

Abstract
Isoform-selective agonists and antagonists of the lysophosphatidic acid (LPA) G protein coupled receptors (GPCRs) have important potential applications in cell biology and therapy. LPA GPCRs regulate cancer cell proliferation, invasion, angiogenesis, and also biochemical resistance to chemotherapy- and radiotherapy-induced apoptosis. LPA and its analogues also are feedback inhibitors of the enzyme lysophospholipase D (lysoPLD, a.k.a., autotaxin, ATX), a central regulator of invasion and metastasis. For cancer therapy, the optimal therapeutic profile would be a metabolically stabilized, pan-LPA receptor antagonist that also inhibited lysoPLD. For protection of gastrointestinal mucosa and lymphocytes, LPA agonists would be desirable to minimize or reverse radiation or chemical-induced injury. Analogues of lysophosphatidic acid (LPA) that are chemically modified to be less susceptible to phospholipases and phosphatases show activity as long-lived receptor-specific agonists and antagonists for LPA receptors, as well as inhibitors for the lysoPLD activity of ATX.
Keywords: phosphorothioate, methylene phosphonate, aminooxy, autotaxin, lysophospholipase D inhibitor, receptor isoform selectivity, GPCR antagonist, tumor regression
LPA: Small but Powerful
Among the bounty of phospholipids involved in cell signaling, lysophosphatidic acid (LPA) plays an unusually diverse set of important roles. Though chemically simple, LPA (1- or 2-radyl-sn-glycerol 3-phosphate) elicits a rich array of biological responses, including platelet aggregation, promotion of cell survival, and cell migration [1, 2]. LPA signaling regulates infertility [3, 4] as well as cancer cell proliferation, invasion, angiogenesis, and biochemical resistance to chemotherapy and radiotherapy-induced apoptosis [5, 6]. One mechanism of enhanced tumor cell invasion by LPA is the receptor-mediated activation of the Rho and Rac GTPase pathways, known to be essential for the regulation of the actin cytoskeleton and cell motility [7]. A second pathway is the regulation of matrix metalloproteinase activity by LPA, a key process in metastasis [8]. In addition to cancer, there is an ever expanding series of medicinal chemistry targets for lysolipid signaling [9]. Most of the known effects of LPA are transduced via three G protein-coupled receptors (GPCRs): LPA1, LPA2, LPA3, also known as EDG2, EDG4 and EDG7, respectively [10]. Recently three purinergic cluster GPCRs, LPA4/GPR23 [11], LPA5/GPR92 [12, 13], and LPA4/GPR87 [14] as well as the nuclear transcription factor PPARγ, have been reported to interact with LPA [15], but the biological roles of these receptors remain unclear. The three canonical receptors appear to have a unique structure-activity relationship for acyl groups, backbone, and phosphate analogues [16, 17]. Short and long chain fatty alcohol phosphates also activate the EDG receptors [18, 19].
LPA is produced extracellularly from lysophosphatidylcholine by the lysophospholipase D (lysoPLD) activity of autotaxin (ATX) [4, 20]. Even before its lysoPLD activity was known, ATX was known to be a metastasis-enhancing motogen with potent angiogenic effects in tumors [21]. Thus, in addition to a normal functional role in stabilizing blood vessels during embryonic development [22], local production of LPA by ATX/lysoPLD could support the invasion of tumor cells, promoting metastasis [1]. ATX is feedback inhibited by LPA [23], and the lysoPLD activity can be readily assessed in a high-throughput mode using a fluorogenic substrate [24]. For cancer therapy, therefore, both LPA receptor antagonists and ATX inhibitors have strong potential, by both blocking the growth-supporting and anti-apoptotic effects of LPA and reducing its titer. Indeed, compounds that are simultaneously pan-antagonists and ATX-inhibitors, such as analogues described below, are noteworthy dual-effect candidates for drug development [25].
We previously reviewed a wide variety of metabolically-stabilized LPA and PA analogues [26] with partial selectivity for individual LPA GPCRs and PPARγ [15, 27-32]. This overview will highlight the most recently synthesized analogues and their corresponding biological activities. Specifically, we will focus on four classes of compounds. First, we describe the cyclic carba LPA (ccPA) analogues, which have structure-dependent agonist and antagonist properties, and are also potent ATX inhibitors, Second, we describe the α-hydroxymethylene LPA phosphonate analogues with structure-dependent agonist properties, and the α-halomethylene LPA phosphonate analogues, the majority of which act as pan-LPA GPCR receptor antagonist and potent autotaxin inhibitors [33]. Third, we show that the aminooxy-LPA (AO-LPA) analogues are surprisingly potent, partially selective LPAR agonists. Fourth, we outline the alkoxymethylene phosphonates (MP-LPA) analogues, which exhibit remarkably LPA2-specific agonist effects. Before 2005, none of phosphonate analogues of LPA or PA retained the 3-oxygen functionality of the glyceryl backbone; instead, that oxygen had been replaced with methylene, fluoromethylene, and difluoromethylene moieties [26]. Our efforts to obtain “phosphate-free” stabilized analogues by preparing neutral, phosphomimetic sulfonamide and sulfonamidoxy analogues of LPA, also failed to give a biologically active agent [34].
