosphorylation of insulin receptor substrate 2. J. Biol. Chem. 1996;271:278–282. doi: 10.1074/jbc.271.1.278. [DOI] [PubMed] [Google Scholar]
- 23.Burfoot MS, Rogers NC, Watling D, Smith JM, Pons S, Paonessaw G, Pellegrini S, White MF, Kerr IM. Janus kinase-dependent activation of insulin receptor substrate 1 in response to interleukin-4, oncostatin M, and the interferons. J. Biol. Chem. 1997;272:24183–24190. doi: 10.1074/jbc.272.39.24183. [DOI] [PubMed] [Google Scholar]
- 24.Lekmine F, Uddin S, Sassano A, Parmar S, Brachmann SM, Majchrzak B, Sonenberg N, Hay N, Fish EN, Platanias LC. Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J. Biol. Chem. 2003;278:27772–27780. doi: 10.1074/jbc.M301364200. [DOI] [PubMed] [Google Scholar]
- 25.Lekmine F, Sassano A, Uddin S, Smith J, Majchrzak B, Brachmann SM, Hay N, Fish EN, Platanias LC. Interferon-gamma engages the p70 S6 kinase to regulate phosphorylation of the 40S S6 ribosomal protein. Exp. Cell Res. 2004;295:173–182. doi: 10.1016/j.yexcr.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 26.Kaur S, Lal L, Sassano A, Majchrzak-Kita B, Srikanth M, Baker DP, Petroulakis E, Hay N, Sonenberg N, Fish EN, Platanias LC. Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J. Biol. Chem. 2007;282:1757–1768. doi: 10.1074/jbc.M607365200. [DOI] [PubMed] [Google Scholar]
- 27.Brachmann SM, Yballe CM, Innocenti M, Deane JA, Fruman DA, Thomas SM, Cantley LC. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol Cell Biol. 2005;25:2593–2606. doi: 10.1128/MCB.25.7.2593-2606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur S, Sassano A, Dolniak B, Joshi S, Majchrzak-Kita B, Baker DP, Hay N, Fish EN, Platanias LC. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc. Natl. Acad. Sci. U.S.A. 2008;105:4808–4813. doi: 10.1073/pnas.0710907105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, Young PR, Fish EN, Platanias LC. Activation of the p38 mitogenactivated protein kinase by type I interferons. J. Biol. Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 30.Horvai AE, Xu L, Korzus E, Brard G, Kalafus D, Mullen T-M, Rose DW, Rosenfeld MG, Glass CK. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl. Acad. Sci. U.S.A. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 2004;10:1374–1378. doi: 10.1038/nm1133. [DOI] [PubMed] [Google Scholar]
- 33.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, García-Sastre A, Leib DA, Pekosz A, Knobeloch KP, Horak I, Virgin HW., 4th IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang HM, Yuan J, Cheung P, Chau D, Wong BW, McManus BM, Yang D. Gamma interferon-inducible protein 10 induces HeLa cell apoptosis through a p53-dependent pathway initiated by suppression of human papillomavirus type 18 E6 and E7 expression. Mol. Cell Biol. 2005;25:6247–6258. doi: 10.1128/MCB.25.14.6247-6258.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 36.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 37.Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N. Translational control of the innate immune response through IRF-7. Nature. 2008;452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 38.Platanias LC. Introduction: interferon signals: what is classical and what is nonclassical? J. Interferon Cytokine Res. 2005;25:732. doi: 10.1089/jir.2005.25.732. [DOI] [PubMed] [Google Scholar]
- 39.Platanias LC, Fish EN. Signaling pathways activated by interferons. Exp. Hematol. 1999;27:1583–1592. doi: 10.1016/s0301-472x(99)00109-5. [DOI] [PubMed] [Google Scholar]
- 40.Wang K, Scheel-Toellner D, Wong SH, Craddock R, Caamano J, Akbar AN, Salmon M, Lord JM. Inhibition of neutrophil apoptosis by type 1 IFN depends on cross-talk between phosphoinositol 3-kinase, protein kinase C-delta, and NF-kappa B signaling pathways. J. Immunol. 2003;171:1035–1041. doi: 10.4049/jimmunol.171.2.1035. [DOI] [PubMed] [Google Scholar]
- 41.Yang CH, Murti A, Pfeffer SR, Kim JG, Donner DB, Pfeffer LM. Interferon alpha /beta promotes cell survival by activating nuclear factor kappa B through phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 2001;276:13756–13761. doi: 10.1074/jbc.M011006200. [DOI] [PubMed] [Google Scholar]