Metabolically stabilized LPA analogues
The catabolism of LPA proceeds via three main pathways [35]: (a) lipid phosphate phosphatase enzymes hydrolyze the phosphomonoester to monoacylglycerol; (b) acyltransferases esterify LPA to PA; and (c) LPA-specific lysophospholipases hydrolyze the sn-1 acyl chain to form the corresponding glycerol phosphate. In addition, a nonenzymatic intramolecular acyl chain migration can occur that interconverts the sn-1- and sn-2-acyl species [28]. Earlier, we surveyed the structure-activity relationships (SARs) of a wide array of LPA isosteres [26] in which labile groups have been chemically stabilized. Successful substitutions included replacement of the sn-1 and sn-2 hydroxyl groups with fluorine, methoxy or 2-hydroxyethoxy groups, and replacement of the acyl esters with alkyl ethers [26]. The phosphomimetics include methylene (-CH2-) phosphonates, α-X methylene phosphonates (where X = -CHF-, CHBr-, or -CHOH-), phosphorothioates [32, 36], phosphonothioates, methyl phosphonates and monofluoromethyl phosphonates [26, 33]. In several studies of receptor activation, the activities of phosphomimetic LPA analogues were correlated with the second pKa values [26, 31]. We have now focused our medicinal chemistry efforts on four types of phosphomimetic functional groups that may display higher affinity for LPA receptors, either as agonists or antagonists: α-X methylene phosphonates, alkoxymethylene phosphonates, phosphonothioate and the phosphorothioate. Importantly, two of these three phosphomimetics have the same value of second pKa as phosphate [26].
In this review we will focus on results from 2005-2008, with an emphasis on the biological evaluation of the most potent LPAR agonists, antagonists, and enzyme inhibitors. It will become clear that targeting the LPA signaling pathway may indeed offer therapeutic opportunities for both agonists (radioprotection and wound healing) and antagonists (treatment of metastatic tumors).
Cyclic carba-LPA analogues
The sn-1,2-cyclic phosphate derivative of LPA, commonly known as “cyclic PA (cPA)” affects numerous cellular functions, including anti-mitogenic regulation of the cell cycle, regulation of actin stress fiber formation and rearrangement [37], and inhibition of cancer cells invasion and metastasis [38]. Recently, a meticulous structure-function study of 2-carbacyclic and 3-carbacyclic analogues of cPA (ccPA) showed that both regioisomers inhibited cancer cell invasion and metastasis. Moreover, both isomers were potent, selective inhibitors of the lysophospholipase D activity of ATX but lacked detectable agonist activity for LPA1,2,3 EDG-family receptors [39] . We independently explored the SAR of the 3-carba ccPA compounds, in particular preparing the pKa-matched phosphonothioate and fluoromethylene phosphonate analogues of ccPA [40]. Figure 1 summarizes the pharmacology of these ccPA analogues, which showed subtype-specific agonist and antagonist activity in Rh 7777 or CHO cells expressing the individual EDG-family LPA receptors. Among the oleoyl derivatives, the monofluoromethylene phosphonate and phosphonothionate ccPA analogues were both modest LPA1/3 antagonists. In contrast, the difluoromethylene phosphonate ccPA analogue and ccPA itself showed weak or no LPAR agonist activity [40].
Figure 1.

Structure-activity relationships for cyclic carba LPA analogues. Structures, receptor activation pharmacology, and ATX inhibition of analogues of ccPA. Adapted from Ref. 40.
Figure 2 illustrates the difference between the acyclic α-fluoro and α,α-difluoromethylene phosphonates and the corresponding cyclic ccPA analogues. These differ only by the formation of a cyclic phosphonate ester onto the sn-2 hydroxyl group. Both the 18:1 LPA itself and the 18:1 LPA phosphonate itself are micromolar pan-agonists for LPA1-4 [33], while the corresponding 18:1 ccPA analogue has no agonist or antagonist activity below 10 μM. The acyclic α-fluoromethylene phosphonate was a potent LPA3 agonist, while the cyclic α-fluoromethylene phosphonate was a selective LPA1/3 antagonist [26, 31]. Upon cyclization, the inactive acyclic α,α-difluoromethylene phosphonate became a weak LPA1/2/3 agonist.
Figure 2.

Different pharmacologies for cyclic and acyclic LPA analogues with comparable structures
LPA1 and LPA3 may contribute to the pathogenesis of rheumatoid arthritis (RA), through the modulation of fibroblast-like synoviocytes (FLS) migration and cytokine production [41]. In FLS isolated from synovial tissues of patients with RA, exogenously applied LPA induced FLS migration and secretion of IL-8/IL-6, whereas the phosphorothioate LPA3 agonist OMPT [32, 36] stimulated cytokine synthesis but not cell motility. The LPA-induced motility of FLS and cytokine production were both suppressed by LPA1/3 receptor antagonists. ATX was also abundant in the synovial fluid of RA patients, and synovial fluid-induced cell motility was reduced by compounds with LPA1/3 receptor antagonist activity and/or ATX-inhibitory activity, such as the phosphonothioate analogue of ccPA (ATX inhibition only) as well as the α-bromomethylene phosphonate (both ATX inhibitor and pan-LPA antagonist [41].
Phosphonate LPA analogues -- the importance being alpha
Previously, we stabilized LPA analogues by substitution of the bridging oxygen in the monophosphate by either a α-monofluoromethylene (-CHF-) moiety or an α,α-difluoromethylene (-CF2-) group [31]. As predicted from the hypothesis of matching the second pKa value [26], only the α-monofluoromethylene analogue was an LPAR agonist. Indeed, the α-monofluoromethylene analogue was a potent agonist for LPA3 but not for LPA1 or LPA2. Consistent with the design of these phosphonates to resist degradation by lipid phosphate phosphatases, the α-monofluoromethylene analogue showed a half-life of 17 h when co-incubated with cultured cells, compared to less than 30 min for 1-oleoyl-LPA. [31].
We next synthesized of a more complete series of α-substituted methylene phosphonate analogues of LPA [33]. Each of these analogues contained a hydrolysis-resistant phosphonate mimic of the labile monophosphate of natural LPA, and we prepared both the palmitoyl and oleoyl derivatives of each analogue. The pharmacological properties of the palmitoyl α-X methylene phosphonate analogues (Figure 3) were characterized in terms of LPA receptor subtype-specific agonist and antagonist activity in RH7777 and CHO cells expressing the individual LPA GPCRs. In particular, the parent unsubstituted phosphonate LPA analogue (X = H) was a weak antagonist for LPA1/2/3, but a weak agonist for LPA4. Most importantly, the α-bromomethylene (X = Br) and α-chloromethylene (X = Cl) phosphonates showed pan-LPA receptor subtype antagonist activity. These α-X analogues were initially prepared and tested as diastereomeric mixtures at the α-carbon, but separation and individual testing of the α-hydroxymethylene phosphonate analogues revealed little pharmacological difference between the two single diastereoisomers. Importantly, the α-bromomethylene phosphonates (BrP-LPA) were the first reported antagonists for the LPA4 GPCR. Also important to the utility of these antagonists in cancer therapy, each of the α-substituted methylene phosphonates inhibited the lysoPLD activity of ATX. Finally, unlike many LPA analogues, none of these compounds activated the intracellular LPA receptor PPARγ [33].
Figure 3.

Structure-activity relationships for α-X methylene phosphonate palmitoyl LPA analogues. Adapted from Ref. 33.
In vitro chemotaxis studies, addressing the effects of LPA/ccPA analogues on invasiveness, were performed to evaluate the anti-metastatic potential of the phosphonothioate ccPA (thio-ccPA) and α-bromomethylene phosphonate LPA (BrP-LPA) analogues (Figure 4) (M. Serban, unpublished results). LPA1 is the most important GPCR mediating cell motility and invasion of normal and neoplastic cells [42]. Transformed NIH 3T3 cells expressing ATX and ras were used for this study [21, 43]. For the assay, 24-well Transwell plates with inserts (8 μm membrane pore size) pre-coated with Matrigel (0.35 mg/ml) were used. The culture medium in the lower wells was augmented with 10% fetal bovine serum as the chemoattractant. In the inserts, cells (5 × 105 cells/ml) were plated in serum free conditioned medium (±10 μM of LPA or analogues) and incubated for 24 h. Two inserts per treatment were then processed by staining the insert membranes and counting the migrated cells in five distinct fields/insert at 400X magnification. When treated with BrP-LPA or ccPA, invasiveness of the NIH 3T3 ras ATX cells decreased to 40% and 36 %, respectively, relative to the untreated controls. Compared to the LPA treatment, BrP-LPA decreased chemotaxis by 23%. Analogously, thio-ccPA reduced invasiveness by 30%, relative to ccPA. These results are consistent with previous reports that each of these compounds inhibited ATX [23, 33, 38, 40], which is associated with increased metastatic potential [40].
Figure 4.

Inhibition of migration of NIH 3T3 ras ATX cells by ATX inhibitors and LPA antagonists. Statistical significance is indicated by p < 0.05.
Lipid signaling through phosphatidylinositol 3,4,5-trisphosphate and lysophosphatidic acid (LPA) pathways is aberrant in the majority of cancers. While inhibitors of PI 3-kinase pathway are now being evaluated in human patients, anti-cancer agents that modify LPA receptor signaling and cause regression of tumors or inhibition of metastasis in vivo have not yet been used in the clinic. A comprehensive study of 2-carba and 3-carba ccPA analogues with ATX inhibitory activity demonstrated significant reduction of A2058 melanoma cell invasion in vitro and B16F10 melanoma cell metastasis in vivo [39]. Recently, we found that our pan-antagonist/ATX inhibitory BrP-LPA analogue (as the diastereomeric mixture) reduced metastasis to the lung in normal C57BL/6 mice that were injected with B16F10 murine melanoma cells. The mice were treated twice (Day 3, Day 7) with 10 mg/kg of three LPA antagonist/ATX inhibitors (thio-ccPA, CHF-ccPA, or BrP-LPA). Quantification of the number of lesions on the lungs at Day 21 revealed that both BrP-LPA and thio-ccPA showed statistically reduced lung metastases (M. Murph, Y. Xu, G. Jiang, G. B. Mills, and G. D. Prestwich, unpublished results).