- 42.Ruth K, Carlsson L, Hallberg B, Lundgren E. Interferon-alpha promotes survival of human primary B-lymphocytes via phosphatidylinositol 3-kinase. Biochem. Biophys. Res. Commun. 2001;284:583–586. doi: 10.1006/bbrc.2001.5025. [DOI] [PubMed] [Google Scholar]
- 43.Barca O, Ferre S, Seoane M, Prieto JM, Lema M, Senaris R, Arce VM. Interferon beta promotes survival in primary astrocytes through phosphatidylinositol 3-kinase. J. Neuroimmunol. 2003;139:155–159. doi: 10.1016/s0165-5728(03)00160-7. [DOI] [PubMed] [Google Scholar]
- 44.Navarro A, Anand-Apte B, Tanabe Y, Feldman G, Larner AC. A PI-3 kinase-dependent, Stat1-independent signaling pathway regulates interferon-stimulated monocyte adhesion. J. Leukoc. Biol. 2003;73:540–545. doi: 10.1189/jlb.1002508. [DOI] [PubMed] [Google Scholar]
- 45.Thyrell L, Hjortsberg L, Arulampalam V, Panaretakis T, Uhles S, Dagnell M, Zhivotovsky B, Leibiger I, Grander D, Pokrovskaja K. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J. Biol. Chem. 2004;279:24152–24162. doi: 10.1074/jbc.M312219200. [DOI] [PubMed] [Google Scholar]
- 46.McKay DM, Watson JL, Wang A, Caldwell J, Prescott D, Ceponis PM, Di Leo V, Lu J. Phosphatidylinositol 3′-kinase is a critical mediator of interferon-gamma-induced increases in enteric epithelial permeability. J. Pharmacol. Exp. Ther. 2007;320:1013–1022. doi: 10.1124/jpet.106.113639. [DOI] [PubMed] [Google Scholar]
- 47.Rosner D, Stoneman V, Littlewood T, McCarthy N, Figg N, Wang Y, Tellides G, Bennett M. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am. J. Pathol. 2006;168:2054–2063. doi: 10.2353/ajpath.2006.050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guiducci C, Ghirelli C, Marloie-Provost MA, Matray T, Coffman RL, Liu YJ, Barrat FJ, Soumelis V. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J. Exp. Med. 2008;205:315–322. doi: 10.1084/jem.20070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen H, Ramana CV, Bayes J, Stark GR. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J. Biol. Chem. 2001;276:33361–33318. doi: 10.1074/jbc.M105070200. [DOI] [PubMed] [Google Scholar]
- 50.Uddin S, Fish EN, Sher D, Gardziola C, Colamonici OR, Kellum M, Pitha PM, White MF, Platanias LC. The IRS-pathway operates distinctively from the Stat-pathway in hematopoietic cells and transduces common and distinct signals during engagement of the insulin or interferon-alpha receptors. Blood. 1997;90:2574–2582. [PubMed] [Google Scholar]
- 51.Uddin S, Sassano A, Deb DK, Verma A, Majchrzak B, Rahman A, Malik AB, Fish EN, Platanias LC. Protein kinase C-delta (PKC-delta) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 2002;277:14408–14416. doi: 10.1074/jbc.M109671200. [DOI] [PubMed] [Google Scholar]
- 52.Deb DK, Sassano A, Lekmine F, Majchrzak B, Verma A, Kambhampati S, Uddin S, Rahman A, Fish EN, Platanias LC. Activation of protein kinase C delta by IFN-gamma. J. Immunol. 2003;171:267–273. doi: 10.4049/jimmunol.171.1.267. [DOI] [PubMed] [Google Scholar]
- 53.Choudhury GG. A linear signal transduction pathway involving phosphatidylinositol 3-kinase, protein kinase Cepsilon, and MAPK in mesangial cells regulates interferon-gamma-induced STAT1alpha transcriptional activation. J. Biol. Chem. 2004;279:27399–27409. doi: 10.1074/jbc.M403530200. [DOI] [PubMed] [Google Scholar]
- 54.Saharinen P, Ekman N, Sarvas K, Parker P, Alitalo K, Silvennoinen O. The Bmx tyrosine kinase induces activation of the Stat signaling pathway, which is specifically inhibited by protein kinase Cdelta. Blood. 1997;90:4341–4353. [PubMed] [Google Scholar]
- 55.Jiang X, Borgesi RA, McKnight NC, Kaur R, Carpenter CL, Balk SP. Activation of nonreceptor tyrosine kinase Bmx/Etk mediated by phosphoinositide 3-kinase, epidermal growth factor receptor, and ErbB3 in prostate cancer cells. J. Biol. Chem. 2007;282:32689–32698. doi: 10.1074/jbc.M703412200. [DOI] [PubMed] [Google Scholar]
- 56.Hjortsberg L, Lindvall C, Corcoran M, Arulampalam V, Chan D, Thyrell L, Nordenskjold M, Grandér D, Pokrovskaja K. Phosphoinositide 3 kinase regulates a subset of interferon-alpha-stimulated genes. Exp. Cell Res. 2007;313:404–414. doi: 10.1016/j.yexcr.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 57.Kharas MG, Fruman DA. ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005;65:2047–2053. doi: 10.1158/0008-5472.CAN-04-3888. [DOI] [PubMed] [Google Scholar]