Moreover, we showed that the BrP-LPA diastereomeric mixture reduced cell migration and invasion, and caused regression of orthotopic breast tumors in vivo [44] (Figure 5). In that study, we also synthesized the two separate diastereoisomers of the BrP-LPA. The separate syn and anti diastereomers of BrP-LPA were pan-LPA GPCR antagonists for LPA1-5. Moreover, each diastereomer was a submicromolar inhibitor of the lysoPLD activity of ATX. Computational models correctly predicted the diastereoselectivity of antagonism for three EDG family LPA receptors. The anti isomer was more effective than syn in reducing migration of MDA-MB-231 human breast cancer cells, and the anti isomer was superior in reducing invasion of these cells. Finally, orthotopic engineered tumors [45-47] were established in the mammary fat pads of nude mice by injection of the MB-231 cells in an in situ-crosslinkable extracellular matrix [46]. After two weeks, mice were treated twice per week for two weeks with BrP-LPA alone (10 mg/kg), Taxol alone (10 mg/kg), or Taxol followed by BrP-LPA; tumors from untreated animals served as controls. As shown in Figure 5, BrP-LPA was highly effective in reducing tumor size. In addition, the pan-antagonist/ATX inhibitor BrP-LPA was superior to Taxol in reducing blood vessel density in tumors [44]. Moreover, both the anti and syn diastereomers of BrP-LPA eliminated tumors at 3 mg/kg [44]. Figure 5 also illustrates the reduction of tumor size in a tissue engineering model for liver metastasis of an HCT-116 human colon cancer (G. Yang, unpublished results).
Figure 5.
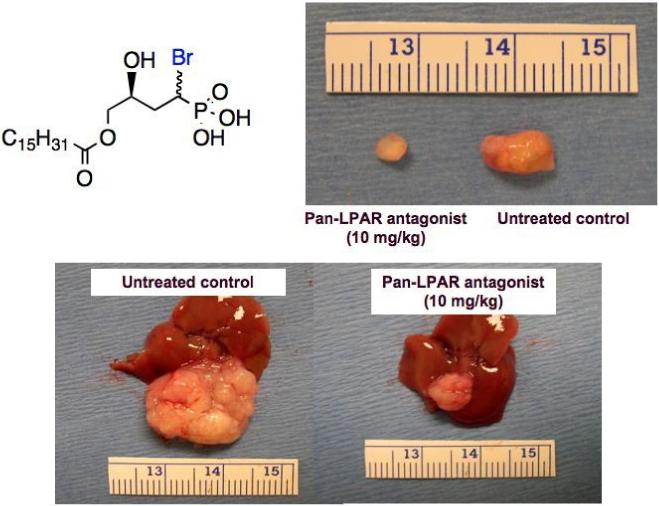
α-Bromomethylene phosphonate LPA analogues cause tumor regression. Above, the structure of the pan-antagonist/ATX inhibitor BrP-LPA and its effect on reducing tumor size for a tissue engineered orthotopic MDA-MB-231 human breast cancer tumor. Below, reduction of tumor size in a tissue engineering model for liver metastasis of a HCT-116 human colon cancer Adapted from Ref. 44.
Aminooxy LPA analogues
We used the Mitsunobu reaction to introduce the aminooxy (AO) functionality in a stereocontrolled manner to complete an efficient synthetic approach to the enantiomerically pure sn-2 AO-LPA analogues [48]. The aminooxy function appears in a toxic amino acid analogue, the leguminous anticancer natural product agent L-canaline [49], and its toxicology is attributed to its high nucleophilicity, particularly in reactions with carbonyl groups. Both the palmitoyl and oleoyl acyl derivatives were prepared. The AO-LPA analogues were evaluated for pharmacological activity against G protein-coupled receptors, inhibition of the lysophospholipase activity of autotaxin (ATX), and the ability to stimulate migration of intestinal epithelial cells in a scratch wound assay.
Figure 6 summarizes the pharmacological properties of the 16:0 and 18:1 AO-LPA analogues on LPA1-4 GPCRs. Both palmitoyl and oleoyl sn-2-AO LPA analogues were agonists for the LPA1, LPA2, and LPA4 receptors, but were antagonists for the LPA3 receptor. The 18:1 analogue was the most potent (382 nM) compound and was a full agonist for LPA2. This same analogue was also the most potent (56 nM) partial antagonist for LPA3. Both AO-LPA analogues inhibited recombinant ATX activity at 10 μM with potencies similar to those of LPA itself. Moreover, the AO-LPA analogues enhanced of migration of IEC-6 human intestinal endothelial cells. This effect is congruent with the agonist activity towards the LPA2 receptor, which is required for protection against radiation injury [50].
Figure 6.

Aminooxy LPA pharmacology and cell biology. Left, structure of AO-LPA and pharmacological results. Right, AO-LPA enhanced migration of intestinal epithelial cells in a scratch wound assay. Adapted from Ref. 48.
Methylenephosphonate LPA analogues
An efficient stereocontrolled synthesis afforded alkoxymethylenephosphonate (MP) analogues of lysophosphatidic acid (LPA) and phosphatidic acid (PA) [51]. Previous efforts had identified the MP analogues as potentially useful phosphatase-resistant ligands in signaling by phosphatidylinositol 3-phosphate [52], phosphatidylinositol 5-phosphate [53], and phosphatidylinositol 3,4,5-trisphosphate [54]. The pharmacological properties of MP-LPA analogues were characterized for LPA receptor subtype-specific agonist and antagonist activity using Ca2+-mobilization assays in RH7777 cells expressing the individual LPA1-LPA3 receptors, CHO cells expressing LPA4, and activation of a PPARγ reporter gene construct expressed in CV-1 cells (Figure 7).
Figure 7.

Pharmacology of oleoyl derivative of MP-LPA. Adapted from Ref. 51.
The MP-LPA analogues exhibited an unanticipated pattern of partial agonist/antagonist activity for the LPA G protein-coupled receptor family and the intracellular LPA receptor PPARγ. Activation of downstream signaling in HT29 colon cancer cells, which exclusively express LPA2, was determined for MP-LPA and 18:1 LPA and compared with activation in SKOV3 and OVCAR3 ovarian cancer cells, which express LPA1, LPA2, and LPA3. Unexpectedly, reverse-phase protein arrays showed that four MP-LPA and MP-PA analogues selectively activated downstream signaling in HT29 cells with greater potency than LPA. In particular, the oleoyl MP-LPA analogue showed a concentration-dependent increase in phosphorylation and activation of AKT, MEK, and pS6 in HT29 cells. In contrast, the four MP-LPA analogues were equipotent with LPA for pathway activation in the SKOV3 and OVCAR3 cells. Thus, the MP analogues appear to selectively activate signaling via the LPA2 receptor subtype, while simultaneously suppressing signaling through the LPA1 and LPA3 subtypes [51].
Summary
The chemical synthesis and SAR of metabolically-stabilized LPA analogues have revealed novel isoform-selective agonists and antagonists for the LPA GPCRs and nuclear receptors. These LPA-selective analogues may well prove to be useful new reagents to characterize cellular and physiological roles of LPA receptor subtypes both in vitro and in vivo. These tools will thus facilitate the unraveling of the fundamental biology of LPA receptor signaling and the development of human therapies based upon targeting these receptors.
Acknowledgements
We thank the National Institutes of Health for grant NS29632 to G.D.P. We also thank our colleagues, G. Tigyi (U Tennessee, Memphis), J. Aoki (U Tokyo), A. Parrill (U Memphis), M. Murph and G. B. Mills (M. D. Anderson Cancer Center) for ongoing collaborative studies on biological activity in literature cited herein, and S. W. Nam (Catholic U, Korea) for providing the NIH 3T3 ras ATX cell line.
Abbreviations used
- LPA
lysophosphatidic acid
- GPCR
G protein coupled receptor
- EDG
endothelial differentiation gene
- PA
phosphatidic acid
- SAR
structure-activity relationship
- ATX
autotaxin
- lysoPLD
lysophospholipase D
- MP-LPA
alkoxymethylene phosphonates
- AO-LPA
aminooxy-LPA
- SAR
structure-activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- [1].Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nature Review. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- [2].Feng L, Mills GB, Prestwich GD. Modulators of lysophosphatidic acid signaling. Expert Opin. Therap. Patents. 2003;13:1619–1634. [Google Scholar]
- [3].Ye X, Hama K, Contos JJ, Anliker B, Inoue A, Skinner MK, Suzuki H, Amano T, Kennedy G, Arai H, Aoki J, Chun J. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature. 2005;435:104–108. doi: 10.1038/nature03505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tokumura A, Kanaya Y, Miyake M, Yamano S, Irahara M, Fukuzawa K. Increased production of bioactive lysophosphatidic acid by serum lysophospholipase D in human pregnancy. Biol. Reprod. 2002;67:1386–1392. doi: 10.1095/biolreprod.102.004051. [DOI] [PubMed] [Google Scholar]
- [5].Bektas M, Payne SG, Liu H, Goparaju S, Milstien S, Spiegel S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J. Cell Biol. 2005;169:801–811. doi: 10.1083/jcb.200407123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mills GB, Eder A, Fang X, Hasegawa Y, Mao M, Lu Y, Tanyi J, Tabassam FH, Wiener J, Lapushin R, Yu S, Parrott JA, Compton T, Tribley W, Fishman D, Stack MS, Gaudette D, Jaffe R, Furui T, Aoki J, Erickson JR. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer. Treat. Res. 2002;107:259–283. doi: 10.1007/978-1-4757-3587-1_12. [DOI] [PubMed] [Google Scholar]
- [7].Fleming IN, Elliott CM, Collard JG, Exton JH. Lysophosphatidic acid induces threonine phosphorylation of Tiam1 in Swiss 3T3 fibroblasts via activation of protein kinase C. J. Biol. Chem. 1997;272:33105–33110. doi: 10.1074/jbc.272.52.33105. [DOI] [PubMed] [Google Scholar]
- [8].Gschwind A, Prenzel N, Ullrich A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002;62:6329–6336. [PubMed] [Google Scholar]
- [9].Gardell SE, Dubin AE, Chun J. Emerging medicinal roles for lysophospholipid signaling. Trends Molec. Med. 2006;12:65–75. doi: 10.1016/j.molmed.2005.12.001. [DOI] [PubMed] [Google Scholar]
- [10].Moolenaar WH. Bioactive lysophospholipids and their G protein-coupled receptors. Exp. Cell Res. 1999;253:230–238. doi: 10.1006/excr.1999.4702. [DOI] [PubMed] [Google Scholar]
- [11].Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- [12].Kortarsky K, Boketoft Å, Bristulf J, Nilsson NE, Norberg Å, Hansson S, Owman C, Sillard R, Leeb-Lundberg LMF, Olde B. Lysophosphatidic Acid Binds to and Activates GPR92, a G Protein-Coupled Receptor Highly Expressed in Gastrointestinal Lymphocytes. J. Pharmacol. Exp. Ther. 2006;318:619–628. doi: 10.1124/jpet.105.098848. [DOI] [PubMed] [Google Scholar]
- [13].Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. GPR92 as a New G12/13- and Gq-coupled Lysophosphatidic Acid Receptor That Increases cAMP, LPA5. J. Biol. Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- [14].Tabata K, Baba K, Shiraishi A, Ito M, Fujita N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem Biophys Res Commun. 2007;363:861–866. doi: 10.1016/j.bbrc.2007.09.063. [DOI] [PubMed] [Google Scholar]
- [15].McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc. Natl. Acad. Sci. USA. 2003;100:131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bandoh K, Aoki J, Hosono H, Kobayashi S, Kobayashi T, Murakami-Murofushi K, Tsujimoto M, Arai H, Inoue K. Molecular cloning and characterization of a novel human G protein-coupled receptor, EDG7, for lysophosphatidic acid. J. Biol. Chem. 1999;274:27776–27785. doi: 10.1074/jbc.274.39.27776. [DOI] [PubMed] [Google Scholar]
- [17].Bandoh K, Aoki J, Taira A, Tsujimoto M, Arai H, Inoue K. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Lett. 2000;478:159–165. doi: 10.1016/s0014-5793(00)01827-5. [DOI] [PubMed] [Google Scholar]
- [18].Virag T, Elrod DB, Liliom K, Sardar VM, Parrill AL, Yokoyama K, Durgam G, Deng W, Miller DD, Tigyi G. Fatty alcohol phosphates are subtype-selective agonists and antagonists of lysophosphatidic acid receptors. Mol. Pharmacol. 2003;63:1032–1042. doi: 10.1124/mol.63.5.1032. [DOI] [PubMed] [Google Scholar]
- [19].Durgam GG, Virag T, Walker M, Tsukahara R, Yasuda S, Liliom K, van Meeteren L, Moolenaar WH, Wilke N, Siess W, Tigyi G, Miller DD. Synthesis, structure-activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARγ, and inhibitors of autotaxin. J. Med. Chem. 2005;48:4919–4930. doi: 10.1021/jm049609r. [DOI] [PubMed] [Google Scholar]
- [20].Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- [21].Nam S, Clair T, Kim Y-S, McMarlin A, Schiffmann E, Liotta L, Stracke M. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 2001;61:6938–6944. [PubMed] [Google Scholar]
- [22].Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, Noji S, Yatomi Y, Aoki J, Arai H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem. 2006;281:25822–25830. doi: 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- [23].van Meeteren LA, Ruurs P, Christodoulou E, Goding JW, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem. 2005;280:21155–21161. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- [24].Ferguson CG, Bigman CS, Richardson RD, Meeteren L.A.v., Moolenaar WH, Prestwich GD. Fluorogenic phospholipid substrate to detect lysophospholipase D/autotaxin activity. Organic Lett. 2006;8:2023–2026. doi: 10.1021/ol060414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tanyi J, Crotzer D, Wolf J, Yu S, Hasegawa Y, Lahad J, Cheng KW, Umezu-Goto M, Prestwich GD, Morris A, Newman RA, Felix EA, Lapis R, Mills GB. In: Functional Lipidomics. Feng L, Prestwich GD, editors. CRC Press/Taylor & Francis; New York: 2006. pp. 101–123. [Google Scholar]
- [26].Prestwich GD, Xu Y, Qian L, Gajewiak J, Jiang G. New metabolically stabilized analogues of lysophosphatidic acid: Agonists, antagonists, and enzyme inhibitors. Biochem. Soc. Trans. 2005;33:1357–1361. doi: 10.1042/BST0331357. [DOI] [PubMed] [Google Scholar]
- [27].Xu Y, Prestwich GD. Synthesis of chiral (α,α-difluoroalkyl)phosphonate analogues of (lyso)phosphatidic acid via hydrolytic kinetic resolution. Organic Lett. 2002;4:4021–4024. doi: 10.1021/ol026684s. [DOI] [PubMed] [Google Scholar]
- [28].Xu Y, Prestwich GD. Concise synthesis of acyl migration-blocked 1,1-difluorinated analogues of lysophosphatidic acid. J. Org. Chem. 2002;67:7158–7161. doi: 10.1021/jo0203037. [DOI] [PubMed] [Google Scholar]
- [29].Xu Y, Qian L, Prestwich GD. Synthesis of a-fluorinated phosphonates from α-fluorovinylphosphonates: a new route to analogues of lysophosphatidic acid. Organic Lett. 2003;5:2267–2270. doi: 10.1021/ol034597+. [DOI] [PubMed] [Google Scholar]
- [30].Xu Y, Qian L, Prestwich GD. Synthesis of monofluorinated analogues of lysophosphatidic acid. J. Org. Chem. 2003;68:5320–5330. doi: 10.1021/jo020729l. [DOI] [PubMed] [Google Scholar]
- [31].Xu Y, Aoki J, Shimizu K, Umezu-Goto M, Hama K, Takanezawa T, Yu S, Mills GB, Arai H, Qian L, Prestwich GD. Structure-activity relationships of fluorinated lysophosphatidic acid analogues. J. Med. Chem. 2005;48:3319–3327. doi: 10.1021/jm049186t. [DOI] [PubMed] [Google Scholar]
- [32].Qian L, Xu Y, Simper T, Jiang G, Aoki J, Umezo-Goto M, Arai H, Yu S, Mills GB, Tsukahara R, Makarova N, Fujiwara Y, Tigyi G, Prestwich GD. Phosphorothioate analogues of alkyl lysophosphatidic acid as LPA3 Receptor-Selective Agonists. Chem. Med. Chem. 2006;1:376–383. doi: 10.1002/cmdc.200500042. [DOI] [PubMed] [Google Scholar]
- [33].Jiang G, Xu Y, Fujiwara Y, Tsukahara T, Tsukahara R, Gajewiak J, Tigyi G, Prestwich GD. α-Substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA. Chem. Med. Chem. 2007;2:679–690. doi: 10.1002/cmdc.200600280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gajewiak J, Prestwich GD. Phosphomimetic sulfonamide and sulfonamidoxy analogues of (lyso)phosphatidic acid. Tetrahedron Lett. 2006;47:7607–7609. [Google Scholar]
- [35].Tigyi G, Parrill AL. Molecular mechanisms of lysophosphatidic acid action. Prog. Lipid. Res. 2003;42:498–526. doi: 10.1016/s0163-7827(03)00035-3. [DOI] [PubMed] [Google Scholar]
- [36].Qian L, Xu Y, Hasegawa Y, Aoki J, Mills GB, Prestwich GD. Enantioselective responses to a phosphorothioate analogue of lysophosphatidic acid with LPA3 receptor-selective agonist activity. J. Med. Chem. 2003;46:5575–5578. doi: 10.1021/jm034207p. [DOI] [PubMed] [Google Scholar]
- [37].Fischer DJ, Liliom K, Guo H, Nusser N, Virag T, Murakami-Murofushi K, Kobayashi S, Erickson JP, Sun G, Miller DD, Tigyi G. Naturally occurring analogs of lysophosphatidic acid elicit different cellular responses through selective activation of multiple receptor subtypes. Mol. Pharmacol. 1998;54:979–988. doi: 10.1124/mol.54.6.979. [DOI] [PubMed] [Google Scholar]
- [38].Mukai M, Imamura F, Ayaki M, Shinkai K, Iwasaki T, Murakami-Murofushi K, Murofushi H, Kobayashi S, Yamamoto T, Nakamura H, Akedo H. Inhibition of tumor invasion and metastasis by a novel lysophosphatidic acid (cyclic LPA) Int. J. Cancer. 1999;81:918–922. doi: 10.1002/(sici)1097-0215(19990611)81:6<918::aid-ijc13>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- [39].Baker D, Fujiwara Y, Pigg KR, Tsukahara R, Kobayashi S, Murofushi H, Uchiyama A, Murakami-Murofushi K, Koh E, Bandle RW, Byun HS, Bittman R, Fan D, Murph M, Mills GB, Tigyi G. Carba analogs of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J. Biol. Chem. 2006;281:22786–22793. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xu Y, Jiang G, Tsukahara R, Fujiwara Y, Tigyi G, Prestwich GD. Phosphonothioate and fluoromethylene phosphonate analogues of cyclic phosphatidic acid: Novel antagonists of lysophosphatidic acid receptors. J. Med. Chem. 2006;49:5309–5315. doi: 10.1021/jm060351+. [DOI] [PubMed] [Google Scholar]
- [41].Zhao C, Fernandez MJ, Prestwich GD, Turgeon M, Battista JD, Clair T, Poubelle PE, Bourgoin SG. Regulation of lysophosphatidic acid receptor expression and function in human synoviocytes: Implications for rheumatoid arthritis? Molec. Pharm. 2008;73:587–600. doi: 10.1124/mol.107.038216. [DOI] [PubMed] [Google Scholar]
- [42].Hama K, Aoki J, Fukaya M, Kishi Y, Sakai T, Suzuki R, Ohta H, Yamori T, Watanabe M, Chun J, Arai H. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J. Biol. Chem. 2004;279:17634–17639. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- [43].Nam S, Clair T, Campo C, Lee H, Liotta L, Stracke M. Autotaxin (ATX), a potent tumor motogen, augments invasive and metastatic potential of ras-transformed cells. Oncogene. 2000;19:241–247. doi: 10.1038/sj.onc.1203263. [DOI] [PubMed] [Google Scholar]
- [44].Zhang H, Xu X, Gajewiak J, Tsukahara R, Fujiwara Y, Liu J, Fells J, Perygin D, Parrill AL, Tigyi G, Prestwich GD. Dual activity lysophosphatidic acid receptor pan-antagonist/lysophospholipase D inhibitor suppresses breast cancer cell migration and invasion in vitro and causes tumor regression in vivo. J. Biol. Chem. 2008 doi: 10.1158/0008-5472.CAN-09-0302. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Prestwich GD. Evaluating drug toxicity and efficacy in three dimensions: using synthetic extracellular matrices in drug discovery. Acc. Chem. Res. 2008;41:139–148. doi: 10.1021/ar7000827. [DOI] [PubMed] [Google Scholar]
- [46].Prestwich GD. Simplifying the extracellular matrix for 3-D cell culture and tissue engineering: A pragmatic approach. J. Cell. Biochem. 2007;101:1370–1383. doi: 10.1002/jcb.21386. [DOI] [PubMed] [Google Scholar]
- [47].Liu Y, Shu XZ, Prestwich GD. Tumor engineering: Orthotopic cancer models in mice using cell-loaded, injectable, cross-linked hyaluronan derived hydrogels. Tissue Eng. 2007;13:1091–1101. doi: 10.1089/ten.2006.0297. [DOI] [PubMed] [Google Scholar]
- [48].Gajewiak J, Tsukahara R, Fujiwara Y, Tigyi G, Prestwich GD. Synthesis, pharmacology and cell biology of sn-2-aminooxy analogues of lysophosphatidic acid. Org. Lett. 2008;10:1111–1114. doi: 10.1021/ol7030747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rosenthal GA. L-Canaline: a potent antimetabolite and anti cancer agent from leguminous plants. Life Sci. 1997;60:1635–1641. doi: 10.1016/s0024-3205(96)00595-4. [DOI] [PubMed] [Google Scholar]
- [50].Deng W, Shuyu E, Tsukahara R, Valentine W, Durgam G, Gududuru V, Balazs L, Manickam B, Arsura M, Vanmiddlesworth L, Johnson L, Parrill A, Miller D, Tigyi G. The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterol. 2007;132:1834–1851. doi: 10.1053/j.gastro.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gajewiak J, Tsukahara R, Tsukahara T, Yu S, Lu Y, Mills GB, Tigyi G, Prestwich GD. Alkoxymethylenephosphonate analogues of (lyso)phosphatidic acid stimulate signaling networks coupled to the LPA2 receptor. Chem. Med. Chem. 2007;2:1789–1798. doi: 10.1002/cmdc.200700111. [DOI] [PubMed] [Google Scholar]
- [52].Gajewiak J, Xu Y, Lee SA, Kutateladze TG, Prestwich GD. Synthesis and molecular recognition of phosphatidylinositol-3-methylenephosphate. Organic Lett. 2006;8:2811–2813. doi: 10.1021/ol060903i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Huang W, Zhang H, Davrazou F, Kutateladze TG, Shi X, Gozani O, Prestwich GD. Stabilized phosphatidylinositol-5-phosphate analogues as ligands for the nuclear protein ING2: Chemistry, biology, and molecular modelling. J. Am. Chem. Soc. 2007;129:6498–6506. doi: 10.1021/ja070195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang H, Markadieu N, Beauwens R, Erneux C, Prestwich GD. Synthesis and biological activity of PTEN-resistant analogues of phosphatidylinositol 3,4,5-trisphosphate. J. Am. Chem. Soc. 2006;128:16464–16465. doi: 10.1021/ja065002j. [DOI] [PMC free article] [PubMed] [Google Scholar]